")
Back to Journals » OncoTargets and Therapy » Volume 10
CXCL12 suppresses cisplatin-induced apoptosis through activation of JAK2/STAT3 signaling in human non-small-cell lung cancer cells
Authors Wang M, Lin T, Wang YC, Gao S, Yang ZY, Hong X, Chen GY
Received 23 January 2017
Accepted for publication 19 May 2017
Published 29 June 2017 Volume 2017:10 Pages 3215—3224
DOI https://doi.org/10.2147/OTT.S133055
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Meng Wang,1 Tie Lin,2 Yicun Wang,3 Song Gao,4 Zhaoyang Yang,1 Xuan Hong,1 Gongyan Chen1
1Department of Respiratory Medicine, Harbin Medical University Cancer Hospital, 2Department of Surgery, The First Affiliated Hospital of Harbin Medical University, Harbin, 3Jilin Provincial Key Laboratory on Molecular and Chemical Genetics, The Second Hospital of Jilin University, Changchun, 4Department of Clinical Oncology, Shengjing Hospital, China Medical University, Shenyang, People’s Republic of China
Aims: Poor efficacy of chemotherapy drugs in non-small-cell lung cancer (NSCLC) is the key reason for the failure of treatment, but the mechanism of this remains largely unknown. Stromal cell-derived factor 1-alpha (SDF-1α/CXCL12) is a small chemotactic cytokine protein that plays an important role in tumor progression. In this study, we investigated the anti-apoptotic mechanism of the CXCL12/CXCR4 axis in response to cisplatin, a commonly used chemotherapeutic drug, in human lung adenocarcinoma A549 cells.
Methods: CXCL12 blocks cisplatin-induced apoptosis in A549, and the results were shown by propidium iodide/annexin V staining in vitro. The mechanism of CXCL12 stimulating phosphorylation of STAT3 through CXCR4/JAK2 was demonstrated by immunofluorescence and Western blotting. The expression of CXCL12 and p-STAT3 in clinical specimens was examined by immunohistochemistry.
Results: CXCL12 significantly decreased the ratio of apoptotic cells and stimulation of phospho-signal transducer and activator of transcription (p-STAT)-3 in a time-dependent manner through interaction with CXCR4. Among the signaling molecules downstream of CXCR4, the JAK2/STAT3 pathway plays a predominant role in the anti-apoptotic effect of CXCL12. Analysis of clinical specimens revealed that increased CXCL12 and p-STAT3 expression correlates with enhanced lung cancer progression.
Conclusion: These data suggest that CXCR4 contributes to CXCL12-mediated anti-apoptosis by activating JAK2/STAT3 pathway in NSCLC cells. Therefore, targeting CXCL12/CXCR4 signaling pathway reveals a potential therapeutic approach for NSCLC.
Keywords: CXCL12, CXCR4, JAK2/STAT3, apoptosis, non-small-cell lung cancer
Introduction
Non-small-cell lung cancer (NSCLC) is the most common malignancy in northern China, and its mortality rate has shown an upward trend.1 Chemotherapy is a mainstay therapy for NSCLC, especially for advanced NSCLC. However, most NSCLC patients will develop chemoresistance following a period of chemotherapy. Genetics plays an essential role in the pathophysiological mechanism of NSCLC, and it leads to non-sensitivity of NSCLC to platinum-based chemotherapy.2 The development of novel drug target requires profound understanding of the molecular mechanisms of lung cancer.
Chemokines are produced by cancer-associated fibroblasts, a component of stromal cells, and influence the metastatic potential and site-specific dissemination of cancer cells.3 Stromal-derived factor-1 (SDF-1/CXCL12), a 68-amino acid protein, belongs to the CXC chemokine family. Previous studies have described the effects of CXCL12 in many cancers, including its role in promoting local invasion and distant metastasis of lung cancer.4–6 Recent results demonstrated that CXCL12, as the sole endogenous ligand for the C-X-C chemokine receptor type 4 (CXCR4) receptor, robustly inhibits apoptotic cell death through an intrinsic pathway in cancer cells.7,8 Endogenous CXCR4 expression on carcinoma cells is correlated with poor prognosis in several cancers.9–11 Knockdown of CXCR4 expression by small interfering RNA in breast carcinoma cells decreases cell invasion and adhesion in vitro and abrogates tumor growth in vivo.12 In small-cell lung cancer (SCLC) cells, CXCR4 antagonists such as plerixafor (AMD3100) and T140 analogs (TN14003/BKT140) disrupt CXCR4-mediated SCLC cell adhesion to stromal cells, thereby sensitizing SCLC cells to cytotoxic drugs such as etoposide and antagonizing cell adhesion-mediated drug resistance.13 All of these results suggest that the CXCL12/CXCR4 axis could be involved in drug resistance.
Cisplatin (CDDP) is a commonly used chemotherapeutic drug in human lung adenocarcinoma. CDDP promotes its cytotoxicity by forming DNA–protein cross-links, DNA mono-adducts, and intrastrand DNA cross-links, which all trigger apoptosis. However, the success of platinum drugs in the treatment of various types of cancer has been challenged by the hamper of intrinsic and acquired resistance.14 At present, the mechanisms involved in cisplatin resistance are not yet fully understood, because the research in platinum drug resistance is limited. Inhibition of apoptosis and cellular proliferation are important mechanisms in drug resistance. Studies identified that CXCL12 also activates multiple signaling pathways related to cell apoptosis and proliferation. Janus kinase (JAK) signal transducer and activator of transcription (STAT) signal pathway is associated with a wide variety of biological processes, including cell apoptosis, proliferation, and tumor progression.15,16 Moreover, the JAK2/STAT3 pathway is an important anti-apoptotic pathway that is frequently activated in different cancer cells.17–20 The phosphorylation of 705 tyrosine residue in STAT3 protein, which is a crucial event for its activation, leads to form STAT3 homodimers and translocation into the nuclei. Nuclear localized STAT3 dimer binds to the promoters of various target genes and regulates their transcriptions, which are involved in cancer cell proliferation, survival and invasion.21 Persistent activation of STAT3 may lead to oncogenesis by promoting tumor angiogenesis and resistance to apoptosis. It has been reported that activation of the JAK2/STAT3 pathway via CXCL12–CXCR4 signaling increases the malignancy and metastasis of breast cancer.22 However, the role of CXCL12 on CDDP-induced apoptosis in lung cancer is not clear, and it is important to improve the efficacy of platinum drugs.
The aims of this study are to explore whether CXCL12 plays an anti-apoptotic role in A549 human lung cancer cells. We further discussed the underlying molecular mechanisms involving CXCR4, JAK2, and STAT3 by examining the effects of CXCL12 on apoptosis-related pathways.
Materials and methods
Reagents and antibodies
Human recombinant chemokine SDF-1α/CXCL12 was obtained from R&D Systems Inc. (Minneapolis, MN, USA). Anti-human CXCR4 antibody was purchased from Abcam (Cambridge, MA, USA). CXCR4 antagonist, AMD3100, was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). AG490 was purchased from DuPont Merck (Hangzhou, China). STAT3 and phospho-STAT3 (Serine 727) antibodies were purchased from Cell Signaling Technology (Boston, MA, USA).
Cell line and culture
Human lung adenocarcinoma cell line (A549) was the generous gift of Heilongjiang Cancer Institute (Harbin, China). Cells were maintained in an Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum, 50 U/mL penicillin and 50 μg/mL streptomycin and then incubated at 37°C in a humidified atmosphere of 5% CO2.
The A549 cell line in this study was donated by Heilongjiang Cancer Institute for research only, which was approved by the Ethical Review Committee of Harbin Medical University.
Assay of apoptosis
The effect of CXCL12 on anti-apoptosis was detected through propidium iodide (PI)//annexin V. According to the results of our preliminary experiment, serum-free A549 cells pretreated with CDDP (20 μg/mL) were incubated with or without CXCL12 (100 ng/mL) and AMD3100 (5 μg/mL) or AG490 (50 μM) for 48 h and collected. The cells were collected and centrifuged at 1,000× g for 10 min. After washing with phosphate-buffered saline (1× PBS), cells were added to 500 μL annexin V-binding buffer (Keygen, Nanjing, China), incubated for 15 min with 2 μL fluorescein isothiocyanate (FITC)-conjugated annexin V and 10 μL PI (Keygen), and then analyzed on a FACSort (Becton-Dickinson, San Jose, CA, USA).
Western blot
A549 cells were treated with CDDP and CXCL12 (100 ng/mL) for various periods of time (from 30 min to 24 h). AMD3100 (5 μg/mL) or AG490 (50 μM) was added 30 min before CXCL12 and CDDP treatment. Cells were lysed in lysis buffer consisting of 20 mM Tris–HCl (pH 7.5), 2 mM ethylenediaminetetraacetic acid (EDTA), 150 mM NaCl, 1% Triton X-100, and protease inhibitors. After centrifugation at 12,000× g/min for 5 min at 4°C, the supernatant was obtained and used as a total cell lysate for analysis of protein concentration by the Bradford method (BioRad, Hercules, CA, USA). Equal amounts of cellular proteins (30 μg/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. After blocking with 5% skimmed milk in Tris-buffered saline/Tween-20 overnight at 24°C, the blots were incubated with rabbit anti-human STAT3 (1:1,000) and p-STAT3 (Serine 727) (1:1,000) for 2 h at room temperature. The blots were subsequently washed three times (10 min for each wash) with Tris-buffered saline/Tween-20 and then treated with the appropriate alkaline phosphatase-conjugated anti-rabbit secondary antibody (1:2,000; Promega Corporation, Fitchburg, WI, USA) for 1 h at room temperature. The bands were visualized using the 5-bromo-4-chloro-3’-indoly phosphate/nitroblue tetrazolium chloride (Promega Corporation) coloration method. Proteins were quantified using an electrochemiluminescence (ECL) system (Pierce Biotechnology, Rockford, IL USA).
Immunofluorescence
Cells were treated with drugs as same as above. After collection, cells were fixed in 4% paraformaldehyde for 15 min. After fixation, cells were washed twice with 1× PBS (0.01 M, pH 7.4) and then incubated with 1% fetal bovine serum in a constant-temperature incubator at 37°C for 20 min. Rabbit anti-human STAT3 (1:1,000) and p-STAT3 (Serine 727) (1:500) were incubated with cells overnight at 4°C. After washing with 1× PBS five times, cells were incubated with rabbit anti-mouse immunoglobulin-FITC secondary antibody (1:1,000) for 1 h. Finally, cells were washed six times with 1× PBS, covered with PBS with 50% glycerol, and viewed under a fluorescence microscope.
Immunohistochemistry
A total of 208 cases with pathologically confirmed NSCLC were involved in this study. Human solid lung cancer specimens were collected from The Tumor Hospital Affiliated Harbin Medical University from 2002 to 2004. The patients received neither chemotherapy nor radiation therapy prior to surgery.
The patients included 128 males and 80 females, of mean age 59.6 years (range, 35–76 years). The study was approved by the ethical review committee of Harbin Medical University, Harbin, China. All patients were provided with written informed consent to participate in the study.
Immunohistochemical staining for CXCL12 and p-STAT3 was performed using the standard streptavidin–peroxidase biotin technique (SP technique) with an SP kit (Zhongshan Co., Beijing, China). Paraffin sections (4 μm) were deparaffinized in xylene and then rehydrated using an alcohol gradient. Hydrated autoclave pretreatment was performed by boiling for 5 min in citrate buffer (10 mM, pH 6.0). After endogenous peroxidase was quenched with 3% hydrogen peroxide and blocked for 10 min, the sections were incubated overnight at 4°C with antibodies against CXCL12 (1:200; R&D Systems Inc.) or p-STAT3 (1:150; Cell Signaling Technology). Biotinylated immunoglobulin and peroxidase-conjugated streptavidin were then added. Finally, 3,3′-diaminobenzidine was added for color development, and hematoxylin was used for counterstaining. The mean percentage of positive tumor cells was determined in at least five fields at ×200 magnification. All slides were evaluated by the consensus of experienced pathologists. For the negative control, PBS was used in place of the primary antibodies. We adopted the German semi-quantitative scoring system to determine the staining intensity and extent, which has been widely accepted and used in previous studies.23 CXCL12 expression was quantified using a visual grading system based on the extent of staining (percentage of positive cells graded on scale from 0 to 4: 0, <5%; 1, 5%–25%; 2, 25%–50%; 3, 50%–75%; 4, >75%) and the intensity of staining (graded on a scale of 0–3: 0, none; 1, weak staining; 2, moderate staining; 3, strong staining). Five representative fields at ×400 magnification were evaluated. A weighted score was assigned to each case by multiplying the score for the percentage of positive cells by the staining intensity score. Cases with a weighted score <1 were considered negative; otherwise, they were considered positive.24 Positive expression of p-STAT3 was defined as >25% nuclear staining with greater than moderate staining intensity of tumor cells.25
Statistical analysis
Data are expressed as the mean ± standard deviation (SD). Statistical analysis was carried out using the Student’s t-test for unpaired samples and analysis of variance (ANOVA). All the tests were two-tailed, and P<0.05 was considered statistically significant.
Results
CXCL12 blocks apoptosis in A549 through CXCR4
The percentage of A549 cells in the apoptotic phase was 55.67%±1.85% after treatment with CDDP. However, the percentage of apoptotic cells was significantly reduced to 39.28%±0.85% by pretreatment with 100 ng/mL CXCL12 (P<0.05). AMD3100, a CXCR4 antagonist, completely suppressed the anti-apoptotic effect of CXCL12 (48.40±0.45 vs 39.28±0.85, P<0.05; Figure 1). These results indicate that CXCL12 exerts its anti-apoptotic effect on A549 cells through CXCR4.
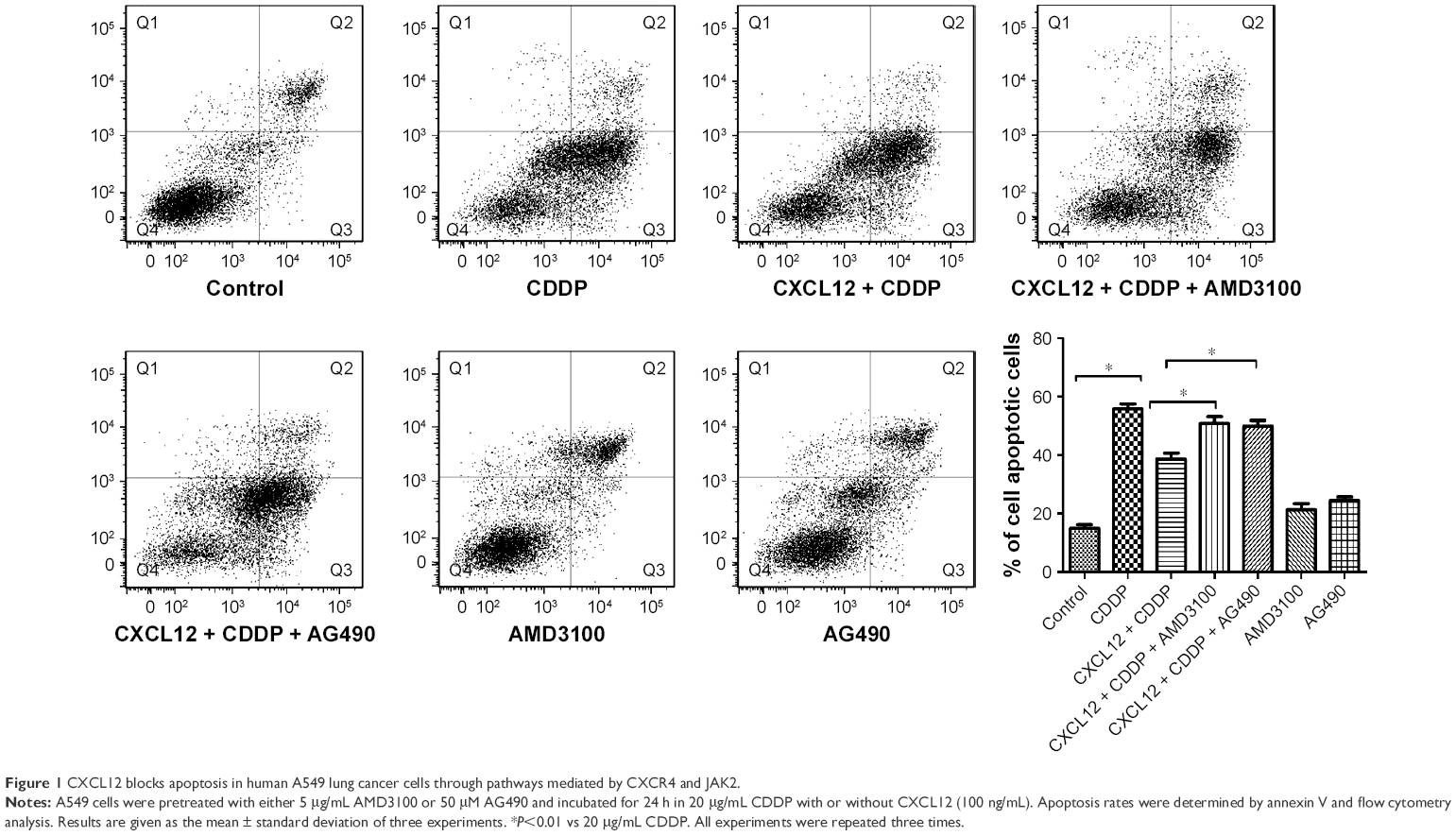 | Figure 1 CXCL12 blocks apoptosis in human A549 lung cancer cells through pathways mediated by CXCR4 and JAK2. |
The JAK2/STAT3 pathway mediates the anti-apoptotic effect of CXCL12 in A549 cells
To further study the mechanisms underlying the anti-apoptotic effect of CXCL12 in A549 cells, we investigated the signaling molecules in pathways downstream of CXCR4. A549 cells were pretreated for 30 min with AG490, a JAK2 inhibitor. Next, A549 cells were stimulated with CXCL12; 12 h after adding 50 μM AG490, the anti-apoptotic effect of CXCL12 was completely reversed (51.57±0.97 vs 39.28±0.85, P<0.05; Figure 1). These data indicated that CXCL12 can decrease cell apoptosis through activation of JAK2/STAT3 that can be reversed by AG490.
Western blot analysis revealed that CXCL12 stimulates p-STAT3 (Serine 727) in A549 cells in a time-dependent manner (Figure 2A). Treatment with 100 ng/mL CXCL12 caused a significant increase in p-STAT3 (Serine 727) at 24 h (Figure 2B). Therefore, these results suggested that CXCL12 exerts its anti-apoptotic effect in A549 cells through JAK2 and subsequent STAT3 activation. To elucidate the molecular mechanism underlying anti-apoptotic induced by CXCL12, the expressions of BCL-2 family members and related proteins, such as BCL-2 and BCL-XL, were determined by Western blot in A549 cells. As shown in Figure 2C, expression levels of BCL-2 and BCL-XL were increased by treatment with CXCL12, while expression levels of BCL-2 and BCL-XL were reduced by treatment with AG490 or AMD3100 in A549 cells (Figure 2C and D).
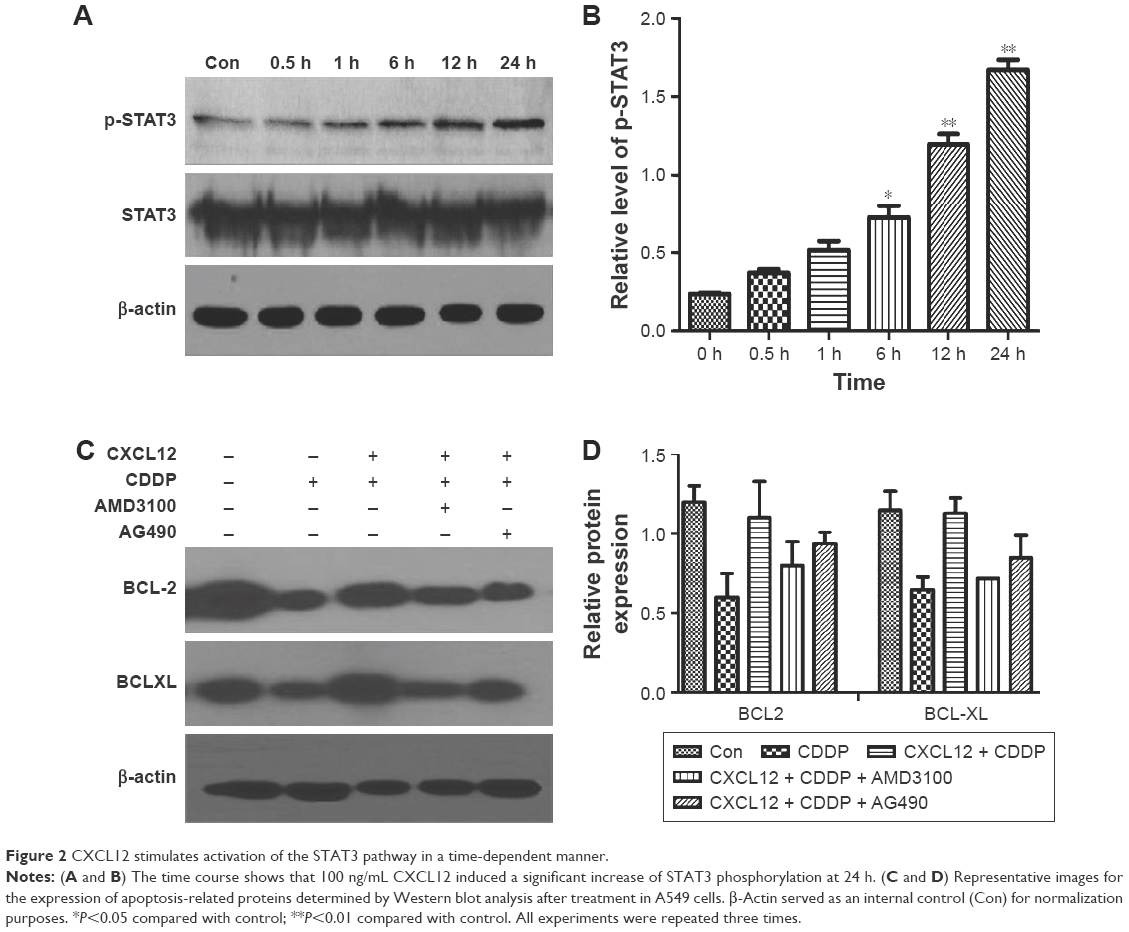 | Figure 2 CXCL12 stimulates activation of the STAT3 pathway in a time-dependent manner. |
CXCL12 stimulates phosphorylation of STAT3 through CXCR4/JAK2
We also found that the CXCR4 inhibitor AMD3100 blocked CXCL12-induced phosphorylation of STAT3 (Figure 3A and B). This indicated that CXCR4 is responsible for CXCL12-induced STAT3 activation. Because JAK2 mediates STAT3 activation, we tested the effects of AG490. JAK2 inhibitors abolished STAT3 phosphorylation in response to CXCL12 (Figure 3A and B). Cells were further subjected to immunofluorescence staining analysis. CDDP-treated cancer cells were pretreated with 100 ng/mL CXCL12, and immunofluorescence studies showed increased levels of p-STAT3 (Serine 727) in the nucleus. Furthermore, addition of AG490 or AMD3100 showed decreased p-STAT3 (Serine 727) in the nucleus (Figure 3C).
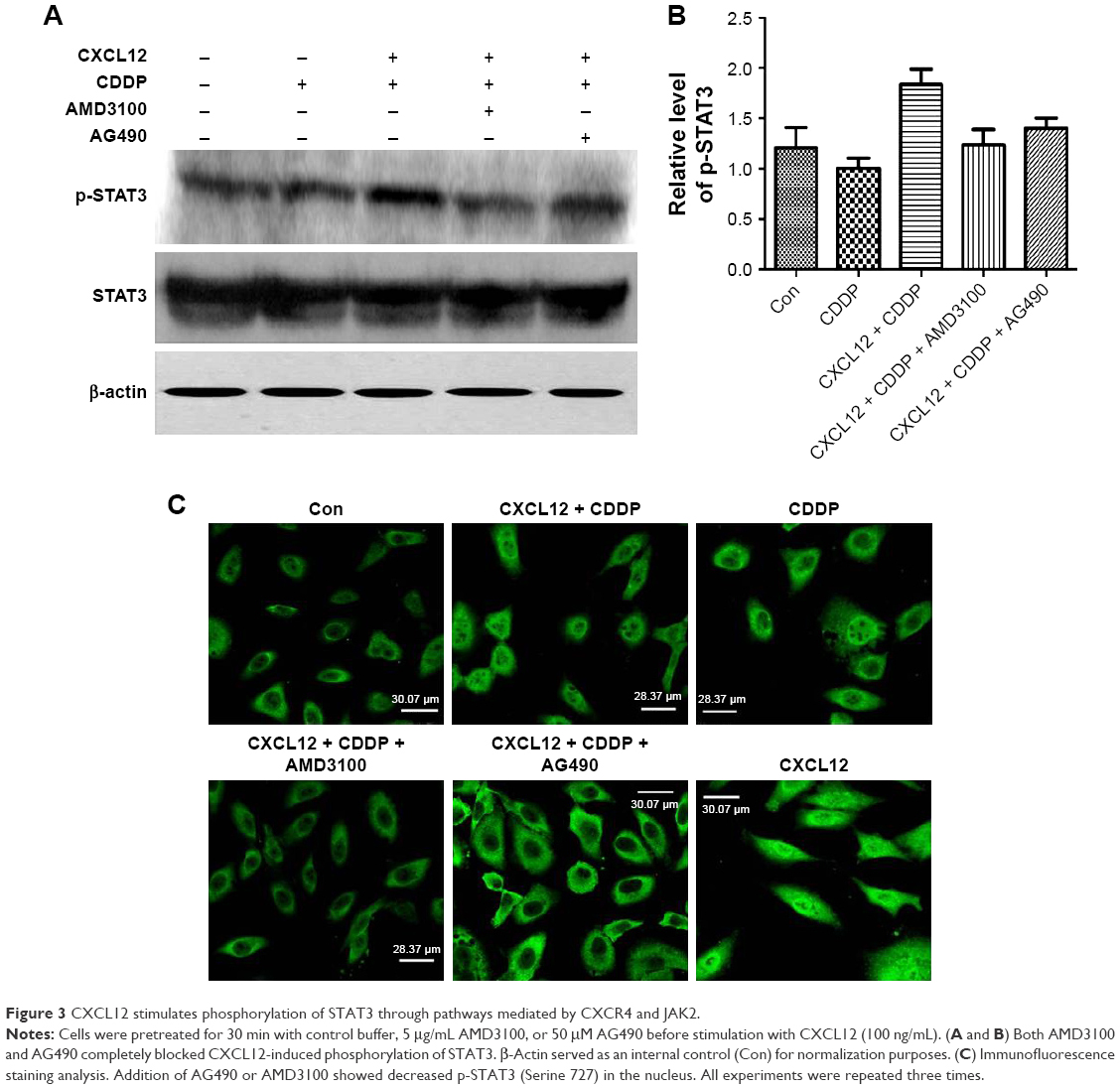 | Figure 3 CXCL12 stimulates phosphorylation of STAT3 through pathways mediated by CXCR4 and JAK2. |
Correlation of CXCL12 and p-STAT3 expression in NSCLC
The expressions of CXCL12 and p-STAT3 in tumor tissues were detected by immunohistochemistry using their respective antibodies and analyzed by an expert oncopathologist. Representative immunohistochemical staining of CXCL12 and p-STAT3 is provided in Figure 4. Expression of CXCL12 and p-STAT3 was detected in 115 (55.3%) and 96 (46.2%) samples, respectively, and the detail is shown in Table 1. CXCL12 expression significantly correlated with tumor classification, lymph node metastasis, stage, and tumor size (P<0.05), and the expression of p-STAT3 was significantly associated with lymph node metastasis (P<0.05). Moreover, a weak association was found between p-STAT3 and tumor classification (P=0.09), and correlation between CXCL12 and p-STAT3 was noted (r=0.136, P=0.027) (Table 2).
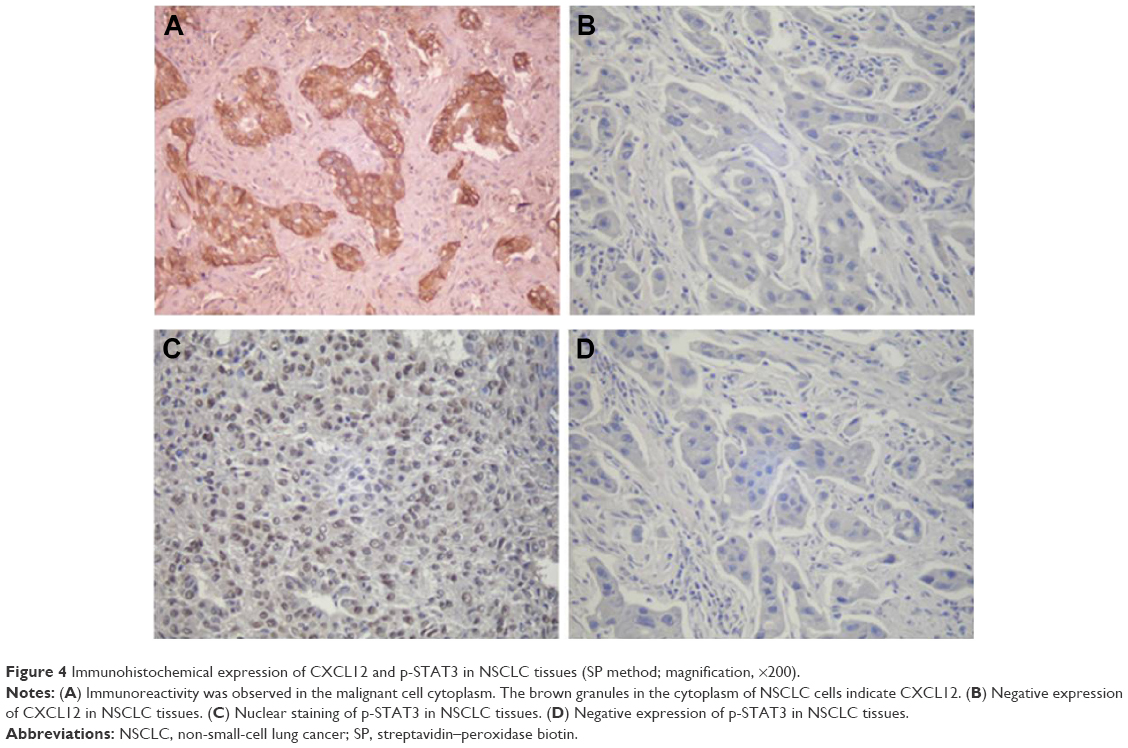 | Figure 4 Immunohistochemical expression of CXCL12 and p-STAT3 in NSCLC tissues (SP method; magnification, ×200). |
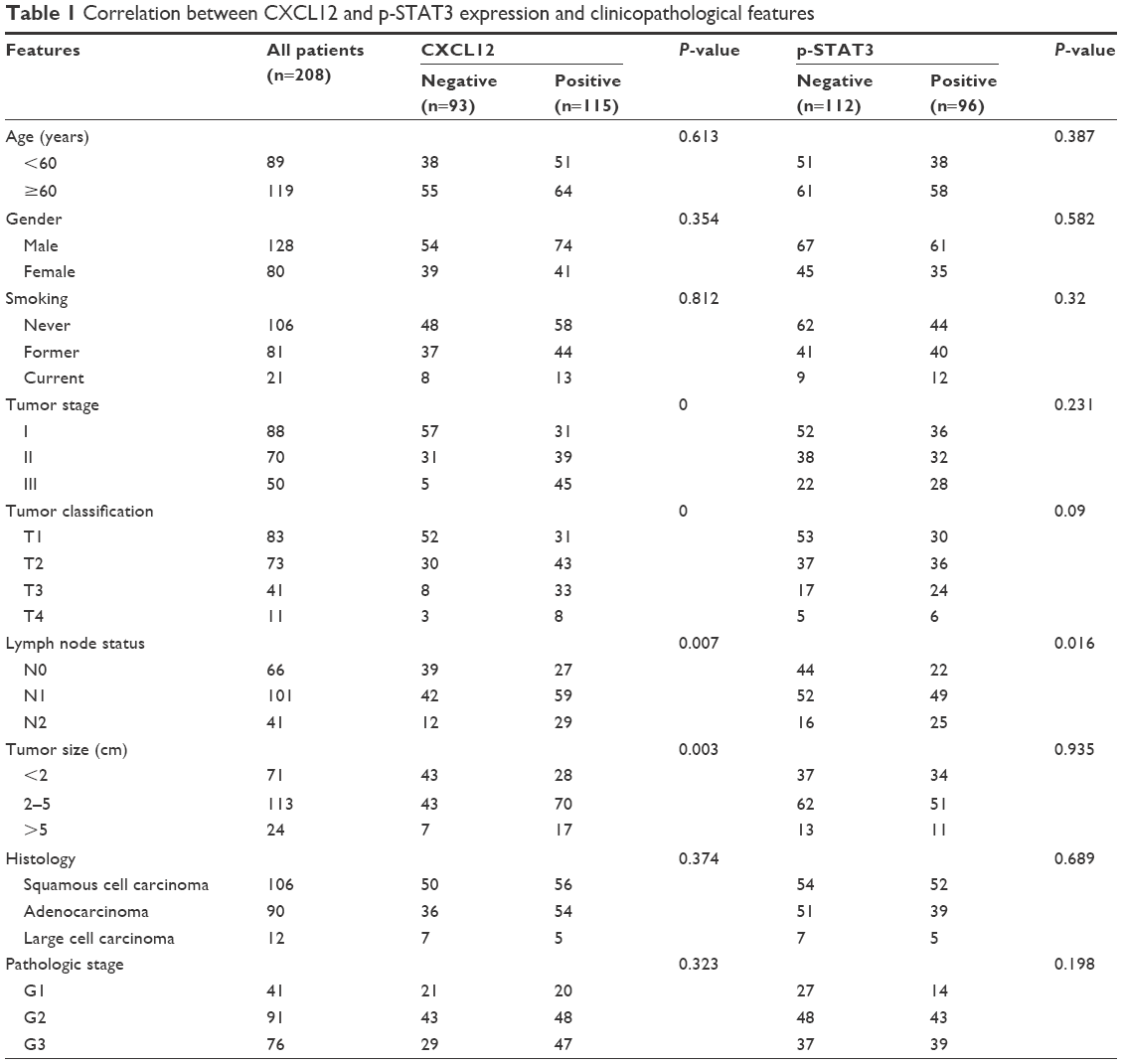 | Table 1 Correlation between CXCL12 and p-STAT3 expression and clinicopathological features |
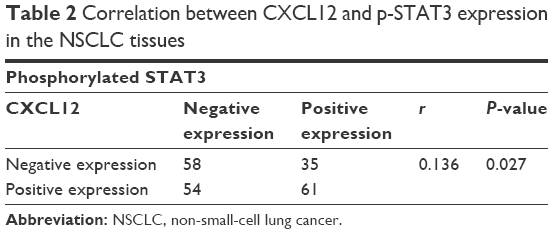 | Table 2 Correlation between CXCL12 and p-STAT3 expression in the NSCLC tissues |
Discussion
To our knowledge, there are few reports about the anti-apoptosis effect of CXCL12 on human lung cancer cells. Additionally, it was found that CXCL12 induced the resistance of the NSCLC cell to cisplatin via JAK2/STAT3 pathway. Moreover, there was a correlation of expression between CXCL12 and p-STAT3 in patients with NSCLC, indicating tumor invasion. Our data might provide new insights into the pathophysiological mechanism of NSCLC and suggest a novel drug target for NSCLC.
Recent studies showed that CXCL12 could act as a key factor for tumor growth, invasion, and angiogenesis via CXCR4. CXCR4 has been found to be overexpressed in many types of solid tumors, including liver cancer, breast cancer and colorectal cancer. Furthermore, expression of CXCR4 plays an important role in lung cancer cells.26 Our previous studies demonstrated that the positive expression rate of CXCR4 in human lung cancer was 56.3% and correlated with tumor classification, lymph node metastasis, stage, and tumor size.27 Therefore, CXCL12/CXCR4 axis could be involved in abnormal lung function.28
Previous studies suggest that the CXCL12/CXCR4 axis could be involved in drug resistance. In chronic myelogenous leukemia, CXCL12 could enhance the resistance of K562 cells to adriamycin (ADM) by increasing the expression of CXCR4, up-regulating the downstream PI3K/Akt pathway, and promoting translocation of nuclear factor kappa B (NF-κB) dimers into nucleus and subsequently decreasing the expression of apoptosis-related proteins in K562 cells.29 CDDP is a commonly used chemotherapeutic drug in human lung adenocarcinoma. However, the role of CXCL12 on CDDP-induced apoptosis in lung cancer is not clear. In this study, we investigated whether activation of the JAK2/STAT3 cascade is involved in CXCL12-mediated suppression of CDDP-induced apoptosis in lung cancer cells by using flow cytometry analysis of PI/annexin V and found that CXCL12 protects A549 cells from CDDP-induced apoptosis. Moreover, our results demonstrate that the anti-apoptotic role of CXCL12 in A549 cells is mediated by CXCR4 (P<0.05).
STAT3 is constitutively activated by numerous cytokines, growth factors, and oncogenic proteins in many types of human cancers, and it participates in the regulation of malignant processes. Moreover, STAT3 activation is involved in chemoresistance in human NSCLC.30 STAT3 is a major downstream target of JAK2 involved in CXCL12-induced proliferation of SCLC and bladder cancer cells.31,32 In our study, treatment with the JAK2 antagonist AG490 completely reversed the anti-apoptotic effect of CXCL12 in A549 cells (P<0.05). We determined the phosphorylation status of STAT3 using Western blot analysis and found that CXCL12 increased p-STAT3 (Serine 727) levels in A549 cells in a time-dependent manner. These results indicate that the JAK2/STAT3 pathway contributes to CXCL12-mediated anti-apoptosis signaling (Figure 5). These results are consistent with previous reports showing the behavior of CXCL12 in drug resistance.33
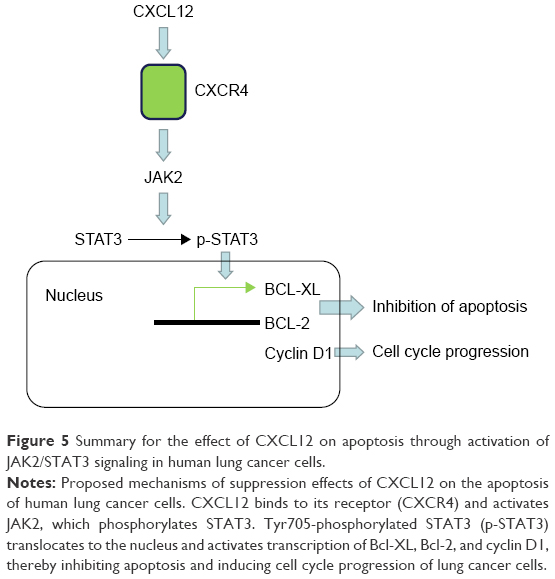 | Figure 5 Summary for the effect of CXCL12 on apoptosis through activation of JAK2/STAT3 signaling in human lung cancer cells. |
A recent study revealed that activation of latent cytoplasmic STAT3 occurred upon phosphorylation, which induces STAT protein dimerization, translocation to the nucleus, and transcriptional regulation of downstream gene targets.34 In this study, we showed that CXCL12 induces STAT3 phosphorylation and nuclear localization in A549 cells. Treatment with AG490 or AMD3100 suppressed CXCL12-induced STAT3 nuclear localization, suggesting involvement of JAK2 and CXCR4 in this process. In the present study, we found that CXCL12 markedly suppressed CDDP-induced apoptosis in lung cancer cells with an increase of BCL-2 and BCL-XL in NSCLC cells. In addition, targeting BCL-2 family proteins is an effective approach to inducing intrinsic apoptosis in cancer cells, which contributes to the cytotoxic therapies of chemotherapeutic drugs in NSCLC patients.
Clinical specimen analysis further confirmed co-expression of CXCL12 and p-STAT3. We observed expression of CXCL12 protein in 55.3% of the 208 NSCLC tissue samples analyzed. In the current study, 46.2% of patients expressed p-STAT3, consistent with previous studies.35 Overexpression of CXCL12 in the NSCLC tumor tissues was significantly correlated with tumor classification, lymph node metastasis, stage, and tumor size. These results suggest that CXCL12 may contribute to tumor progression and angiogenesis. We further identified a connection between CXCL12 and p-STAT3, and our results indicate that CXCL2 may suppress apoptosis through the STAT3 signaling pathway. Together, our results indicate that CXCL12 is an anti-apoptotic molecule in A549 lung cancer cells and that the underlying molecular mechanism for this effect involves CXCR4 and JAK2/STAT3. Moreover, Yu et al36 demonstrated that RNAi targeting of CXCR4 inhibits tumor growth by inducing cell cycle arrest and apoptosis of malignant cells in oral squamous cell carcinoma. Thus, the observations of this study suggest that the clinical application of appropriate combinations of novel CXCR4 inhibitors and conventional chemotherapeutic drugs could be beneficial for treating patients with lung cancer.
However, there are some limitations in our study. For instance, we need to perform animal experiments to verify if the anti-apoptotic mechanism of the CXCL12/CXCR4 axis in response to cisplatin treatment in vitro is the same in vivo. In addition, the small cohort of lung cancer patients in this study was a limitation. Also, we only used one cell line to confirm our conclusion. Moreover, interaction between CXCR4 pathways and PI3K/AKT was identified by a recently published study. These findings suggest a novel mechanism in which CXCR4 signaling is involved in PI3K/AKT pathway repression. In combination with our findings and recently published studies, we will focus future studies on the association of CXCL12 with lung cancer and the related pathway in different cell lines.
Acknowledgments
This work was supported by the Natural Science Foundation of Heilongjiang Province (QC2012C005), the Education Department Foundation of Heilongjiang Province (12521192), and the Health Department Foundation of Heilongjiang Province (2010-115).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. | ||
Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–550. | ||
Wald O, Shapira OM, Izhar U. CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic roles and therapeutic potential. Theranostics. 2013;3(1):26–33. | ||
Franco R, Pirozzi G, Scala S, et al. CXCL12-binding receptors expression in non-small cell lung cancer relates to tumoral microvascular density and CXCR4 positive circulating tumoral cells in lung draining venous blood. Eur J Cardiothorac Surg. 2012;41(2):368–375. | ||
Rodriguez-Lara V, Peña-Mirabal E, Baez-Saldaña R, et al. Estrogen receptor beta and CXCR4/CXCL12 expression: differences by sex and hormonal status in lung adenocarcinoma. Arch Med Res. 2014;45(2):158–169. | ||
Kremer KN, Dudakovic A, McGee-Lawrence ME, et al. Osteoblasts protect AML cells from SDF-1-induced apoptosis. J Cell Biochem. 2014;115(6):1128–1137. | ||
Qin L, Gong C, Chen AM, et al. Peroxisome proliferator-activated receptor γ agonist rosiglitazone inhibits migration and invasion of prostate cancer cells through inhibition of the CXCR4/CXCL12 axis. Mol Med Rep. 2014;10(2):695–700. | ||
Kim J, Takeuchi H, Lam ST, et al. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J Clin Oncol. 2005;23(12):2744–2753. | ||
Maréchal R, Demetter P, Nagy N, et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br J Cancer. 2009;100(9):1444–1451. | ||
Masuda T, Nakashima Y, Ando K, et al. Nuclear expression of chemokine receptor CXCR4 indicates poorer prognosis in gastric cancer. Anticancer Res. 2014;34(11):6397–6403. | ||
Lapteva N, Yang AG, Sanders DE, Strube RW, Chen SY. CXCR4 knockdown by small interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther. 2005;12(1):84–89. | ||
Burger JA, Stewart DJ. CXCR4 chemokine receptor antagonists: perspectives in SCLC. Expert Opin Investig Drugs. 2009;18(4):481–490. | ||
Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Pérez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7(1):3–18. | ||
Chen CL, Loy A, Cen L, et al. Signal transducer and activator of transcription 3 is involved in cell growth and survival of human rhabdomyosarcoma and osteosarcoma cells. BMC Cancer. 2007;7:111. | ||
Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21(55):8404–8413. | ||
Jorvig JE, Chakraborty AL. Zerumbone inhibits growth of hormone refractory prostate cancer cells by inhibiting JAK2/STAT3 pathway and increases paclitaxel sensitivity. Anticancer Drugs. 2015;26(2):160–166. | ||
Siveen KS, Nguyen AH, Lee JH, et al. Negative regulation of signal transducer and activator of transcription-3 signalling cascade by lupeol inhibits growth and induces apoptosis in hepatocellular carcinoma cells. Br J Cancer. 2014;111(7):1327–1337. | ||
Swiatek-Machado K, Mieczkowski J, Ellert-Miklaszewska A, et al. Novel small molecular inhibitors disrupt the JAK/STAT3 and FAK signaling pathways and exhibit a potent antitumor activity in glioma cells. Cancer Biol Ther. 2012;13(8):657–670. | ||
Du W, Hong J, Wang YC, et al. Inhibition of JAK2/STAT3 signalling induces colorectal cancer cell apoptosis via mitochondrial pathway. J Cell Mol Med. 2012;16(8):1878–1888. | ||
Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers (Basel). 2014;6(2):926–957. | ||
Liu X, Xiao Q, Bai X, et al. Activation of STAT3 is involved in malignancy mediated by CXCL12-CXCR4 signaling in human breast cancer. Oncol Rep. 2014;32(6):2760–2768. | ||
Kok LF, Lee MY, Tyan YS, et al. Comparing the scoring mechanisms of p16INK4a immunohistochemistry based on independent nucleic stains and independent cytoplasmic stains in distinguishing between endocervical and endometrial adenocarcinomas in a tissue microarray study. Arch Gynecol Obstet. 2010;281(2):293–300. | ||
Falleni M, Pellegrini C, Marchetti A, et al. Survivin gene expression in early-stage non-small cell lung cancer. J Pathol. 2003;200(5):620–626. | ||
Hatiboglu MA, Kong LY, Wei J, et al. The tumor microenvironment expression of p-STAT3 influences the efficacy of cyclophosphamide with WP1066 in murine melanoma models. Int J Cancer. 2012;131(1):8–17. | ||
Qiu X, Guo S, Wu H, Chen J, Zhou Q. Identification of Wnt pathway, uPA, PAI-1, MT1-MMP, S100A4 and CXCR4 associated with enhanced metastasis of human large cell lung cancer by DNA microarray. Minerva Med. 2012;103(3):151–164. | ||
Wang M, Chen GY, Song HT, Hong X, Yang ZY, Sui GJ. Significance of CXCR4, phosphorylated STAT3 and VEGF-A expression in resected non-small cell lung cancer. Exp Ther Med. 2011;2(3):517–522. | ||
Kamlah F, Hänze J, Arenz A, et al. Comparison of the effects of carbon ion and photon irradiation on the angiogenic response in human lung adenocarcinoma cells. Int J Radiat Oncol Biol Phys. 2011;80(5):1541–1549. | ||
Wang Y, Miao H, Li W, et al. CXCL12/CXCR4 axis confers adriamycin resistance to human chronic myelogenous leukemia and oroxylin A improves the sensitivity of K562/ADM cells. Biochem Pharmacol. 2014;90(3):212–225. | ||
Harada D, Takigawa N, Kiura K. The role of STAT3 in non-small cell lung cancer. Cancers (Basel). 2014;6(2):708–722. | ||
Pfeiffer M, Hartmann TN, Leick M, Catusse J, Schmitt-Graeff A, Burger M. Alternative implication of CXCR4 in JAK2/STAT3 activation in small cell lung cancer. Br J Cancer. 2009;100(12):1949–1956. | ||
Shen HB, Gu ZQ, Jian K, Qi J. CXCR4-mediated Stat3 activation is essential for CXCL12-induced cell invasion in bladder cancer. Tumour Biol. 2013;34(3):1839–1845. | ||
Hoellenriegel J, Zboralski D, Maasch C, et al. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood. 2014;123(3):1032–1039. | ||
Shain KH, Yarde DN, Meads MB, et al. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69(3):1009–1015. | ||
Kim HS, Park YH, Lee J, et al. Clinical impact of phosphorylated signal transducer and activator of transcription 3, epidermal growth factor receptor, p53, and vascular endothelial growth factor receptor 1 expression in resected adenocarcinoma of lung by using tissue microarray. Cancer. 2010;116(3):676–685. | ||
Yu T, Wu Y, Huang Y, et al. RNAi targeting CXCR4 inhibits tumor growth through inducing cell cycle arrest and apoptosis. Mol Ther. 2012;20(2):398–407. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.