")
Back to Journals » International Journal of Nanomedicine » Volume 14
CXCR4-targeted liposomal mediated co-delivery of pirfenidone and AMD3100 for the treatment of TGFβ-induced HSC-T6 cells activation
Authors Ullah A , Wang K, Wu P, Oupicky D, Sun M
Received 16 May 2018
Accepted for publication 19 March 2019
Published 26 April 2019 Volume 2019:14 Pages 2927—2944
DOI https://doi.org/10.2147/IJN.S171280
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Aftab Ullah,1 Kaikai Wang,1 Pengkai Wu,1 David Oupicky,1,2 Minjie Sun1
1State Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, Nanjing 210009, People’s Republic of China; 2Center for Drug Delivery and Nanomedicine, Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE, 68198, USA
Background: Liver fibrosis is a chronic liver disease associated with an excessive accumulation of extracellualr matrix (ECM) proteins which ultimately lead to cirrohosis and hepatocellular carcinoma.
Purpose: Liver fibrosis therapies that use combination approaches with the ability to affect multiple disease pathways have proven higher efficacies. This study aimed at optimizing and characterizing the co-encapsulation of pirfenidone (PF) and AMD3100 (AMD) into CXCR4-targeted combination liposomes (CTC liposome) for CXCR4 targeting, and the inhibition of major molecular culprits ie α-SMA, CXCR4, TGFβ, and P-p38 involved in liver fibrosis in-vitro.
Methods: The CTC liposomes were prepared using the thin-film hydration method. The concentration of encapsulated AMD and PF was measured by HPLC and UV spectrophotometry, respectively. Tramsmission electron microscopy (TEM) was used to determine the liposomal morphology. The CXCR4 targeting ability was determined by CXCR4 redistribution assay. Confocal microscopy and flowcytometry were used to determine the CXCR4 mediated cell uptake. The apoptosis inducing and protein downreguating ability of CTC liposomes were determined by apoptosis assay and western blot analysis, respectively. In-vivo biodistribution and Hoechst staining were used to confirm the feasibility of CTC liposome for the in-vivo applications and drug targeted accumulation, respectively.
Results: The TEM studies revealed that CTC liposomes were spherical in shape. The cumulative release of AMD and PF from CTC liposome was 67% and 84%, respectively, at 48 h. Compared to the free drug counterparts, encapsulated drugs displayed higher cell viability. The CXCR4 redistribution assay confirmed the CXCR4 targeting and antagonistic ability of CTC liposomes. The CTC liposomes were internalized more effectively via caveolae-mediated endocytic pathways. CTC liposomes displayed aggressive apoptosis (87.3%) in TGFβ-induced activated HSC-T6 cells suggesting a propensity to fibrosis regression. Also, CTC liposomes significantly reduced α-SMA (65%), CXCR4 (77%), TGFβ (89%), and P-p38 (66%) expressions, better than free drugs. CTC@IR780 liposomes (CTC liposomes incorporating IR780 dye) were more accumulated in fibrotic livers compared to free IR780, as judged by in-vivo imaging, biodistribution analysis, and Hoechst staining. These findings suggest that this simple and stable CTC liposomal system holds a great promise for the treatment and prevention of liver fibrosis.
Keywords: AMD3100, pirfenidone, liposome, liver fibrosis
Introduction
Liver fibrosis is a scarring process associated with excessive accumulation of extracellular matrix (ECM) proteins.1 It is caused by autoimmune diseases, autoimmune hepatitis, viral hepatitis (eg, hepatitis B virus, hepatitis C virus), nonalcoholic steatohepatitis and alcohol-induced liver injury.2 Liver fibrosis ultimately leads to cirrhosis, and hepatocellular carcinomas (3–5% worldwide).2–4 Liver fibrosis is a leading cause of global premature morbidities and mortalities.2 Great progress has been made in understanding the cellular mechanisms of liver fibrosis but activation of hepatic stellate cells (HSC) is the main event, and HSC are considered as primary therapeutic targets.5,6 During liver injury of any etiology, HSCs are transformed to myofibroblasts with ECM overproduction.
The existing treatment strategies to treat liver fibrosis include caffeine, ursodeoxycholic acid (UDCA), chenodeoxycholic (CDCA), pioglitazone, vitamin E, and silymarin7 Efforts have been made in recent years to establish upcoming treatment strategies like gene therapies using nanoparticles, viral vectors, non viral vectors and ligand modified liposomes for liver fibrosis.8–13 However, for clinical applications of gene therapies, there are still many barriers like immunogenicity and toxicity vectors, and their interaction with the biological materials.14 Currently, among the latest treatment strategies for the treatment of liver fibrosis, suppression of the fibrogenesis by blocking certain receptors/signaling pathways in HSC could help in the fibrosis resolution.5 Numerous new anti-fibrotic drugs eg, colchicine, angiotensin-converting enzyme inhibitors (ACEs) and PPAR-δ agonists blocking inflammatory pathways, and inhibiting profibrogenic cytokines. These drugs have proven greater impact in pre-clinical studies.15–19 However, they were failed in inhibiting disease progression in clinical practice.20–23 Pirfenidone (5-methyl-1-phenylpyridin-2[H-1]-one), is a new an antifibrotic drug approved by the US FDA (United States Food and Drug Administration) for IPF (idiopathic pulmonary fibrosis).24 PF was relatively well tolerated in phase III clinical trials.25–27 PF has been used in animal models for treating chronic hepatitis C, hypertrophic cardiomyopathy and fibrotic renal disorders.28–30 PF can be administered intratracheally, orally and intravenously as microspheres, aerosols, and liposomes respectively.31–33 Clinically, PF has been used for the treatment of lung fibrosis.34,35 It is orally administered causing sever GIT (gastrointestinal tract) side effects.36 Also, using PF alone had no targeting tendency.37 Taken all together, we need a new drug delivery system to decrease the side effects and improve its targeting ability. So, we developed CTC liposomes for targetted delivery of PF to CXCR4 overexpressed cells.
Phosphorylated-p38 mitogen-activated protein kinase (P-p38) activates HSC and results in fibrosis.38 Inhibition of P-p38 may prevent activation of HSC.39 PF down-regulated P-p38 level in renal fibrosis, decreasing collagen and α-SMA expressions.40,41 Transforming growth factor β (TGFβ) is one of the most powerful and widely distributed profibrogenic mediators and a dominant stimulator of ECM overproduction.42,43 PF significantly reduced TGFβ expression in TGFβ-induced activated HSC and renal fibrosis.38,44
The upregulated stromal derived growth factor alpha (SDF-1α) /CXCR4 axis causes HSC activation.45,46 Plerixafor (AMD3100) is a bicyclam small molecule CXCR4 antagonist approved by US FDA and European Medicines Agency (EMA) clinically for use in hemopoietic stem cells mobilization for autologous transplant for patients with lymphoma and multiple myeloma (MM).47 Previously, AMD inhibited binding of SDF-1α with CXCR4 on hemopoietic stem cells and ameliorated liver fibrosis.48–50 Similarly, inhibition of SDF-1α/CXCR4 axis in activated HSC may be a potential treatment option for liver fibrosis.45,51,52 Previously, AMD has been used in pulmonary fibrosis.53 AMD can be administered by intravenous, subcutaneous, and intratracheal routes of administration.53–55 Different drug delivery systems like polymeric nanoparticles, liposomes, and NLCS are used for AMD delivery to treat various diseases.56–58 In this study, an AMD3100 equipped CTC liposomal system was developed which helps to localize PF and AMD for joint pharmacodynamics effect.
Targeted co-delivery of drugs to show the combination effects is a major issue, and liposomes may be an effective tool.59–62 Liposomes are suitable nanocarriers for multiple drug deliveries .63,64 Combination therapy could greatly improve the outcomes of patients with liver fibrosis.65,66
AMD functionalized nano-carriers accumulates within CXCR4 receptor-positive cells.67 Herein, CTC liposomal system equipped with the AMD was developed and evaluated for its CXCR4 antagonistic ability, and its antifibrotic effects (Scheme 1). Taken together, the CTC liposomal system successfully targeted CXCR4, and overcome the upregulated SDF-1α/CXCR4 axis, α-SMA, P-p38, and TGFβ expressions.
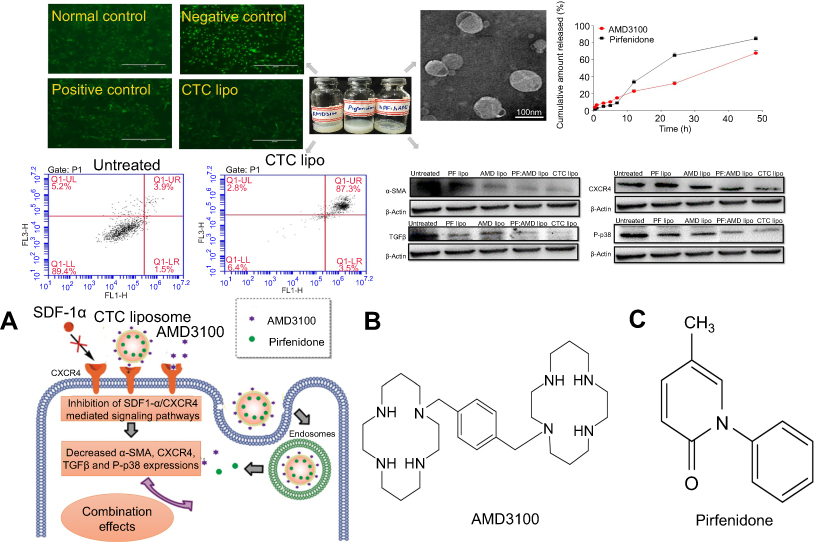 | Scheme 1 CXCR4 antagonistic activity of CTC liposome and the joint pharmacodynamic effects of PF and AMD (A) Mechanism of CXCR4 antagonism and the combined effects of PF and AMD (B) Chemical structure of AMD (C) Chemical structure of PF. |
Materials and methods
Materials
AMD3100 was purchased from Shanghai Biochempartner Co., Ltd. PF (>98%), acetonitrile, hydrochloric acid, tetra butyl ammonium hydrogen sulfate (TBAHS), high purity cholesterol, and egg phosphatidylcholine (PC) were obtained from Aladdin Industrial Corporation (China). Methanol was purchased from CINC High Purity Solvents Co. (Shanghai, China). Human SDF-1α was from Shenandoah Biotechnology, Inc. (Warwick, PA). G418 reagent was bought from Life Technologies (USA). Dulbecco’s modified Eagle medium (DMEM), penicillin/streptomycin (Pen-Strep), and fetal bovine serum (FBS) were purchased from Hyclone (Waltham, MA). Phosphate buffer solution (PBS), Trypsin EDTA solution and Annexin V-FITC/PI apoptosis detection kit were from KeyGen bio-tech (Nanjing, China). IR780 was purchased from Sigma Aldrich (Saint Louis, Missouri, USA). TGFβ, α-SMA, CXCR4, and P-p38 antibodies were obtained from Abcam (Cambridge, MA). Ethanol, sodium hydroxide, and sodium chloride were all analytical grade and were purchased from Shanghai Ling Feng Chemical Reagent Co., Ltd (China). Water was purified by deionization, distillation, and reverse osmosis (Milli-Q plus). U2OS cells stably expressing functional EGFP-CXCR4 fusion protein were purchased from Fisher Scientific and provided by Dr. David Oupicky (University of Nebraska medical center, Omaha Nebraska, USA). Immortalized rat HSC-T6 cell line was purchased from Shanghai FuMeng Gene Biotechnology Co., LTD. (Shanghai, China).
Methods
Preparation of liposomes
Liposomes were prepared by thin-film hydration method combined with probe sonication.33,68,69 Briefly, PC, cholesterol, and AMD were dissolved in dichloromethane and ethanol (1/4, v/v). Coumarin-6 (C6) was dissolved in the lipid part to obtain coumarin 6-encapsulated-AMD liposomes (C6-AMD liposomes), and coumarin 6-encapsulated liposomes (C6-liposomes). The PC, cholesterol, and drug mass ratio were 60:7.5:1.5.33,70 The solvent was evaporated under vacuum at 37 °C for 40 mins. The dried lipid thin film was hydrated with PBS containing PF at 37 °C for 1h, and ultra-sonicated at 100W for 600s in an ice bath. Unincorporated PF and AMD were removed by centrifugation at 12,000 rpm at 4 °C for 30 min using ultrafiltration tubes (50kDa) (Millipore, USA).33
AMD (0.1 mg) was adsorbed at the surface of liposomes. The mixture was vortexed to obtain CTC liposomes through the electrostatic interactions between positively charged AMD and negatively charged liposomes.67 The following different liposomal formulations were prepared. PF liposome (PF lipo; PF=5 mg/mL), AMD liposome (AMD lipo; AMD=5 mg/mL), PF:AMD liposome (PF:AMD lipo; PF=2.5 mg/mL, AMD=2.5 mg/mL), and CTC liposomes (PF=5 mg /mL, AMD=5 mg /mL). Free drug solutions were prepared by dissolving AMD and PF in PBS (pH 7.4). PF (Free PF=5 mg/mL), AMD (AMD=5 mg/mL), PF:AMD (PF=2.5 mg/mL, AMD=2.5 mg/mL), and hPF:hAMD (PF=5 mg/mL, AMD=5 mg/mL). Free drug solutions and liposome were filtered using pyrogen-free 0.22 μM membrane filter (Millipore, USA).
Particle size and zeta potential measurement
Particle size and zeta potential of liposomes were determined by using Brookhaven Instruments-Zeta Plus (Brookhaven, USA). Briefly, liposomes were diluted with distilled water and shaken thoroughly at 25 °C before measurement.
Transmission electron microscopy (TEM)
The liposomal morphology was visualized by using TEM (TEM, H-600, Hitachi, Japan). A drop of the water-diluted liposomal suspension was placed onto a 200 mesh carbon coated copper grid forming a thin liquid film which was air-dried before observing at room temperature.
Drug loading and encapsulation efficiency
The drug loading (DL) and encapsulation efficiency (EE) of PF and AMD were determined by HPLC (Waters e2695, USA) with an attached UV detector, and UV- spectrophotometer (Agilent Technologies, USA) at wavelengths 315 and 215 nm, respectively. A mixture of TBAHS (10 mM, pH 3.37) and acetonitrile (58:42, v/v) was used as a mobile phase with a flow rate of 1 mL/min (diamond ODS C18 column, 150×4.6 mm, and 5 µm) for HPLC . The concentration of PF and AMD was determined from their respective standard curves. The DL, EE of AMD and PF was calculated using the following equations:
(A.1) 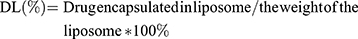
(A.2) 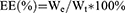
Where We were the analyzed amount of encapsulated AMD or PF; Wt was the analyzed amount of total AMD or PF.
In-vitro drug release of CTC liposomes
The dialysis bag method was employed to evaluate the in-vitro release of drugs from CTC liposomes. Briefly, 2.5 ml of liposome suspension was transferred into a clamped dialysis bag (MWC 3500). The bag was immersed in a beaker containing 100 mL of PBS (pH=7.4) guided with mechanical shaking (250 rpm) at 37±0.5℃. At specified time intervals, 1 ml of the dissolution medium was withdrawn and replaced with the same volume of pre-warmed fresh PBS immediately. The collected samples were then centrifuged at 12,000 rpm for 15 min and the supernatant layer was analyzed for drug detection.
In-vitro stability evaluation of CTC liposomes
The CTC liposomes were resuspended in isotonic PBS and FBS at 1:9 at 37 °C. The particle size was determined by a Brookhaven Instruments-Zeta Plus at certain intervals of time. While for storage stability, the change in particle size of liposomes was monitored at 4 °C for 15 days.
Animals and cell lines
Mice (seven weeks old, 20–25 g) were supplied by the experimental animal center of Yangzhou University (Yangzhou, China). The animals used in this study were treated according to the standard protocols evaluated and approved by the ethical committee of China Pharmaceutical University.
Immortalized rat hepatic stellate cell (HSC-T6 cells) were cultured in DMEM supplemented with 10% FBS and 1% PS at 37 °C in a 5% CO2 atmosphere. U2OS cells stably expressing functional EGFP-CXCR4 fusion protein were cultured in DMEM supplemented with 2 mM L-Glutamine, 10% FBS, 1% Pen-Strep and 0.5 mg/mL G418. The cells were maintained at 37°C in an incubator supplemented with 5% CO2.
Examination of cell viability
Methyl thiazolyl tetrazolium (MTT) assay was performed to determine the cell viability of encapsulated drugs and free drugs. HSC-T6 cells were seeded (1x104 cells/well) in 96-well plate for 24 h. The old medium was replaced with fresh medium containing liposomes and free drugs. After 24 h incubation at 37 °C, cells were washed twice with PBS. DMEM contained MTT was added to each well of the culture plate and followed by an additional 4 h incubation at 37 °C. After dissolving the dark blue formazan crystals in DMSO, the plates were read on a microtiter plate reader, using Synergy-2 multifunctional microplate reader (BioTek, USA) at a wavelength of 490 nm.71 The following equation was used for the calculation of cell viability.
(A.3) 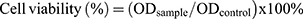
Where OD sample was the absorbance intensity of cells against various treatments and OD control was the absorbance intensity of the normal control cells.
CXCR4 antagonism
The CXCR4 antagonistic ability of CTC liposomes was determined using CXCR4 redistribution assay. U2OS cells expressing EGFP-CXCR4 receptors were seeded (8.0x103 cells/well) in a 96-well plate for 24 h at 37 °C. The cells were washed twice with 100 µL assay buffer (DMEM supplemented with 2 mM L-Glutamine, 1% FBS, 1% Pen-Strep and 10 mM HEPES). The cells were incubated with free AMD, CTC liposome and SDF-1α at 37 °C for 30 min. Cells treated with SDF-1α alone were used as the negative control. AMD (300 nM) was used as the positive control. CTC liposome (AMD final concentration 300nM) was used as a test sample for evaluating its CXCR4 targeting/antagonistic ability. Then, SDF-1α was then added to each well (final concentration of 10 nM). After 1h incubation at 37 °C, cells were fixed with 4% formaldehyde at room temperature for 20 min and washed 4 times with PBS, and the nuclei were stained with 1 μM Hoechst 33258 for 30 mins. Cells were imaged using EVOS FL microscope at 20x.
Cellular uptake and internalization mechanisms
The cellular uptake and internalization mechanisms of CTC liposomes were investigated by combining flow cytometry and confocal laser scanning microscopy. To verify the cell uptake and internalization mechanisms, a hydrophobic fluorescent probe, C6 was loaded in liposomes for the formation of C6-liposomes and C6-AMD liposomes72,73
For flow cytometry analyses, HSC-T6 cells were seeded (5x104 cells/well) into 24-well plates at 37 °C. After the cells reached 80–90% confluence, the old culture medium was replaced with a fresh medium containing C6-solution, C6-liposomes, and C6-AMD liposomes. After 2 h incubation, cellular uptake was stopped immediately with ice-cold PBS. Finally, the cells were collected by trypsinization, and fluorescence intensity for C6 in the cells was measured using flow cytometer. While for fluorescence microscopy, experiments were performed as described above, and HSC-T6 cells were washed with PBS thrice and fixed with 4% paraformaldehyde for 10 min. After the cell nuclei were stained with 4‘,6-diamidino-2-phenylindole (DAPI) for 15 min, the cellular uptake was visualized with confocal laser scanning microscope (Zeiss LSM 700, Oberkochen, Germany).72,73 The cellular uptake mechanism of C6-AMD liposomes was investigated using C6-liposomes as a control.
For the internalization mechanism, HSC-T6 cells were first pre-treated with the endocytic inhibitors at 37℃ for 30 min.74 Chlorpromazine (10 μg/mL) as a clathrin-mediated endocytosis inhibitor, nystatin (30 μg/mL) as a caveolae-mediated endocytosis inhibitor, NaNO3 (50 μg/mL) as an energy metabolism inhibitor, cyclodextrin (300 μg/mL) as clathrin caveolae-mediated endocytosis inhibitor and amiloride (13 μg/mL) as a macropinocytosis inhibitor were used as endocytic inhibitors.75 Cells were incubated with liposomes in the presence of these endocytic inhibitors for 2 h. The fluorescence intensity was measured by flow cytometry and the relative uptake index (RUI) was calculated using the following equation:
(A.4) 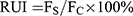
Where FS is the fluorescence intensity of liposomes treated with different kinds of endocytic inhibitors, and FC is the fluorescence intensity of liposomes without inhibitor treatment.
To investigate the cell uptake of CTC liposomes mediated by CXCR4 targeting, HSC-T6 cells were pre-incubated with different concentrations of free AMD for 1h before incubating with C6-AMD-liposomes and C6-liposomes. After 2 h incubation, cells were washed with cold PBS thrice and were collected by trypsinization. The fluorescence intensity was measured using a flow cytometer. The relative fluorescence intensity of C6-AMD liposomes was obtained by comparing to C6-liposomes.
Flow cytometric analysis of apoptosis
The apoptosis-inducing effects of CTC liposomes in activated HSC-T6 cells was determined using an Annexin V-FITC/PI apoptosis detection kit. HSC-T6 cells were seeded (0.5x105 cells/well) into 12-well plates at 37 °C for 24 h. Then, cells were treated with a series of liposomes and free drugs. Cells treated with liposomes were randomly divided into six groups as: (1) Normal control, (2) Untreated, (3) PF lipo (PF=125μg/mL), (4) AMD lipo (AMD=62.5 μg/mL), (5) PF:AMD lipo (PF=31.25 μg/mL, AMD=15.62 μg/mL), and (6) CTC lipo (PF=62.5 μg/mL, AMD=31.25 μg/mL). Also, cells treated with free drugs were divided into six groups as: (1) Normal control, (2) untreated, (3) PF (PF=125μg/mL), (4) AMD (AMD=62.5 μg/mL), (5) PF:AMD (PF=31.25 μg/mL, AMD=15.62 μg/mL), and (6) hPF:hAMD (PF=62.5 μg/mL, AMD=31.25 μg/mL). After 24 h, medium containing TGFβ 10 ng/mL was added to each well except the normal control then cells were washed with cold PBS followed by double-staining with Annexin V-FITC/PI. The apoptosis was analyzed in different experimental groups by flow cytometer (FACS-Calibur, BD Biosciences).
CTC liposomal inhibition of TGFβ-induced activated HSC-T6 cells
HSC-T6 cells were seeded (5x105 cells/well) in 12-well plates. After 80–90% confluence, cells were incubated with free drugs and liposomes. Cells treated with liposomes were randomly divided into five groups as: (1) Untreated, (2) PF lipo (PF=125μg/mL), (3) AMD lipo (AMD=62.5 μg/mL), (4) PF:AMD lipo (PF=31.25 μg/mL, AMD=15.62 μg/mL), and (5) CTC lipo (PF=62.5 μg/mL, AMD=31.25 μg/mL). Also, cells treated with free drugs were divided into five groups as: (1) Untreated, (2) PF (PF=125μg/mL), (3) AMD (AMD=62.5 μg/mL), (4) PF:AMD (PF=31.25 μg/mL, AMD=15.62 μg/mL), and (5) hPF:hAMD (PF=62.5 μg/mL, AMD=31.25 μg/mL). After 24 h, cells were then exposed to TGFβ 2ng/mL for 6 h. Media was then aspirated and cells were washed with PBS. The cells were homogenized using ice-cold RIPA buffer (Beyotime, China) and the whole cells lysate was analyzed for protein expressions using western blot.
Western blot analysis
The whole cell lysates were centrifuged at 12,000 rpm at 4 °C for 15 mins, and the supernatant was collected for protein expression using western blot analysis. 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins. The separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, USA). The membranes were blocked in 5% skimmed milk and then probed with the primary antibodies at 4 °C with gentle shaking, overnight. After 5 times washing with TBS, secondary HRP-conjugated antibody was incubated for 1 h at room temperature. After washing as before, a positive signal was detected using Beyo ECL Plus (Beyotime, China) with an enhanced chemiluminescence system (Tanon, 5200 Multi, China).
In-vivo imaging and biodistribution
The liposomes were systemically administered into healthy and fibrotic mice for the in-vivo imaging and biodistribution analysis to confirm the feasibility of using CTC liposomes for in-vivo applications. Briefly, IR780@CTC liposome and free IR780 were i.v. injected. Images were taken at 0.25, 1, 6, 12, and 24 h after injection using an in-vivo imaging system (IVIS Lumina imaging system, USA). Also, for targeted drug accumulation in the fibrotic liver, mice were divided into free IR780 (0.3 mg/kg), IR780@liposomes (0.3 mg/kg IR780 equivalent), and IR780@CTC liposome (0.3 mg/kg IR780 equivalent), respectively. To verify the IR780 accumulation in livers, mice were sacrificed by cervical dislocation at 24 h after i.v. injection and the livers were harvested and washed in PBS. IR780 accumulation and retention in livers were imaged and analyzed using Carestream Molecular Imaging Software V 5.3.5. The excitation and emission wavelengths for IR780 were fixed at 720 nm and 790 nm, respectively. In addition, the livers were sectioned, stained with Hoechst staining for IR780@CTC liposome accumulation due to possible CXCR overexpression.
Statistical analysis
Graph Pad Prism (GraphPad Software, SanDiego, CA) was used for statistical analysis using two-tailed Student’s t-test and one-way ANOVA. A p-value of <0.05 was considered statistically significant. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001
Results
Physicochemical characterization of CTC liposomes
Liposomes are relatively safe nanocarriers.,76 The CTC liposomes had a spherical shape and uniform particle size of 100 nm (Figure 1A). The mean diameter was 75 nm and had a narrow particle size distribution from about 75~125 nm (Figure 1B). The Zeta potential of CTC liposomes was measured as −18.88 mV. The coating efficiency of AMD was proportional to the alteration on the surface charge. Briefly, AMD was added (0-100 µg/mL) to the blank liposomes and the recorded zeta potential was −20 mV, suggesting the successful adsorption of AMD on the surface of liposomes (Figure 1C). The strong negative zeta potential demonstrates electrostatic repulsion between particles, thus reducing particle aggregation, and hence promoting stability.77 CTC liposomes had a stable particle size over 15 days at 4 °C reflecting the storage stability (Figure 1D). Only a slight change in particle size of CTC liposomes was observed (from 85 to 113 nm over 96 hrs) in PBS and FBS confirming its suitability for in-vivo applications (Figure 1E). The cumulative release of AMD and PF was 67% and 84%, respectively, at 48 h showing stable entrapment of drugs in a simulated physiological environment (Figure 1F). The EE% of PF and AMD in CTC lipo were 81% and 54%, respectively. The DL% of PF and AMD was 6.4% and 3.5%, respectively, in the CTC liposomal system (Table 1).
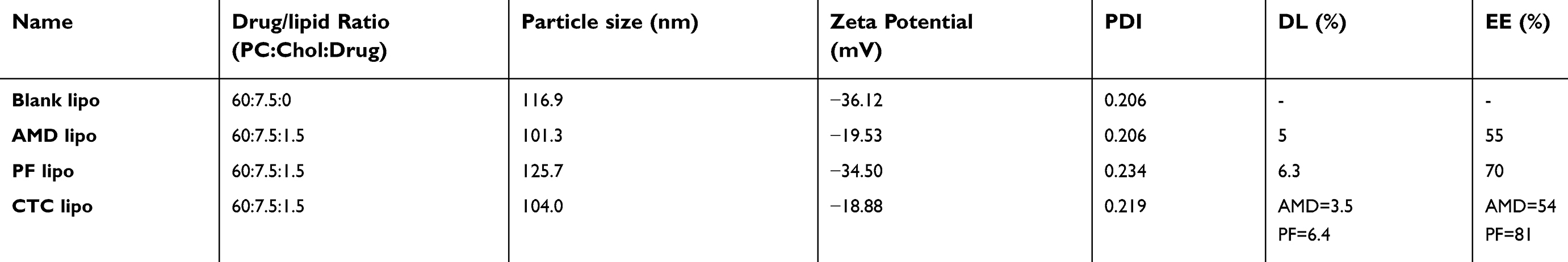 | Table 1 Characteristics of different formulations |
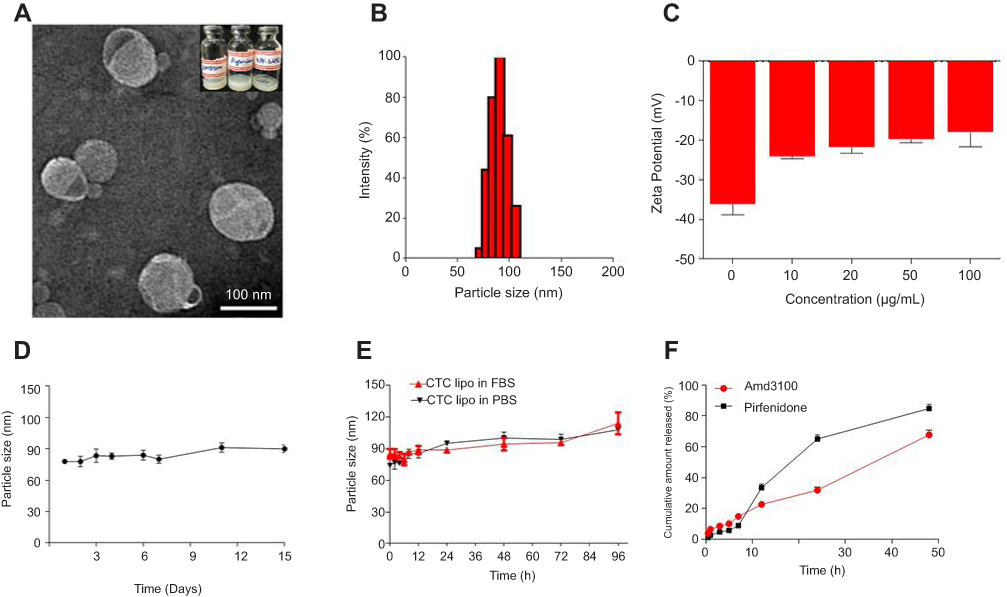 | Figure 1 Physicochemical characterization of CTC liposomes (A) TEM image of CTC liposome (B) Size distribution histogram of CTC liposome (C) Zeta potential of blank liposomes and effect of different concentrations of AMD (D) Storage stability of CTC liposomes (E) PBS and FBS stability of CTC liposomes (F) Cumulative release of AMD and PF. |
Effect of liposomes on cell viability
Compared with free drugs, liposome-encapsulated drugs improved cell viability. Thus, liposomes reduced the drug toxicity and increased drug safety. These findings display that liposomes can be used as a safe vehicle for drug delivery (Figure 2A–C).
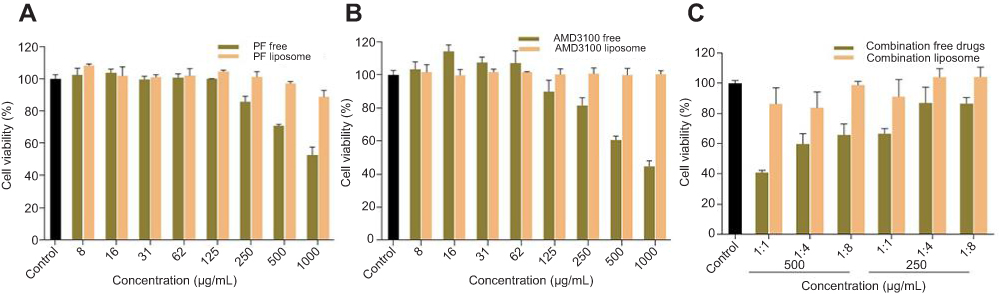 | Figure 2 Cell viability of liposome encapsulated drugs and free drugs (A) PF free and PF liposome (B) AMD free and AMD liposome (C) Combination free drugs and combination liposome. |
CXCR4 antagonism
Next, we investigated the CXCR4 antagonistic ability of the CTC liposomes using a phenotypic CXCR4 receptor redistribution assay.78 This assay can be applied to track and visualize the translocation of EGFP-tagged CXCR4 receptors on the cell membrane to endosomes upon ligand (SDF-1α) stimulation, which is typical behavior for G-protein coupled receptors (GPCRs). Normal control did not show any intracellular change (Figure 3A). As shown in Figure 3B, negative control cells exhibited CXCR4 translocation as indicated by the enhanced green fluorescent signals inside the cells. Positive control cells exhibited a diffused pattern of green fluorescence, indicating the inhibition of CXCR4 translocation after SDF-1α stimulation (Figure 3C). CTC liposomes (Test sample) displayed strong CXCR4 inhibition when compared with positive and negative controls (Figure 3D). Our results confirmed that CTC liposomes function as CXCR4 antagonists. So, the CTC liposomes can be used as an appropriate delivery system for applications aimed for CXCR4 targeted delivery.
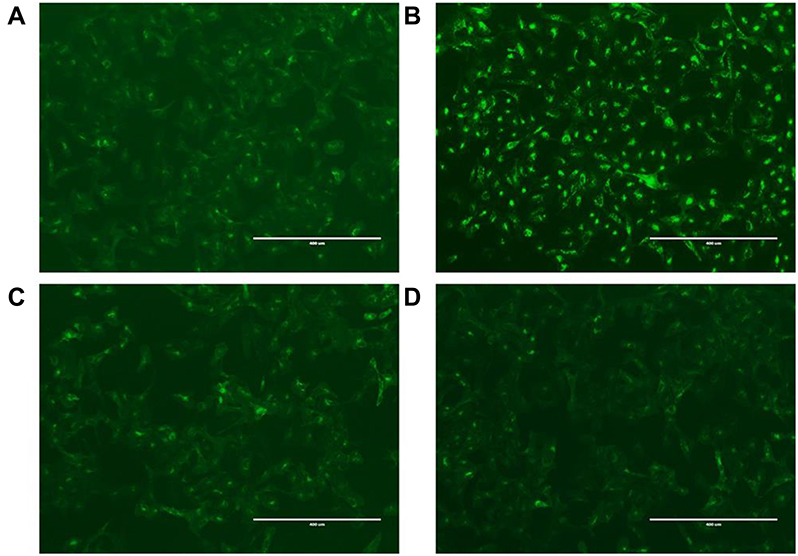 | Figure 3 CXCR4 antagonism (A) Normal control; U2OS cells were treated with PBS (B) Negative control; U2OS cells treated with SDF-1α only. (C) Positive control; U2OS cells treated with SDF-1α and free AMD (D) Test sample; U2OS cells were treated with SDF-1α and CTC liposomes. |
Cellular uptake and internalization mechanisms
From the fluorescence imaging results, the C6-AMD liposomes were internalized more efficiently than C6-liposomes and C6 solution (Figure 4A). Furthermore, the uptake of C6-AMD liposomes was inhibited significantly in the presence of increasing concentrations of free AMD probably due to its CXCR4 antagonistic effect (Figure 4B). Thus, AMD pre-treatment reduced the affinity of the cells for C6-AMD liposomes. While AMD pre-treatment had no significant effects on the uptake of C6-liposomes. The Fluorescence imaging results (Figure 4A) also displayed more uptake of C6-AMD liposomes than C6-liposomes, which is consistent with the flow cytometry results (Figure 4B). These findings demonstrate that adding AMD to the liposomes prompted the uptake of the nanoparticles, displaying that AMD plays a critical role in the uptake.
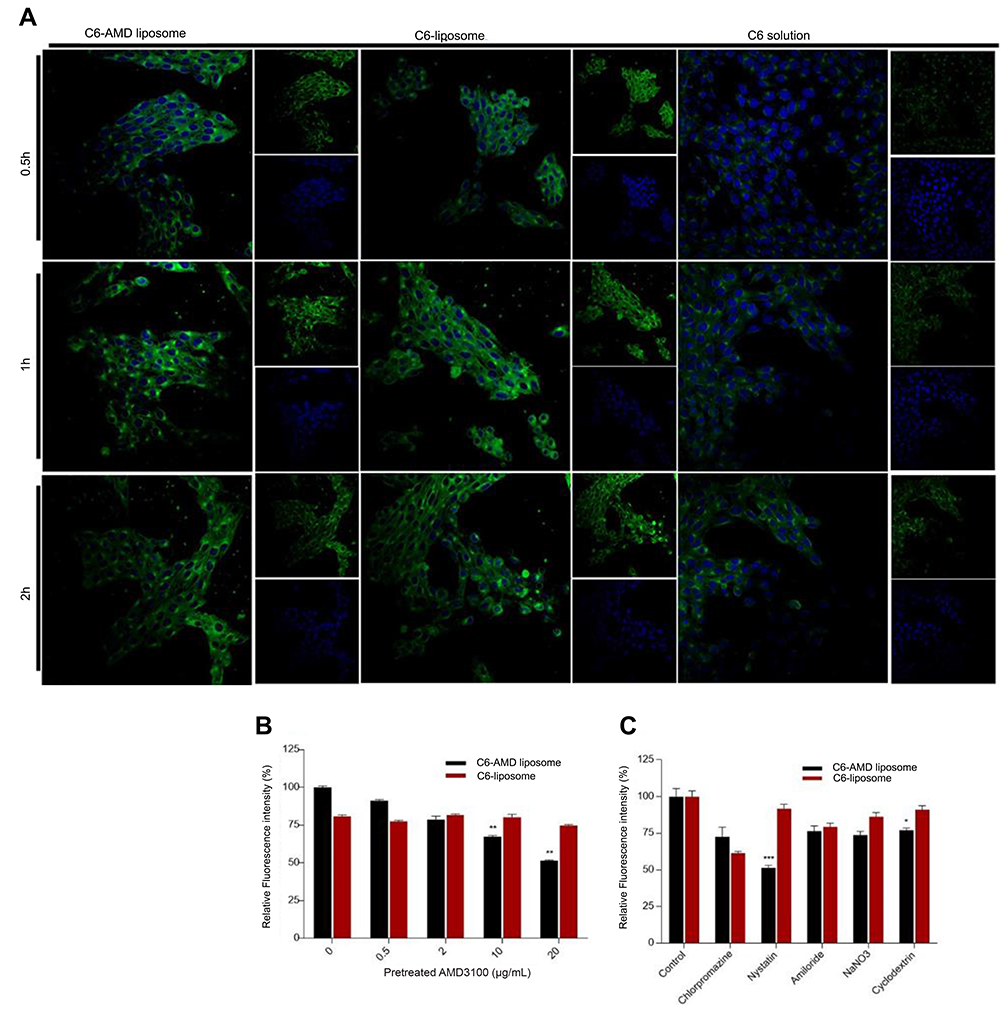 | Figure 4 (A) Cellular uptake of C6-AMD-liposomes by HSC-T6 cells at a different time (0.5, 1, 2 h). The images were taken under 40x (magnification) using a confocal laser scanning microscope. Blue signal: DAPI; Green signal: C6 (B) The influence of AMD pre-treatment on cellular uptake of liposomes (C) Analysis of the endocytosis pathway of C6-AMD liposomes determined by flow cytometry and cells were treated with different endocytosis inhibitors. Student’s t-test was used to estimate the statistical significance of the differences in different groups. A two-sided “p” value less than 0.05 indicated statistical significance. *p<0.05, **p<0.01. |
The understanding of specific endocytosis pathways is necessary to elucidate the intracellular trafficking of C6 liposomes and C6-AMD liposomes. The internalization mechanisms were evaluated by using several endocytosis pathways inhibitors. HSC-T6 cells were treated with different endocytosis inhibitors like nystatin, chlorpromazine, amiloride, NaNO3, and cyclodextrin reduced the cellular uptake of both C6-liposomes and C6-AMD liposomes. However, nystatin (p=0.0003) and cyclodextrin (p=0.028) reduced the cellular uptake significantly. The cellular uptake of C6-AMD liposomes was markedly decreased compared to C6-liposomes in the presence of nystatin (p=0.0003), a caveolae-mediated endocytosis inhibitor. The results indicate that adding AMD to liposomes provoked the uptake of the nanoparticles by the route of caveolin-mediated endocytosis. The results also show that liposomes entered the cells by multiple pathways. However, the nystatin has a significant effect in decreasing the internalization of liposomes, suggesting that caveolae-mediated-endocytosis pathway is mainly involved in the liposome internalization.
Effect of CTC liposomes on apoptosis
Next, we investigated the effects of CTC liposomes on inducing apoptosis in TGFβ-induced activated HSC-T6 cells. The results in Figure 5A show that, in liposomal treatment, PF lipo, AMD lipo, and PF:AMD lipo significantly increased the proportion of activated HSC-T6 cells undergoing late apoptosis by 63.7%, 66.6%, and 75.5%, respectively, as compared with free drug solutions (Figure 5B). However, CTC liposomes exhibited superior apoptosis (87.3%) inducing ability.
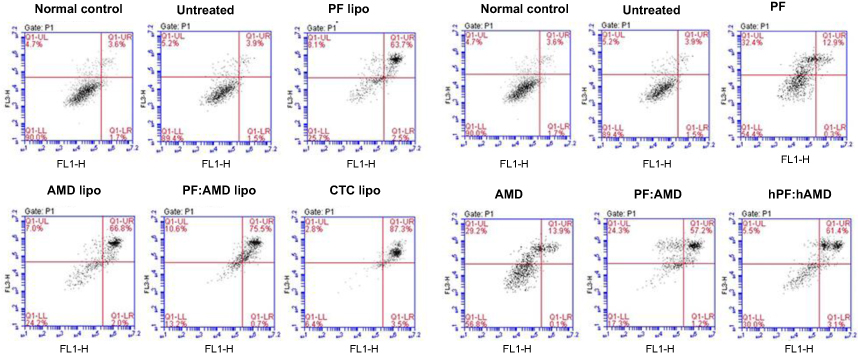 | Figure 5 Effect of encapsulated and free drugs on apoptosis (A) Effect of liposomes on apoptosis in activated HSC-T6 cells (B) Effect of free drugs on apoptosis in activated HSC-T6 cells. |
Inhibition of α-sma expression
The up-regulated SDF-1α/CXCR4 axis activates HSC.45,46 AMD blocks SDF-1α /CXCR4 axis, thus preventing HSC activation.48,50,79 PF inhibits P-p38 leading to inhibited expression of α-SMA.38,44 In this study, we evaluated the effects of CTC liposomes on the inhibition of α-SMA expression. Compared to free drugs (Figure 7A), liposomal treatment indicated a remarkable decrease in α-SMA expression (Figure 6A). Compared to free drugs, HSC-T6 cells treated with PF lipo, AMD lipo, PF:AMD lipo, and CTC lipo significantly reduced expression of α-SMA by 31%, 45%, 53%, and 65%, respectively (Figures 6E and 7E). Comparatively, the CTC lipo showed significant α-SMA downregulation, displaying a maximum decrease of HSC-T6 cells activation (Figure 6A and E). Free drugs did not show significant results except PF:AMD reducing 31% of the elevated α-SMA expression (Figure 7E).
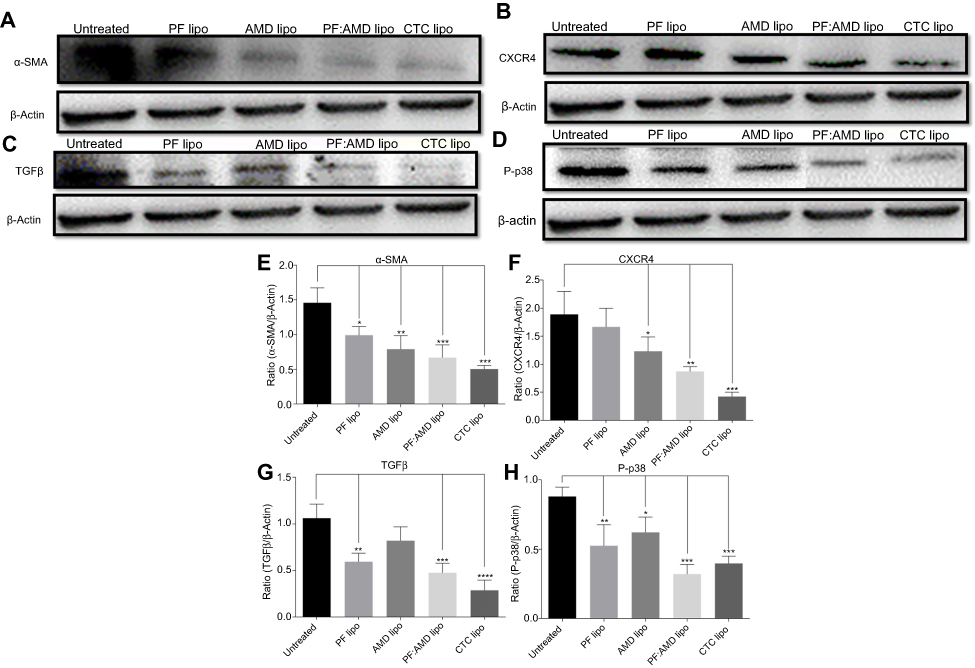 | Figure 6 Effect of liposomes on α-SMA, CXCR4, TGFβ, and P-p38 expressions. Protein expression was determined by western blotting analyses (A) α-SMA expression (B) CXCR4 expression (C) TGFβ expression (D) P-p38 expression. The α-SMA, CXCR4, TGFβ expression and phosphorylation of p38 were determined as a ratio of the total α-SMA, CXCR4, TGFβ and P-p38 expressions to β-Actin, respectively (E) Relative α-SMA expression (F) Relative CXCR4 expression (G) Relative TGFβ expression (H) Relative P-p38 expression. One-Way ANOVA analysis was used to estimate the statistical significance in different groups in comparison with the untreated group. A two-sided “p” value of less than 0.05 indicated statistical significance. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. |
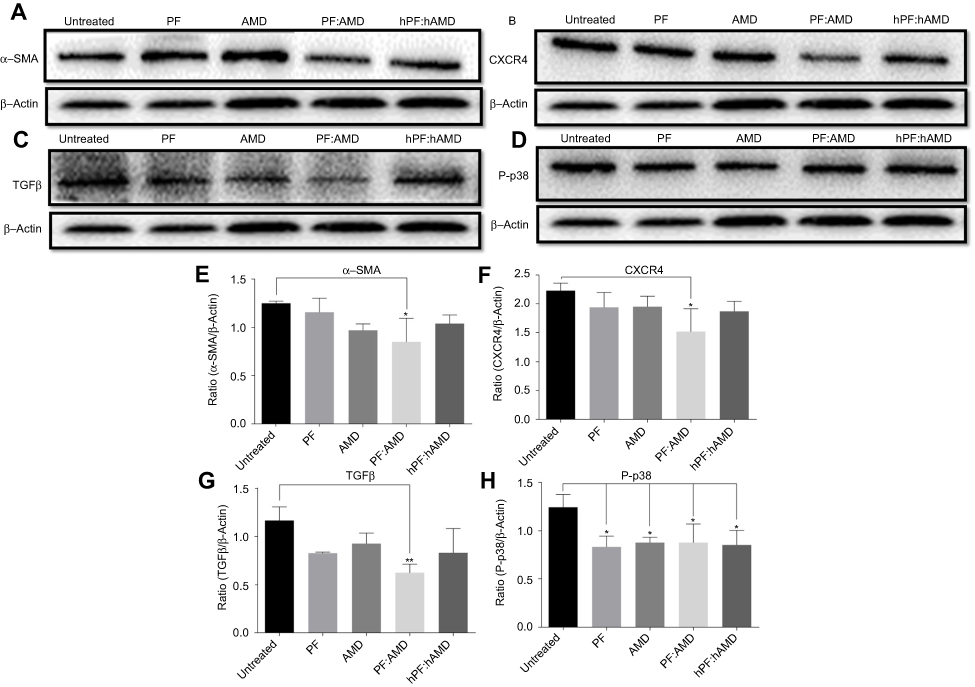 | Figure 7 Effect of free drugs on α-SMA, CXCR4, TGFβ, and P-p38 expressions. Protein expressions were determined by western blotting analyses (A) α-SMA expression (B) CXCR4 expression (C) TGFβ expression (D) P-p38 expression. The α-SMA, CXCR4, TGFβ expression and phosphorylation of p38 were determined as a ratio of the total α-SMA, CXCR4, TGFβ and P-p38 expressions to β-Actin, respectively (E) Relative α-SMA expression (F) Relative CXCR4 expression (G) Relative TGFβ expression (H) Relative P-p38 expression. One-Way ANOVA analysis was used to estimate the statistical significance of the differences in different groups in comparison with the untreated group. A two-sided “p” value of less than 0.05 indicated statistical significance. *p<0.05, **p<0.01. |
Inhibition of CXCR4 expression
SDF-1α/CXCR4 axis and TGFβ were highly upregulated in fibrosis.45,46,80,81 Previously, AMD reduced SDF1 mRNA and CXCR4 expression in lung fibrosis.79 AMD also reduced HSC activation and ameliorated liver fibrosis.45,46 In this study, the enhanced CXCR4 expression was observed in the untreated group, however, the liposomal treatment decreased CXCR4 expression in AMD lipo, PF:AMD lipo and CTC lipo by 34%, 53%, and 77%, respectively. CTC liposomal treatment caused significant CXCR4 reduction in TGFβ-induced activated HSC-T6 cell (Figure 6B and F). None of the free drugs except PF:AMD treatment showed significant reduction of CXCR4 expression (Figure 7B and F).
Inhibition of TGFβ expression
TGFβ activates P-p38 MAPK signaling pathway, promoting HSC activation.82,83 In previous reports, PF significantly diminished TGFβ expression and inhibited P-p38 expression in kidney fibrosis.38 SDF-1α have a synergistic effect on TGFβ expression.45,46,80,81 Previously, AMD down-regulated SDF-1α mRNA expression.79 In this study, TGFβ was highly upregulated in the untreated group while liposomal treatments reduced it significantly (Figure 6C). PF lipo, AMD lipo, PF:AMD lipo and CTC lipo reduced the upregulated TGFβ expression by 46%, 43%, 65%, and 89%, respectively (Figure 6G). Also, PF:AMD group reduced the elevated TGFβ expression level by 46% (Figure 7G). These findings show that compared to free drugs, CTC lipo had the highest down-regulating effect on TGFβ expression (Figures 6C and 7C).
Inhibition of P-p38 expression
Next, we investigated the effect of CTC liposome on the elevated P-p38 expression in TGFβ-induced activated HSC-T6 cells. Based on our findings, PF lipo, AMD lipo, PF:AMD lipo and CTC lipo significantly reduced P-p38 expression by 40%, 29%, 63%, and 66%, respectively (Figure 6D and H). Similarly, PF, AMD, PF:AMD and hPF:hAMD also significantly reduced P-p38 expression by 32%, 29%, 29%, and 31%, respectively (Figure 7D and H). The results in Figure 6D and H indicates that compared to free drugs, CTC lipo has remarkably inhibited the elevated P-p38 expression (Figure 7D and H).
In summary, compared to free drugs, CTC liposomes decreased the elevated expression level of α-SMA, CXCR4, TGFβ, and P-p38 (Figures 6A–D and 7A–D).
In-vivo biodistribution analysis and drug accumulation in the fibrotic liver
To confirm the feasibility of using CTC liposomes in-vivo, the biodistribution of the nanoparticles was estimated in healthy and fibrotic mice. Using the NIR characteristics of IR780 dye, the in-vivo biodistribution at a series of time intervals was monitored with a NIR imaging system. Compared with free IR780 dye, the IR780@CTC liposomes displayed the higher fluorescence intensity in the untreated group (fibrotic mice) at 24 h post-injections (Figure 8A). Furthermore, the IR780@CTC liposome showed higher fluorescence intensities in the fibrotic liver compared with free IR780, IR780@CTC liposome, and IR780@liposome in healthy liver (Figure 8B). The fluorescence intensity of IR780@CTC liposomes was 3.5-fold higher in fibrotic liver compared with free IR780 treated group (Figure 8C). Moreover, Hoechst staining revealed that the red IR780 fluorescence was stronger in fibrotic liver sections compared with the healthy group suggesting the increased accumulation of the IR780@CTC liposomes and displaying possible CXCR4 targeted drug accumulation (Figure 8 D-E). Thus, the AMD decoration showed enhanced targeting ability of CTC liposomes explained by the possible CXCR4 targeting in fibrotic livers.
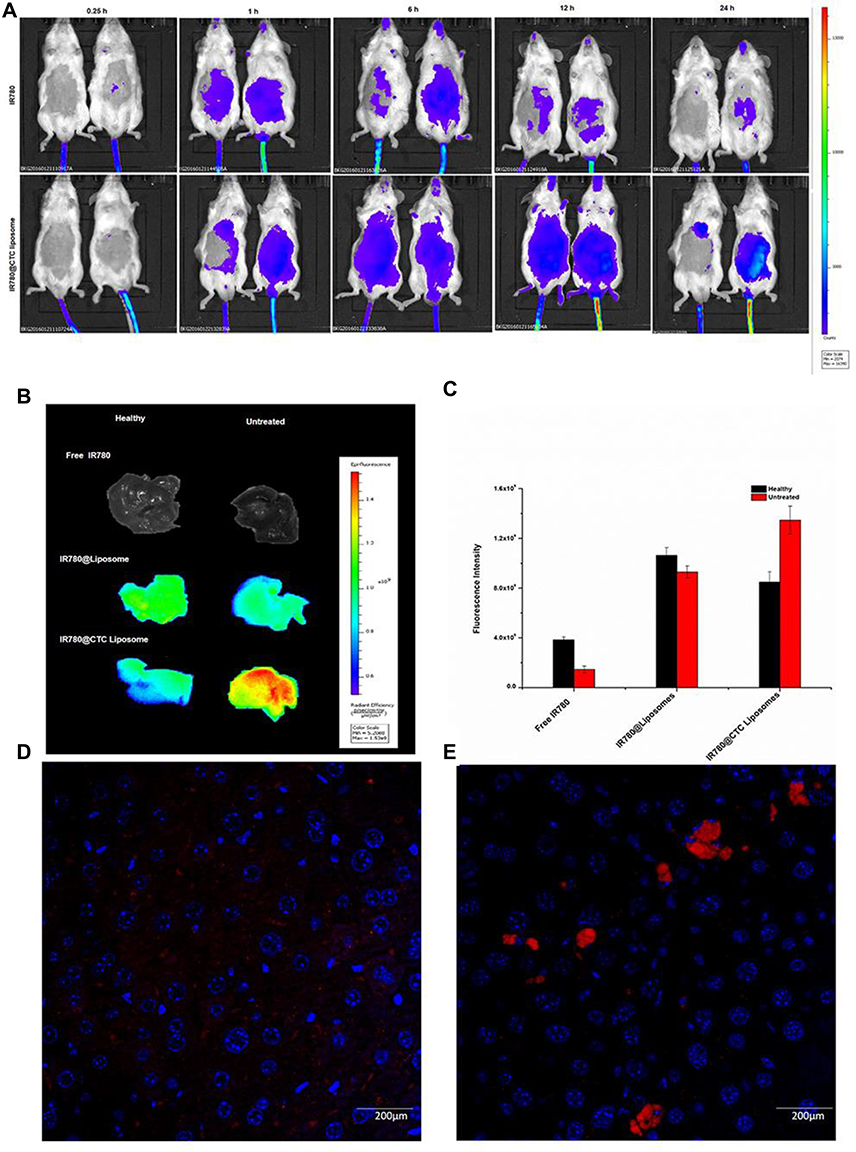 | Figure 8 In-vivo biodistribution and drug accumulation (A) In-vivo biodistribution of IR780@CTC liposomes and free IR780 in healthy and fibrotic mice (B) Accumulation of IR780 in liver detected by ex-vivo imaging at 24 h post-injection in the livers of healthy and fibrotic mice (C) Fluorescence intensities of free IR780, IR780@liposomes, and IR780@CTC liposomes in the healthy and untreated groups (D-E) Detection of IR780 accumulation in healthy and untreated mice using Hoechst staining. Scale bar=200μm. |
Discussion
Liver fibrosis is the ultimate form of chronic liver diseases progressing to liver cirrhosis. Presently, there is no complete cure for liver cirrhosis except liver transplantation.84 So, there is a need for a new treatment strategy to reverse liver fibrosis before cirrhosis. Since decades, animal models are used for screening new investigational drugs in preclinical studies. Currently, in-vitro cell models represent an alternative to animal models, a complementary approach to predict the antifibrotic properties of new investigational drugs.85 Here in this study, we evaluated the antifibrotic activities of PF and AMD using in-vitro TGFβ-induced activated HSC-T6 cells model offering direct access to ECM producing cells in the liver as the key target .86 Anti-fibrotic drugs are not only expected to prevent or treat liver fibrosis but also to produce additional synergic effects regarding inhibition of key players involved in the disease progression like TGFβ, P-p38, CXCR4, and α-SMA.
AMD was adsorbed on the surface of liposomes to play dual functions: CXCR4 targeting, and inhibition of HSC activation. Our findings confirmed that CTC liposomes have significant CXCR4 targeting and inhibited the TGFβ-induced HSC-T6 cell activation, and downregulated the associated α-SMA, CXCR4, TGFβ, and P-p38 expressions.
The morphology and size of the liposomes were visualized directly by TEM, revealing that the CTC liposomes had a spherical shape. The observed size of the CTC liposomes was approximately 100 nm, which was similar to the hydrodynamic diameter obtained from DLS (Figure 1A–B).
AMD caused a noticeable increase in the surface charge of the liposomes. The surface charge changed from −38 mV to −20 mV at the maximum concentration (Figure 1C). The negative zeta potential was attributed to the phospholipids in the liposomes conferring an anionic charge to the nanoparticles.75 The positively charged AMD binds to the negatively charged surface of the liposomes by electrostatic interactions to form CTC liposomes.
The phospholipids in liposomes confer a negative surface charge, which may enhance serum stability by reducing nonspecific interactions with anionic serum components.75 Also, the negative zeta potential suggests strong electrostatic repulsion between particles reducing particle aggregation, and hence promoting stability.77 The chemical stability of liposomes in PBS and FBS predicts the efficacy of drugs for in-vitro and in-vivo applications.87 In this study, the stability of CTC liposomes was evaluated in PBS and FBS and in DW at 4°C . We observed only a slight alteration in the particle size within 15 days and 96 h reflecting its stability and appropriateness for in-vivo applications (Figure 1D–E).
The release profile of liposome predicts the in-vivo fate and efficacy of liposome.87 In- vitro release profiles of PF and AMD in PBS is shown in Figure 1F. The cumulative release of PF and AMD was more than 67% at 48 h reflecting the enhance in-vivo fate and efficacy.
Liposomes reduce the toxicity of encapsulated drugs.88,89 The liposomal encapsulation of PF and AMD reduced the toxicity, in terms of higher cell viability (Figure 2A–C).
In previous reports, AMD was used as a targeting ligand, and AMD functionalized nanocarriers preferentially accumulated within CXCR4 receptor-positive cells.67 In this study, we used AMD decorated CTC liposomes, and the CXCR4 redistribution assay confirmed its CXCR4 targeting ability (Figure 3D).
AMD had a critical role in the uptake of the nanoparticles.75 C6-AMD liposomes were internalized more effectively compared to C6-liposomes (Figure 4A) while the uptake was decreased with AMD pretreatment (Figure 4B). Furthermore, nanoparticles can be internalized by various pathways.75 From our results, C6-AMD and C6 liposomes were internalized by caveolae-mediated endocytosis pathway (Figure 4C).
Apoptosis is a critical factor in fibrosis. Induction of apoptosis of activated HSC improves liver fibrosis.90 Multiple mechanisms show that the TGFβ ameliorates activated HSC-T6 cells apoptosis.91 SDF-1α have a synergistic effect on TGFβ expressions.45,46,50,80,81 While AMD down-regulated SDF-1α mRNA expression.79 Thus, AMD decreases TGFβ level, and hence indirectly augmenting apoptosis of HSC-T6 cells. Previously, TGFβ prevented the apoptosis of activated HSC.91 The augmentation of activated HSC apoptosis promotes the resolution of fibrosis.92 PF significantly inhibited TGFβ expression.38 The inhibitory effect of PF on TGFβ level leads to enhanced apoptosis of activated HSC-T6 cells. In this study, the CTC liposomes induced the apoptotic effects in TGFβ-induced activated HSC-T6 cells by the joint TGFβ inhibitory effects of PF and AMD (Figure 5A).
SDF-1α/CXCR4 axis is up-regulated in liver injury causing activation of HSC.45,46 In previous reports, AMD reduced SDF1 mRNA and CXCR4 expression in lung fibrosis.79 PF inhibits P-p38 expression with a resultant decrease in α-SMA and collagen expressions.38,44 TGFβ level and SDF-1α/CXCR4 axis were highly upregulated in fibrosis, and SDF-1α had a synergistic effect on TGFβ expressions.45,46,80,81 From our findings, the joint pharmacodynamics effect of PF and AMD inhibited the SDF-1α/CXCR4 axis, α-SMA, CXCR4, and TGFβ expressions, respectively (Figures 3D and 6A–C). Previously, AMD down-regulated P-p38 expression and SDF1 mRNA expression in lung fibrosis.79,93 Also, AMD reduced HSC activation and ameliorated liver fibrosis.45,46 PF down-regulated P-p38 expression in renal fibrosis.38 In the current study, CTC liposomes significantly down-regulated the P-p38 expressions (Figure 6D). Collectively, as shown in the Figure 6A–D, CTC liposomes compared to free drugs, displayed better inhibitory effects on α-SMA, CXCR4, TGFβ, and P-p38 expressions (Figure 7A–D)
The in-vivo biodistribution studies and Hoechst staining demonstrated the maximum accumulation of IR780@CTC liposomes in the fibrotic livers displaying the enhanced possible CXCR4-targeted drug accumulation in fibrotic livers (Figure 8A–E). These findings suggest that these nanoparticles could be employed as an effective drug delivery system for the treatment of liver fibrosis in-vitro and in-vivo.
Conclusion
We developed CTC liposomes for combination therapy of TGFβ-induced activated HSC-T6 cell activation. CTC liposomes showed excellent PBS and FBS stability at 37 °C with additional storage stability at 4 °C which was favorable for further long-term in-vivo applications. We successfully evaluated the CXCR4 antagonistic ability of CTC liposomes using CXCR4 redistribution assay. C6-AMD liposomes were internalized better than C6 liposomes and C6-solution reflecting CXCR4 mediated uptake while AMD pretreatment decreased nanoparticles uptake explaining the role of AMD in uptake. The caveolae-mediated endocytosis pathway internalized the liposomes. CTC liposomes induced apoptosis of the TGFβ-induced activated HSC-T6 cells suggesting its role in fibrosis regression. CTC liposomes blocked the upregulated SDF-1α/CXCR4 axis, and significantly decreased the elevated α-SMA, CXCR4, TGFβ, and P-p38 expressions. From the in-vivo biodistribution studies and Hoechst staining, CTC liposomes were better retained in fibrotic mice compared to healthy mice. Our results suggest that the CTC liposomal therapy can be a novel treatment strategy that can effectively be used in combating liver fibrosis.
Acknowledgments
This work was financially supported by the National Science and Technology Major Project (2017YFA0205400), Changjiang Scholar program from the Chinese Ministry of Education, China National Science Foundation (81373983, 81573377), and China Postdoctoral Science Foundation (No. 2016M601923).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Eng FJ, Friedman SL, Fibrogenesis I. New insights into hepatic stellate cell activation: the simple becomes complex. Am J Physiol-Gastr L. 2000;279(1):G7–G11.
2. Nusrat S, Khan MS, Fazili J, Madhoun MF. Cirrhosis and its complications: evidence-based treatment. World J Gastroenterol. 2014;20(18):5442–5460. doi:10.3748/wjg.v20.i18.5442
3. Blachier M, Leleu H, Peck-Radosavljevic M, Valla D-C, Roudot-Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol. 2013;58(3):593–608. doi:10.1016/j.jhep.2012.12.005
4. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5(167):167sr161–167sr161. doi:10.1126/scitranslmed.3004700
5. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Seminars in liver disease. 2001;21(3):351-372. doi:10.1055/s-2001-17556
6. Reeves HL, Friedman SL. Activation of hepatic stellate cells—a key issue in liver fibrosis. Front Biosci. 2002;7(4):808–826. doi:10.2741/reeves
7. Altamirano-Barrera A, Barranco-Fragoso B, Mendez-Sanchez N. Management strategies for liver fibrosis. Ann Hepatol. 2017;16(1):48–56. doi:10.5604/16652681.1226814
8. Iimuro Y, Brenner DA. Matrix Metalloproteinase gene delivery for liver fibrosis. Pharm Res-Dordr. 2008;25(2):249–258. doi:10.1007/s11095-007-9311-7
9. Siller-Lopez F, Sandoval A, Salgado S, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126(4):
10. Sato Y, Murase K, Kato J, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26(4):431–442. doi:10.1038/nbt1396
11. Sobrevals L, Enguita M, Rodriguez C, et al. AAV vectors transduce hepatocytes in vivo as efficiently in cirrhotic as in healthy rat livers. Gene Ther. 2012;19(4):411–417. doi:10.1038/gt.2011.119
12. Kamimura K, Kanefuji T, Yokoo T, et al. Safety assessment of liver-targeted hydrodynamic gene delivery in dogs. PLoS One. 2014;9(9):e107203. doi:10.1371/journal.pone.0107203
13. Terai S, Tanimoto H, Maeda M, et al. Timeline for development of autologous bone marrow infusion (ABMi) therapy and perspective for future stem cell therapy. J Gastroenterol. 2012;47(5):491–497. doi:10.1007/s00535-012-0580-5
14. Garcia-Guerra A, Dunwell TL, Trigueros S. Nano-scale gene delivery systems: current technology, obstacles, and future directions. Curr Med Chem. 2018;25(21):2448–2464. doi:10.2174/0929867325666180108100723
15. Akhmetshina A, Dees C, Pileckyte M, et al. Dual inhibition of c-able and PDGF receptor signaling by dasatinib and nilotinib for the treatment of dermal fibrosis. FASEB J. 2008;22(7):2214–2222. doi:10.1096/fj.07-105627
16. Akhmetshina A, Venalis P, Dees C, et al. Treatment with imatinib prevents fibrosis in different preclinical models of systemic sclerosis and induces regression of established fibrosis. Arthritis Rheumatol. 2009;60(1):219–224. doi:10.1002/art.24186
17. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. New Engl J Med. 2006;354(25):2655–2666. doi:10.1056/NEJMoa055120
18.
19. Rankin AL, Mumm JB, Murphy E, et al. IL-33 induces IL-13–dependent cutaneous fibrosis. J Immunol. 2010;184(3):1526–1535. doi:10.4049/jimmunol.0903306
20. Li Z, Wei W, Chen B, et al. The effect of rhCygb on CCl 4-induced hepatic fibrogenesis in rat. Sci Rep-Uk. 2016;6:23508. doi:10.1038/srep23508
21. Rockey DC. Current and future anti-fibrotic therapies for chronic liver disease. Clin Liver Dis. 2008;12(4):939–962, xi. doi:10.1016/j.cld.2008.07.011
22. Inagaki Y, Higashiyama R, Higashi K. Novel anti-fibrotic modalities for liver fibrosis: molecular targeting and regenerative medicine in fibrosis therapy. J Gastroenterol Hepatol. 2012;27 Suppl 2:85–88. doi:10.1111/j.1440-1746.2011.07006.x
23. Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti-fibrotic therapies. Biochim Biophys Acta. 2013;1832(7):1088–1103. doi:10.1016/j.bbadis.2012.12.007
24. King TE
25. Meyer KC, Danoff SK, Lancaster LH, Nathan SD. Management of idiopathic pulmonary fibrosis in the elderly patient: addressing key questions. Chest. 2015;148(1):242–252. doi:10.1378/chest.14-2475
26. King CS, Nathan SD. Practical considerations in the pharmacologic treatment of idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2015;21(5):479–489. doi:10.1097/MCP.0000000000000190
27. Spagnolo P, Maher TM, Richeldi L. Idiopathic pulmonary fibrosis: recent advances in pharmacological therapy. Pharmacol Ther. 2015;152:18–27. doi:10.1016/j.pharmthera.2015.04.005
28. Macías-Barragán J, Sandoval-Rodríguez A, Navarro-Partida J, Armendáriz-Borunda J. The multifaceted role of pirfenidone and its novel targets. Fibrogenesis Tissue Repair. 2010;3(1):16.
29. Cho ME, Kopp JB. Pirfenidone: an anti-fibrotic therapy for progressive kidney disease. Expert Opin Inv Drug. 2010;19(2):275–283. doi:10.1517/13543780903501539
30. Jose A, Mandapalli PK, Venuganti VVK. Liposomal hydrogel formulation for transdermal delivery of pirfenidone. J Liposome Res. 2016;26(2):139–147. doi:10.3109/08982104.2015.1060611
31. Li D, Gong L. Preparation of novel pirfenidone microspheres for lung-targeted delivery: in vitro and in vivo study. Drug Des Devel Ther. 2016;10:2815–2821. doi:10.2147/DDDT.S113670
32. Rasooli R, Rajaian H, Pardakhty A, Mandegary A. Preference of aerosolized pirfenidone to oral intake: an experimental model of pulmonary fibrosis by paraquat. J Aerosol Med Pulm Drug Deliv. 2018;31(1):25–32. doi:10.1089/jamp.2016.1342
33. Meng H, Xu Y. Pirfenidone-loaded liposomes for lung targeting: preparation and in vitro/in vivo evaluation. Drug Des Devel Ther. 2015;9:3369–3376.
34. Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–19. doi:10.1016/j.ejps.2014.02.014
35. Meyer KC, Decker CA. Role of pirfenidone in the management of pulmonary fibrosis. Ther Clin Risk Manag. 2017;13:427–437. doi:10.2147/TCRM.S81141
36. Bando M, Yamauchi H, Ogura T, et al. Clinical experience of the long-term use of pirfenidone for idiopathic pulmonary fibrosis. Intern Med. 2016;55(5):443–448. doi:10.2169/internalmedicine.55.5272
37. Macías-Barragán J, Sandoval-Rodríguez A, Navarro-Partida J, Armendáriz-Borunda J. The multifaceted role of pirfenidone and its novel targets. Fibrogenesis Tissue Repair. 2010;3(1):1–11.
38. Li Z, Liu X, Wang B, et al. Pirfenidone suppresses MAPK signaling pathway to reverse epithelial‐mesenchymal transition and renal fibrosis. Nephrology. 2017;22(8):589–597. doi:10.1111/nep.12831
39. Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446. doi:10.1038/nm1388
40. Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–829. doi:10.1183/09031936.00005209
41. Lasky J. Pirfenidone. Idrugs. 2004;7(2):166–172.
42. Kanzler S, Lohse AW, Keil A, et al. TGF-β1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol-Gastr L. 1999;276(4):G1059–G1068.
43. Chen MH, Chen JC, Tsai CC, et al. The role of TGF-beta 1 and cytokines in the modulation of liver fibrosis by Sho-Saiko-to in rat‘s bile duct ligated model. J Ethnopharmacol. 2005;97(1):7–13. doi:10.1016/j.jep.2004.09.040
44. Matsuoka H, Arai T, Mori M, et al. A p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2002;283(1):L103–L112. doi:10.1152/ajplung.00187.2001
45. Chen Y, Huang Y, Reiberger T, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59(4):1435–1447. doi:10.1002/hep.26790
46. Wald O, Pappo O, Safadi R, et al. Involvement of the CXCL12/CXCR4 pathway in the advanced liver disease that is associated with hepatitis C virus or hepatitis B virus. Eur J Immunol. 2004;34(4):1164–1174. doi:10.1002/eji.200324441
47. Fruehauf S. Current clinical indications for plerixafor. Transfus Med Hemother. 2013;40(4):246–250. doi:10.1159/000354229
48. De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther. 2010;128(3):509–518. doi:10.1016/j.pharmthera.2010.08.009
49. Liesveld JL, Bechelli J, Rosell K, et al. Effects of AMD3100 on transmigration and survival of acute myelogenous leukemia cells. Leuk Res. 2007;31(11):1553–1563. doi:10.1016/j.leukres.2007.02.017
50. Broxmeyer HE, Orschell CM, Clapp DW, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201(8):1307–1318. doi:10.1084/jem.20041385
51. Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7(8):425–436. doi:10.1038/nrgastro.2010.97
52. Lin TT, Gao DY, Liu YC, et al. Development and characterization of sorafenib-loaded PLGA nanoparticles for the systemic treatment of liver fibrosis. J Control Release. 2016;221:62–70. doi:10.1016/j.jconrel.2015.11.003
53. Makino H, Aono Y, Azuma M, et al. Antifibrotic effects of a CXCR4 antagonist in bleomycin-induced pulmonary fibrosis in mice. J Med Invest. 2013;60(1–2):127–137.
54. de Nigris F, Schiano C, Infante T, Napoli C. CXCR4 inhibitors: tumor vasculature and therapeutic challenges. Recent Pat Anticancer Drug Discov. 2012;7(3):251–264.
55. Welschinger R, Liedtke F, Basnett J, et al. Plerixafor (AMD3100) induces prolonged mobilization of acute lymphoblastic leukemia cells and increases the proportion of cycling cells in the blood in mice. Exp Hematol. 2013;41(3):293–302.e291. doi:10.1016/j.exphem.2012.11.004
56. Mei L, Liu Y, Zhang Q, Gao H, Zhang Z, He Q. Enhanced antitumor and anti-metastasis efficiency via combined treatment with CXCR4 antagonist and liposomal doxorubicin. J Control Release. 2014;196:324–331. doi:10.1016/j.jconrel.2014.10.017
57. Liu C-H, Chan K-M, Chiang T, et al. Dual-functional nanoparticles targeting CXCR4 and delivering antiangiogenic siRNA ameliorate liver fibrosis. Mol Pharm. 2016;13(7):2253–2262. doi:10.1021/acs.molpharmaceut.5b00913
58. Liu CH, Chan KM, Chiang T, et al. Dual-functional nanoparticles targeting CXCR4 and delivering antiangiogenic siRNA ameliorate liver fibrosis. Mol Pharm. 2016;13(7):2253–2262. doi:10.1021/acs.molpharmaceut.5b00913
59. Hu K, Zhou H, Liu Y, et al. Hyaluronic acid functional amphipathic and redox-responsive polymer particles for the co-delivery of doxorubicin and cyclopamine to eradicate breast cancer cells and cancer stem cells. Nanoscale. 2015;7(18):8607–8618. doi:10.1039/c5nr01084e
60. Zhang P, Li J, Ghazwani M, et al. Effective co-delivery of doxorubicin and dasatinib using a PEG-Fmoc nanocarrier for combination cancer chemotherapy. Biomaterials. 2015;67:104–114. doi:10.1016/j.biomaterials.2015.07.027
61. Wu J, Tang C, Yin C. Co-delivery of doxorubicin and interleukin-2 via chitosan-based nanoparticles for enhanced antitumor efficacy. Acta Biomater. 2017;47:81–90. doi:10.1016/j.actbio.2016.10.012
62. Xiao B, Si X, Han MK, Viennois E, Zhang M, Merlin D. Co-delivery of camptothecin and curcumin by cationic polymeric nanoparticles for synergistic colon cancer combination chemotherapy. J Mater Chem B. 2015;3(39):7724–7733.
63. Madrigal-Carballo S, Lim S, Rodriguez G, et al. Biopolymer coating of soybean lecithin liposomes via layer-by-layer self-assembly as a novel delivery system for ellagic acid. J Funct Foods. 2010;2(2):99–106. doi:10.1016/j.jff.2010.01.002
64. Park SN, Jo NR, Jeon SH. Chitosan-coated liposomes for enhanced skin permeation of resveratrol. J Ind Eng Chem. 2014;20(4):1481–1485. doi:10.1016/j.jiec.2013.07.035
65. Meng S, Su B, Li W, et al. Integrin-targeted paclitaxel nanoliposomes for tumor therapy. Med Oncol. 2011;28(4):1180–1187. doi:10.1007/s12032-010-9621-1
66. Wuyts WA, Antoniou KM, Borensztajn K, et al. Combination therapy: the future of management for idiopathic pulmonary fibrosis? Lancet Respir Med. 2014;2(11):933–942. doi:10.1016/S2213-2600(14)70232-2
67. Zevon M, Ganapathy V, Kantamneni H, et al. CXCR-4 targeted, Short Wave Infrared (SWIR) emitting nanoprobes for enhanced deep tissue imaging and micrometastatic cancer lesion detection. Small. 2015;11(47):6347–6357. doi:10.1002/smll.201502202
68. Maestrelli F, Gonzalez-Rodriguez ML, Rabasco AM, Mura P. Preparation and characterization of liposomes encapsulating ketoprofen-cyclodextrin complexes for transdermal drug delivery. Int J Pharm. 2005;298(1):55–67. doi:10.1016/j.ijpharm.2005.03.033
69. Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13(1):238–252.
70. Wei M, Xu Y, Zou Q, et al. Hepatocellular carcinoma targeting effect of PEGylated liposomes modified with lactoferrin. Eur J Pharm Sci. 2012;46(3):131–141. doi:10.1016/j.ejps.2012.02.007
71. Guo G, Kuai D, Cai S, et al. Knockdown of FRAT1 expression by RNA interference inhibits human glioblastoma cell growth, migration, and invasion. PLoS One. 2013;8(4):e61206. doi:10.1371/journal.pone.0061206
72. Bicho A, Peça IN, Roque A, Cardoso MM. Anti-CD8 conjugated nanoparticles to target mammalian cells expressing CD8. Int J Pharm. 2010;399(1–2):80–86. doi:10.1016/j.ijpharm.2010.08.005
73. Simard P, Leroux J-C. pH-sensitive immunoliposomes are specific to the CD33 cell surface antigen of leukemic cells. Int J Pharm. 2009;381(2):86–96. doi:10.1016/j.ijpharm.2009.05.013
74. Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37. doi:10.1038/nature01451
75. Li H, Wang K, Yang X, et al. Dual-function nanostructured lipid carriers to deliver IR780 for breast cancer treatment: anti-metastatic and photothermal anti-tumor therapy. Acta Biomater. 2017;53:399–413. doi:10.1016/j.actbio.2017.01.070
76. Mohammed A, Weston N, Coombes A, Fitzgerald M, Perrie Y. Liposome formulation of poorly water-soluble drugs: optimization of drug loading and ESEM analysis of stability. Int J Pharm. 2004;285(1–2):23–34. doi:10.1016/j.ijpharm.2004.07.010
77. Dong Y, Feng -S-S. Methoxy poly (ethylene glycol)-poly (lactide)(MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25(14):2843–2849. doi:10.1016/j.biomaterials.2003.09.055
78. Li J, Zhu Y, Hazeldine ST, Li C, Oupický D. Dual-function CXCR4 antagonist polyplexes to deliver gene therapy and inhibit cancer cell invasion. Angew Chem Int Ed Engl. 2012;51(35):8740–8743. doi:10.1002/anie.201203463
79. Song JS, Kang CM, Kang HH, et al. Inhibitory effect of CXC chemokine receptor 4 antagonist AMD3100 on bleomycin-induced murine pulmonary fibrosis. Exp Mol Med. 2010;42(6):465–472. doi:10.3858/emm.2010.42.6.048
80. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123(5):1902–1910. doi:10.1172/JCI66369
81. Moreno M, Bataller R. Cytokines and renin-angiotensin system signaling in hepatic fibrosis. Clin Liver Dis. 2008;12(4):825–852. doi:10.1016/j.cld.2008.07.013
82. Hanafusa H, Ninomiya-Tsuji J, Masuyama N, et al. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-β-induced gene expression. J Biol Chem. 1999;274(38):27161–27167.
83. Engel ME, McDonnell MA, Law BK, Moses HL, Interdependent SMAD. JNK signaling in transforming growth factor-β-mediated transcription. J Biol Chem. 1999;274(52):37413–37420.
84. Detlef S, Ook KY. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–1901. doi:10.1172/JCI66028
85. Aoudjehane L, Boelle P-Y, Bisch G, et al. Development of an in vitro model to test antifibrotic drugs on primary human liver myofibroblasts. Lab Invest. 2016;96(6):672. doi:10.1038/labinvest.2016.108
86. Song Y, Zhan L, Yu M, et al. TRPV4 channel inhibits TGF-β1-induced proliferation of hepatic stellate cells. PLoS One. 2014;9(7):e101179. doi:10.1371/journal.pone.0101179
87. Shao X-R, Wei X-Q, Zhang S, et al. Effects of micro-environmental pH of liposome on chemical stability of loaded drug. Nanoscale Res Lett. 2017;12(1):504. doi:10.1186/s11671-017-2256-9
88. Olusanya OT, Haj Ahmad RR, Ibegbu MD, Smith RJ, Elkordy AA. Liposomal drug delivery systems and anticancer drugs. Molecules. 2018;23(4):907. doi:10.3390/molecules23040907
89. Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6:286. doi:10.3389/fphar.2015.00286
90. Dekel R, Zvibel I, Brill S, Brazovsky E, Halpern Z, Oren R. Gliotoxin ameliorates the development of fibrosis and cirrhosis in a thioacetamide rat model. Dig Dis Sci. 2003;48(8):1642–1647.
91. Fu M-Y, He Y-J, Lv XIA, et al. Transforming growth factor-β1 reduces apoptosis via autophagy activation in hepatic stellate cells. Mol Med Rep. 2014;10(3):1282–1288. doi:10.3892/mmr.2014.2383
92. Wright MC, Issa R, Smart DE, et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology. 2001;121(3):685–698.
93. Jeong Sup S, Chun MK, Hyeon Hui K, et al. Inhibitory effect of CXC chemokine receptor 4 antagonist AMD3100 on bleomycin-induced murine pulmonary fibrosis. Exp Mol Med. 2010;42(6):465. doi:10.3858/emm.2010.42.6.048
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.