")
Back to Journals » Journal of Pain Research » Volume 14
CXCL10 and CXCR3 in the Trigeminal Ganglion Contribute to Trigeminal Neuropathic Pain in Mice
Authors Ju YY, Jiang M, Xu F , Wang D, Ding B, Ma LJ, Wu H
Received 23 October 2020
Accepted for publication 23 December 2020
Published 12 January 2021 Volume 2021:14 Pages 41—51
DOI https://doi.org/10.2147/JPR.S288292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Michael Schatman
Yuan-Yuan Ju,1,2 Ming Jiang,2 Feifei Xu,1,3 Dongqin Wang,1,3 Bixiao Ding,1,3 Ling-Jie Ma,2 Hao Wu1
1Department of Otolaryngology, Head, and Neck Surgery, Affiliated Hospital of Nantong University, Nantong, Jiangsu, People’s Republic of China; 2Institute of Pain Medicine, Institute of Nautical Medicine, Nantong University, Nantong, Jiangsu, People’s Republic of China; 3Medical School of Nantong University, Nantong, Jiangsu, People’s Republic of China
Correspondence: Hao Wu
Department of Otolaryngology, Head, and Neck Surgery, Affiliated Hospital of Nantong University, Xisi Road 20#, Nantong, Jiangsu Province 226001, People’s Republic of China
Email [email protected]
Ling-Jie Ma
Institute of Pain Medicine, Institute of Nautical Medicine, Nantong University, Seyuan Road 9#, Nantong, Jiangsu Province 226019, People’s Republic of China
Email [email protected]
Purpose: Trigeminal neuropathic pain is very common clinically, but effective treatments are lacking. Chemokines and their receptors have been implicated in the pathogenesis of chronic pain. This study explored the role of the chemokine CXCL10 and its receptor, CXCR3, in trigeminal neuropathic pain in mice.
Materials and Methods: Trigeminal neuropathic pain was established by partial infraorbital nerve ligation (pIONL) in wild-type and Cxcr3−/− mice. Facial mechanical allodynia was evaluated by behavioral testing. A lentivirus containing Cxcr3 shRNA (LV-Cxcr3 shRNA) was microinjected into the trigeminal ganglion (TG) to knock down Cxcr3 expression. Quantitative polymerase chain reaction assays and immunofluorescence staining were used to examine Cxcl10/Cxcr3 mRNA expression and protein distribution. Western blotting was performed to examine activation of extracellular signal-regulated kinase (ERK) and AKT in the TG. Intra-TG injection of an AKT inhibitor was performed to examine the role of AKT in trigeminal neuropathic pain.
Results: pIONL induced persistent trigeminal neuropathic pain, which was alleviated in Cxcr3−/− mice. Intra-TG injection of LV-Cxcr3 shRNA attenuated pIONL-induced mechanical allodynia. Furthermore, pIONL increased the expression of CXCR3 and its major ligand, CXCL10, in TG neurons. Intra-TG injection of CXCL10 induced pain hypersensitivity in wild-type mice but not in Cxcr3−/− mice. CXCL10 also induced activation of ERK and AKT in the TG of wild-type mice. Finally, pIONL-induced activation of ERK and AKT was reduced in Cxcr3−/− mice. Intra-TG injection of the AKT inhibitor alleviated pIONL-induced mechanical allodynia in WT mice but not in Cxcr3−/− mice.
Conclusion: CXCL10 acts on CXCR3 to induce ERK and AKT activation in TG neurons and contributes to the maintenance of trigeminal neuropathic pain.
Keywords: CXCR3, CXCL10, trigeminal ganglion, neuropathic pain, mice
Introduction
Trigeminal neuropathic pain is the most common form of refractory craniofacial neuropathic pain following injury to the trigeminal system, including facial trauma, compression, and virus infection.1,2 Trigeminal neuropathic pain has a high prevalence, ranging from 4 to 29 people per 100,000;1 however, it is difficult to treat in clinical practice, and the pathophysiological mechanisms are poorly understood.2
Accumulating evidence indicates that chemokine-mediated neuroinflammation plays an important role in the development and maintenance of chronic pain.3–5 Several chemokines, including CX3CL1, CCL2, CXCL1, CXCL13, and CXCL10, are increased in the spinal cord after tissue inflammation or nerve injury and contribute to inflammatory and neuropathic pain via different forms of neuron-glial interaction.6–10 In addition, recent studies have demonstrated that CCL2, CCL4, CX3CL1, and CXCL12 and their cognate receptors in the dorsal root ganglion (DRG) are involved in neuropathic pain via neuron-to-neuron interactions.11–15 In contrast to the well-studied role of chemokines in the DRG and spinal cord in chronic pain, the involvement of chemokines and their receptors in the trigeminal ganglion (TG) in orofacial neuropathic pain has been less frequently investigated.
The chemokines CXCL9, CXCL10, and CXCL11 belong to the same subfamily of chemokines and bind to CXCR3 to exert functions. CXCR3 is expressed in various cells, including endothelial cells, monocytes, T cells, and dendritic cells.16 Meanwhile, CXCR3 is involved in a variety of human diseases, including chronic inflammation, immune dysfunction, cancer, metastasis, and pruritus.16,17 Recent studies have shown that CXCR3 is expressed in spinal neurons and increased after spinal nerve ligation (SNL)8 or inoculation of cancer cells into the tibia.18 CXCL10 is also increased in spinal astrocytes after SNL.8,19 Perfusion of spinal cord slices with CXCL10 enhances excitatory synaptic transmission of lamina II neurons in the dorsal horn via CXCR3.8,19 Importantly, inhibition of CXCR3 by gene knockout, intraspinal injection of lentivirus expressing shRNA, or intrathecal injection of the antagonists NBI-774330 or AMG487 attenuates neuropathic pain.19,20 However, the role of CXCR3 and CXCL10 in trigeminal neuropathic pain and the involved mechanisms remain unknown.
Chemokine receptors are seven transmembrane G-protein coupled receptors. The binding of chemokines with their cognate receptors leads to activation of intracellular signaling pathways, such as the mitogen-activated protein kinase (MAPK) pathway and the phosphatidyl inositol-3 kinase (PI3K) pathway.5 MAPKs, including extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK) are activated in the DRG and spinal cord by peripheral nerve injury.21 The PI3K/AKT pathway in the central and peripheral nervous system also mediates pain hypersensitivity induced by nerve injury, incision, or inflammation.22,23 Previous studies have demonstrated that CXCR3 activates several intracellular kinases, such as Ras/ERK and PI3K/AKT.24,25 Intrathecal injection of CXCL10 induces rapid ERK activation in the spinal cord.8 However, whether MAPKs and AKT can be activated by CXCL10/CXCR3 in the TG has not been investigated.
In this study, using the partial infraorbital nerve ligation (pIONL) model, we studied the role of CXCL10/CXCR3 in the TG in the pathogenesis of trigeminal neuropathic pain. Our results demonstrated that CXCR3 was activated by CXCL10 and contributed to the maintenance of trigeminal neuropathic pain via activation of ERK and AKT in the TG.
Materials and Methods
Animals and Surgery
ICR and C57BL/6 mice (6–8 weeks, male) were purchased from the Experimental Animal Center of Nantong University. Cxcr3−/− mice were purchased from Jackson Laboratory (stock No. 005796). All animals were free to access food and water and housed in the animal facility at a room temperature of 23 ± 1 °C on a 12:12 h light-dark cycle. All experimental procedures were performed in accordance with the UK animals Scientific Procedures Act (1986) and approved by the Animal Care and Use Committee of Nantong University.
The pIONL surgery was performed as described previously.26,27 In brief, the mice were anesthetized with an anesthetic compound (components: 4.25% chloral hydrate, 0.886% sodium pentobarbital, 14.25% anhydrous alcohol, 33.8% propylene glycol, and 2.12% MgSO4) and placed in a supine position. The oral cavity was opened to locate the left tendon of the masseter muscle on the top wall of the oral cavity. A 1-mm incision was made in front of the tendon to expose the infraorbital nerve (ION), and one-half of the ION was ligated with 6–0 silk suture. For the sham operation, an incision was made on the mucous membrane, and the ION was exposed but not ligated. Facial pain behavior was assessed the day before the surgery and at 3, 7, 10, 14, and 21 days after surgery.
Lentiviral Vector Production and Intra-TG Microinjection
Recombinant lentiviruses containing Cxcr3 shRNA (LV-Cxcr3 shRNA, 5′-CTG AAC TTT GAC AGA ACC T-3′) or negative control shRNA (LV-NC, 5′-TTC TCC GAA CGT GTC ACG T-3′) were produced using the pGCSIL-GFP vector by Shanghai GeneChem. Recombinant CXCL10 (murine) was purchased from PeproTech (Rocky Hills, NJ, USA). AKT inhibitor IV was purchased from Sigma-Aldrich (St Louis, MO, USA).
For intra-TG microinjection, the mice were deeply anesthetized with isoflurane. The head was stabilized with one hand, and the needle was positioned at an approximately 10° angle relative to the midline of the head and was inserted medial (1–2 mm) to the palpated portion of the zygomatic process through the infraorbital foramen, infraorbital canal, and foramen rotundum and reached the medial wall of the fossa where the TG is located.28 LV-Cxcr3 shRNA (2 µL), CXCL10 (100 ng in 5 µL), or AKT inhibitor IV (0.2 µg in 5 µL) was slowly injected.
Facial Pain Behavioral Test
Before assessing the facial pain baseline, the mice were habituated to the behavioral test cage for 30 min every day for 3 days in the behavioral test environment. As previously reported,29,30 two von Frey filaments (0.02 g and 0.16 g) were used to stimulate the whisker pad innervated by the ION, and the response of the mice was recorded. The filaments were applied to the ipsilateral whisker pad three times. The mean nocifensive behavior score of three measurements was calculated according to the following criteria: 0 points, no response; 1 point, exploratory behavior - the mouse detected the von Frey filament; 2 points, slight withdrawal response - the mouse slowly moved its face from the stimulation; 3 points, quick and intense withdrawal response with paw lifting; 4 points, the mouse wiped its face with the forepaw toward the stimulated facial area less than three times; 5 points, the mouse wiped its face with the forepaw toward the stimulated facial area more than three times.29,30
Real-Time Quantitative Polymerase Chain Reaction
Total RNA was extracted from the TG using Trizol reagent (Invitrogen, Carlsbad, CA, USA) as described previously.6 Quantitative polymerase chain reaction (qPCR) analysis was performed using a real-time detection system (RotorGene 6000, Qiagen) with SYBR green I dye detection (Takara, Japan). The following primers were used: Cxcr3 forward, 5′-TAC CTT GAG GTT AGT GAA CGT CA-3′; Cxcr3 reverse, 5′-CGC TCT CGT TTT CCC CAT AAT C-3′; Cxcl10 forward, 5′-TGA ATC CGG AAT CTA AGA CCA TCA A-3′; Cxcl10 reverse, 5′-AGG ACT AGC CAT CCA CTG GGT AAA G-3′; Gapdh forward, 5′-AAA TGG TGA AGG TCG GTG TGA AC-3′; Gapdh reverse, 5′-CAA CAA TCT CCA CTT TGC CAC TG-3′. PCR amplification was performed at 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Gapdh was used as an internal control for normalization. The mRNA levels were calculated using the −ΔΔCt method (2−ΔΔCt).
Immunohistochemistry
For immunofluorescence staining, mice were deeply anesthetized and transcardially perfused with 4% paraformaldehyde. TG sections (15 μm) were cut with a cryostat and processed for immunostaining as previously described.31 The following primary antibodies were used: CXCR3 (rabbit, 1:200, Boster Biological Technology), CXCL10 (goat, 1:100, R&D Systems), β3-tubulin (mouse, 1:500, R&D Systems), GFAP (mouse, 1:5000, Millipore), CD11b (mouse, 1:50, AbD), and IBA-1 (rabbit, 1:3000, Wako). The sections were then incubated with Cy3- or Alexa 488-conjugated secondary antibodies (1:1000, Jackson). For the negative control, the primary antibody was omitted, and the sections were incubated with the secondary antibody only. A Leica SP8 confocal microscope was used to examine the stained sections and capture images.
Western Blotting
The TG tissues were homogenized in a lysis buffer containing protease and phosphatase inhibitors (Sigma-Aldrich). A BCA protein assay was used to determine the protein concentrations. Sodium dodecyl polyacrylamide gel electrophoresis and Western blotting were performed as previously described.27 The following antibodies were used: CXCR3 (rabbit, 1:200, Boster Biological Technology), pERK (rabbit, 1:1000, Cell Signaling), ERK (rabbit, 1:1000, Cell Signaling), pp38 (rabbit, 1:1000, Cell Signaling), p38 (rabbit, 1:1000, Cell Signaling), pJNK (rabbit, 1:1000, Cell Signaling), JNK (rabbit, 1:1000, Cell Signaling), pAKT (rabbit, 1:1000, Cell Signaling), AKT (rabbit, 1:1000, Cell Signaling), and IRDye 800CW donkey anti-rabbit secondary antibody. Images were captured using the Odyssey Imaging System (LI-COR Biosciences).
Quantification and Statistics
All results are presented as the mean ± SEM. The behavioral data were analyzed by two-way repeated measures (RM) ANOVA followed by the Bonferroni test. The qPCR data were analyzed by one-way ANOVA followed by the Bonferroni test. The density of specific bands on Western blots was measured with Image J software (NIH). Differences between two groups were compared using Student’s t-test. GraphPad Prism v5.0 was used for statistical analyses, and P < 0.05 was considered to be significant.
Results
Deletion or Inhibition of Cxcr3 Alleviated pIONL-Induced Mechanical Allodynia
As previously reported,27 pIONL induced persistent mechanical allodynia, which started at3 days and was maintained for more than 21 days (Treatment, F1, 60 = 28.79, P < 0.001; Time, F5, 60 = 12.37, P < 0.001; Interaction, F5, 60 = 3.822, P < 0.01, two-way RM ANOVA, Figure 1A). However, pIONL-induced pain hypersensitivity was significantly attenuated in Cxcr3−/− mice compared with wild-type (WT) mice (Treatment, F1, 80 = 28.31, P < 0.001; Time, F5, 80 = 29.43, P < 0.001; Interaction, F5, 80 = 19.30, P < 0.001, two-way RM ANOVA, Figure 1B). To investigate whether CXCR3 in the TG is involved in trigeminal neuropathic pain, we microinjected a LV-Cxcr3 shRNA or LV-NC into the TG. Behavioral results showed that LV-Cxcr3 shRNA markedly attenuated pIONL-induced mechanical allodynia at day 7, 10, and 14 after pIONL (Treatment, F1, 64 = 28.67, P < 0.001; Time, F4, 64 = 29.96, P < 0.001; Interaction, F4, 64 = 8.574, P < 0.001, two-way RM ANOVA, Figure 1C). qPCR showed that LV-Cxcr3 shRNA significantly reduced the Cxcr3 mRNA level (22 ± 14% of LC-NC, P < 0.01, one-way ANOVA, Figure 1D), confirming the knockdown effect of LV-Cxcr3 shRNA in the TG. Collectively, the results suggest that CXCR3 in the TG is involved in the pathogenesis of trigeminal neuropathic pain.
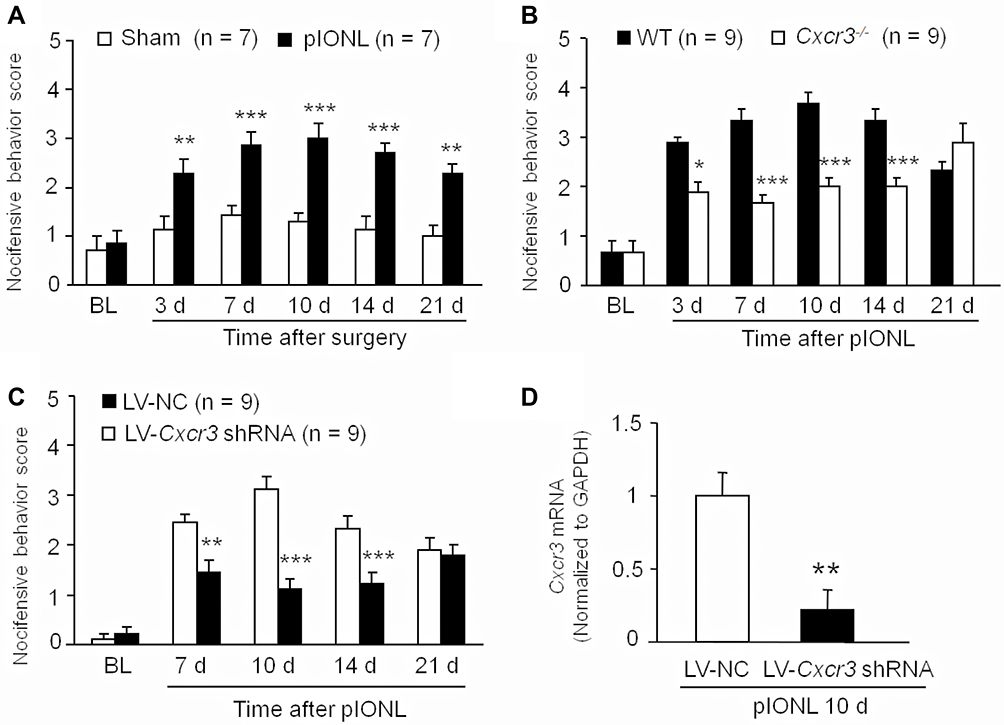 |
Figure 1 Inhibition of CXCR3 in the TG attenuates trigeminal neuropathic pain. (A) pIONL induced mechanical allodynia which started from 3 days and maintained for more than 21 days. ** P < 0.01, *** P < 0.001. Two-way RM ANOVA followed by the Bonferroni test. (B) In Cxcr3−/- mice, the nocifensive behavioral scores were significantly reduced from Day 3 to Day 14. * P < 0.05, *** P < 0.001. Two-way RM ANOVA followed by the Bonferroni test. (C) Intra-TG injection of LV-Cxcr3 shRNA lentivirus alleviated pIONL-induced mechanical allodynia. ** P < 0.01, *** P < 0.001 compared with LV-NC group. Two-way RM ANOVA followed by the Bonferroni test. (D) LV-Cxcr3 shRNA injection reduced Cxcr3 mRNA compared to the LV-NC group. ** P < 0.01, Student’s t-test. |
pIONL Increased CXCR3 Expression in TG Neurons
We then investigated the time-course of Cxcr3 mRNA expression in the TG after pIONL by qPCR. Compared with naïve mice, Cxcr3 mRNA was significantly increased at day 3, 10, and 21 (day 3, P < 0.05; day 10, P < 0.01; day 21, P < 0.05. pIONL vs sham, Figure 2A). Western blots further showed that CXCR3 protein was significantly increased in the TG at 10 days after pIONL (P < 0.01, Student’s t-test, Figure 2B–C). We then examined the CXCR3 distribution in the TG by immunofluorescence staining. CXCR3 exhibited very low expression in the TG of naïve mice (Figure 2D) or sham-operated mice (Figure 2E) but was markedly increased 10 days after pIONL (Figure 2F). To characterize the cellular localization of CXCR3 in the TG, double staining of CXCR3 with the neuronal marker β3-tubulin, satellite cell marker GFAP, or macrophage marker CD11b was performed. The results showed that CXCR3 was largely co-localized with β3-tubulin (Figure 2G), but not with GFAP (Figure 2H) or CD11b (Figure 2I), suggesting neuronal expression of CXCR3 in the TG.
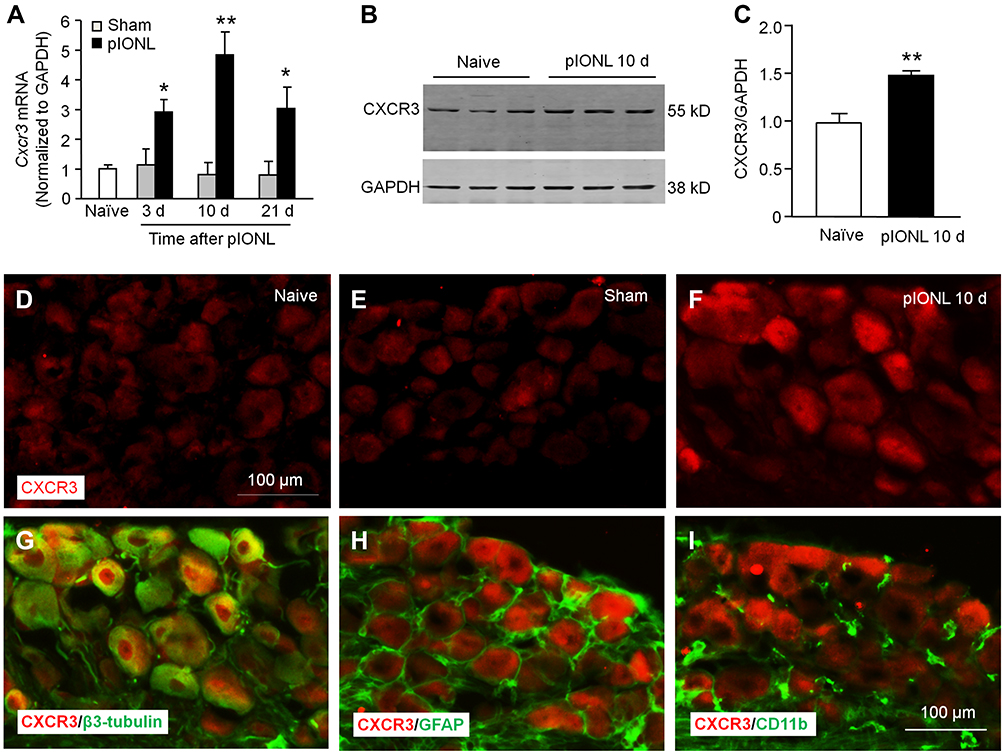 |
Figure 2 pIONL increases CXCR3 expression in TG neurons. (A) The time-course of Cxcr3 mRNA expression in the TG from naïve, sham, and pIONL-operated mice. The mRNA expression of Cxcr3 was increased Day 3, 10, and 21 after pIONL. n = 5 mice per group. * P < 0.05, ** P < 0.01, compared with the corresponding sham group. Student’s t-test. (B–C) Western blot showed that CXCR3 protein was significantly increased 10 days after pIONL. ** P < 0.01, Student’s t-test. (D–F) Representative images of CXCR3 immunofluorescence in the TG from naïve (D), sham (E), and pIONL (F) mice. (G–I) Double immunofluorescence staining shows that CXCR3 was mainly colocalized with the neuronal marker β3-tubulin (G), not with satellite marker GFAP (H) or macrophage marker CD11b (I) in the TG 10 days after pIONL. |
pIONL Increased CXCL10 Expression in TG Neurons
CXCL10 is the major ligand of CXCR3. qPCR showed that Cxcl10 mRNA was increased at day 3 and 10 after pIONL (day 3, P < 0.01; day 10, P < 0.05; day 21, P > 0.05, pIONL vs sham, Figure 3A). We further examined CXCL10 expression and its distribution by immunostaining. No signal was found when the CXCL10 antibody was omitted (Figure 3B). CXCL10 immunoreactivity was low in the TG of naïve mice (Figure 3C) and sham mice (Figure 3D) but increased in the TG of pIONL mice (Figure 3E), confirming the upregulation of CXCL10 after pIONL.
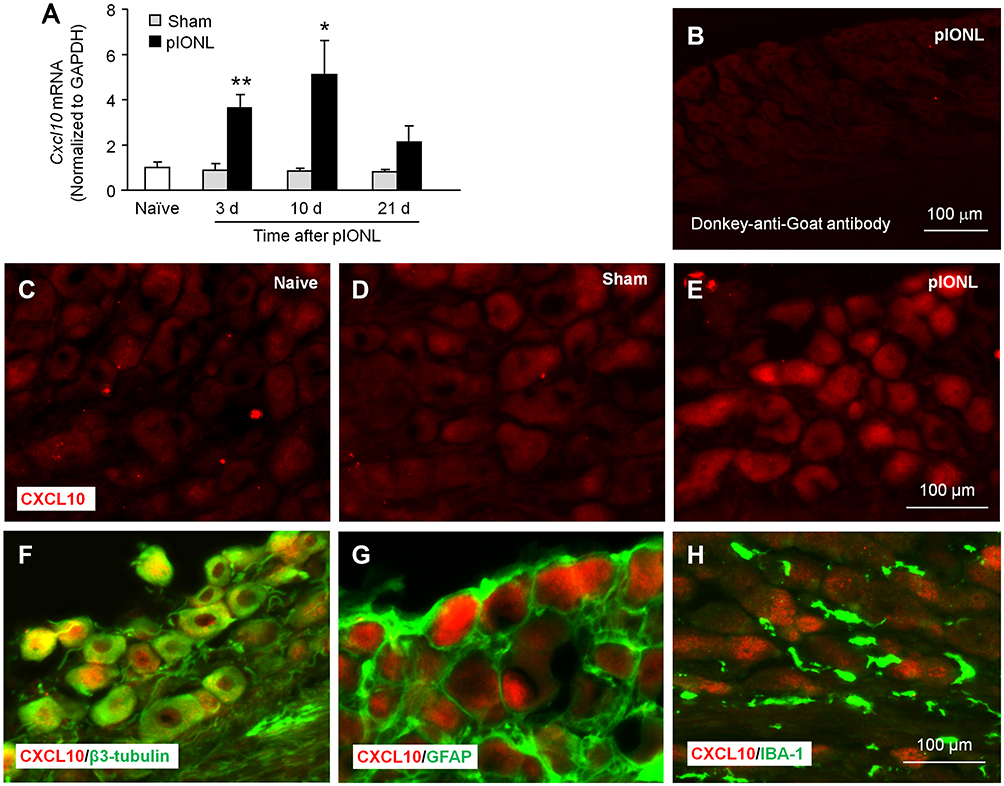 |
Figure 3 pIONL increases CXCL10 expression in TG neurons. (A) The time-course of Cxcl10 mRNA expression in the TG in naïve, sham- and SNL-operated mice. n = 5–6 mice per group. *P < 0.05, **P < 0.01 compared with the corresponding sham group. Student’s t-test. (B) The staining of TG sections with secondary antibody only. (C–E) Fluorescence immunostaining of CXCL10 in the TG of naïve (C), sham (D), and pIONL (E) mice. (F–H) Double immunostaining of CXCL10 with β3-tubulin, GFAP, and IBA-1. CXCL10 was colocalized with β3-tubulin (F), not with GFAP (G) or IBA-1 (H). |
To examine the distribution of CXCL10 in the TG, we performed double staining for CXCL10 and β3-tubulin, GFAP, or IBA-1 (macrophage marker). The immunostaining results showed that CXCL10 was colocalized with β3-tubulin (Figure 3F) but not with GFAP (Figure 3G) or IBA-1 (Figure 3H), indicating the predominant expression of CXCL10 in TG neurons.
CXCL10 Was Sufficient to Induce Pain Hypersensitivity and ERK/AKT Activation in the TG
To examine whether CXCL10 is sufficient to induce pain, we performed intra-TG injection of different doses of CXCL10. Behavioral data showed that 10 ng of CXCL10 did not affect the nociceptive behavioral score. A higher dose (100 ng) increased the nociceptive score at 3 h and 6 h post-injection (Treatment, F2, 63 = 12.88, P < 0.001; Time, F3, 63 = 17.58, P < 0.001; Interaction, F6, 63 = 5.397, P < 0.001, two-way RM ANOVA, Figure 4A). Pain hypersensitivity was diminished 24 h after CXCL10 (100 ng) injection. In contrast, the nocifensive behavioral scores in Cxcr3−/− mice after CXCL10 injection were much lower than those in WT mice (Treatment, F1, 48 = 26.91, P < 0.001; Time, F3, 48 = 30.13, P < 0.001; Interaction, F3,48 = 13.57, P < 0.001, two-way RM ANOVA, Figure 4B). The data suggest that CXCL10 in the TG is sufficient to induce mechanical allodynia via CXCR3.
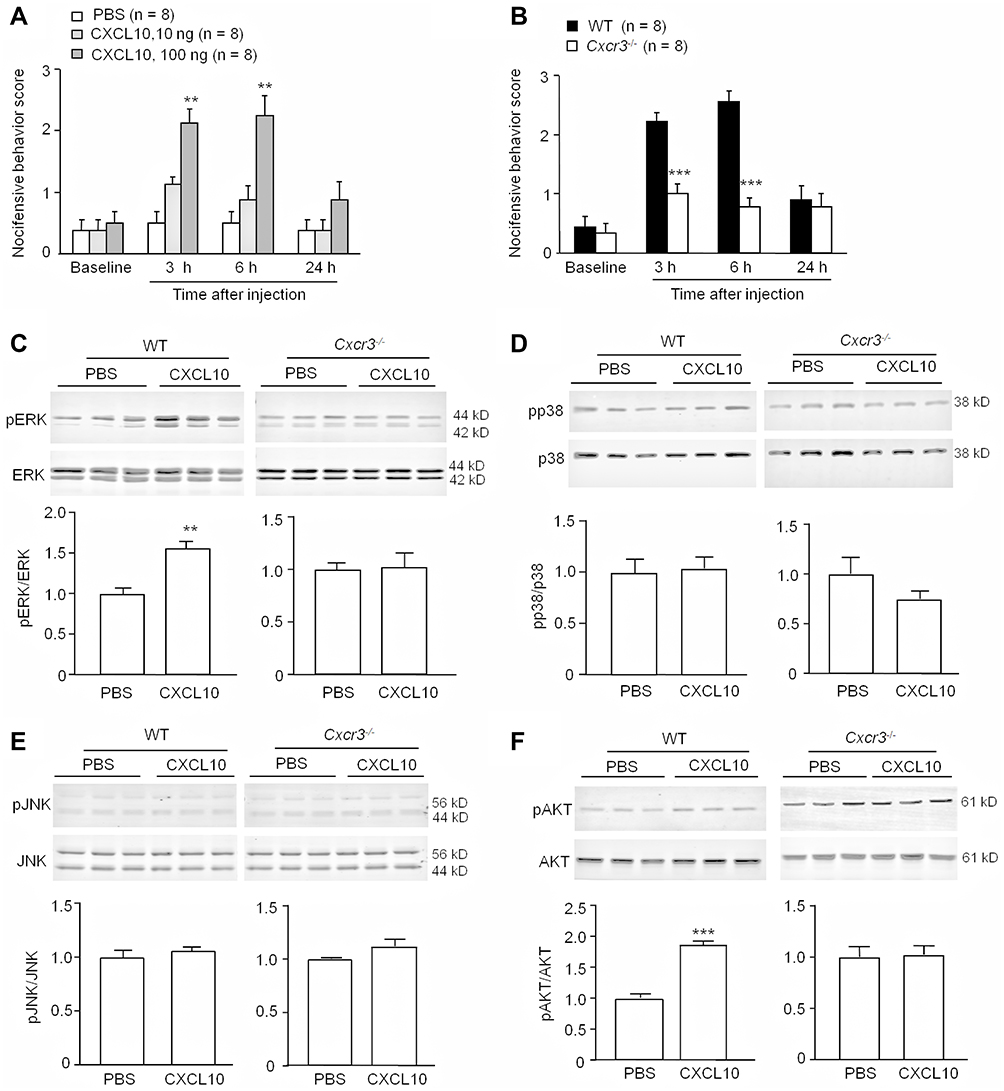 |
Figure 4 CXCL10 induces mechanical allodynia and increases ERK and Akt activation via CXCR3. (A and B) Intra-TG injection of CXCL10 (100 ng) induced mechanical allodynia in WT mice (A), which was reduced in Cxcr3−/- mice (B). ** P < 0.01, *** P < 0.001, compared with PBS group or WT group. Two-way RM ANOVA followed by the Bonferroni test. (C) Intra-TG injection of CXCL10 (100 ng) increased pERK level in WT mice but not in Cxcr3−/- mice. n=3/group. ** P < 0.01 compared with PBS group. Student’s t-test. (D, E) Intra-TG injection of CXCL10 did not affect pp38 (D) or pJNK (E) level in either WT or Cxcr3−/- mice. n=3/group. (F) Intra-TG injection of CXCL10 induced the activation of AKT in WT mice, but not in Cxcr3−/- mice. n=3/group. *** P < 0.001, compared with PBS group. Student’s t-test. |
MAPKs and AKT are important kinases for mediating chronic pain.21 To determine whether CXCL10 can activate these kinases, we performed intra-TG injection of CXCL10 (100 ng) and examined the phosphorylation (p) of MAPKs (ERK, p38, and JNK) and AKT. Three hours after CXCL10 injection, the pERK level was significantly increased in the TG of WT mice (P < 0.01, Student’s t-test) but not in Cxcr3−/− mice (P > 0.05, Student’s t-test, Figure 4C). However, CXCL10 did not affect the activation of p38 or JNK in either WT or Cxcr3−/− mice (P > 0.05, Student’s t-test, Figure 4D-E). In contrast, CXCL10 increased the pAKT level in WT mice (P < 0.05, Student’s t-test) but not in Cxcr3−/− mice (P > 0.05, Student’s t-test, Figure 4F). These data indicate that CXCL10 activates ERK and AKT via CXCR3.
pIONL Induced ERK and AKT Activation via CXCR3
We then compared pERK and pAKT levels in the TG 10 days after pIONL in WT and Cxcr3−/− mice. As shown in Figure 5A, the pERK level was significantly lower in Cxcr3−/− mice than in WT mice (P < 0.05, Student’s t-test). In addition, the pAKT level in the TG of Cxcr3−/− mice was also lower than that in WT mice (P < 0.05, Student’s t-test, Figure 5B). These results suggest that CXCR3 plays a role in pIONL-induced ERK and AKT activation in the TG. Inhibition of ERK has been reported to attenuate pIONL-induced pain hypersensitivity.26 To further examine the role of AKT in the maintenance of pIONL, we performed intra-TG injection of an AKT inhibitor at 10 days after pIONL. The behavioral data showed that AKT inhibitor IV significantly attenuated the nocifensive behavioral score at 1 h, 3 h, and 6 h post-injection (Treatment, F1, 50 = 75.29 = 89.80, P < 0.001; Interaction, F5,50=15.70, P < 0.001, Figure 5C). However, intra-TG injection of AKT inhibitor IV in Cxcr3−/− mice did not affect mechanical allodynia (Figure 5D). These data indicate that AKT is involved in pIONL-induced trigeminal neuropathic pain.
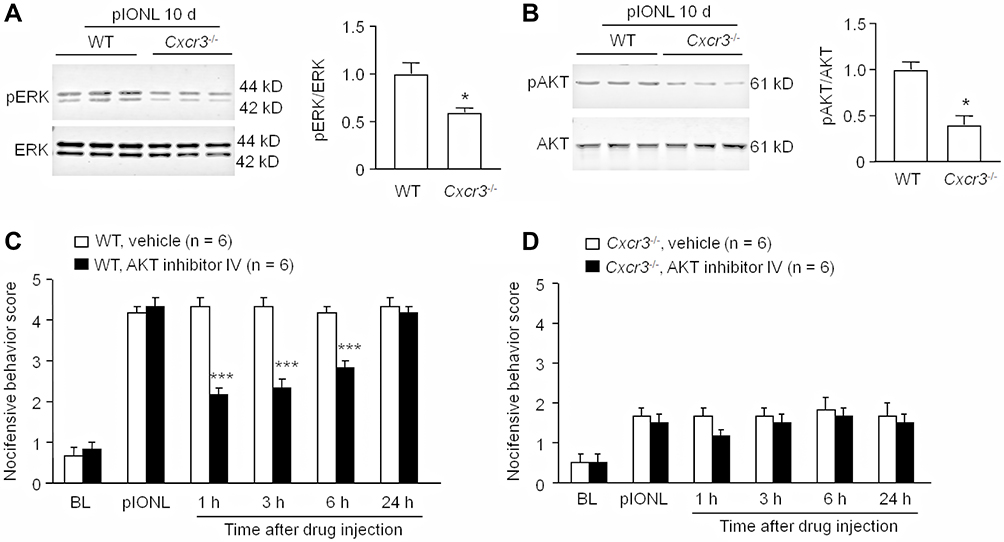 |
Figure 5 pIONL induces ERK and AKT activation via CXCR3. (A) pIONL-induced ERK activation was reduced in Cxcr3−/- mice than that in WT mice. * P < 0.05. Student’s t-test. (B) AKT activation was reduced in Cxcr3−/- mice than that in WT mice. * P < 0.05. Student’s t-test. (C) Intra-TG injection of AKT inhibitor IV in WT mice attenuated pIONL-induced mechanical allodynia at 1 h, 3 h, and 6 h. *** P < 0.001, compared to vehicle. Two-way RM ANOVA followed by the Bonferroni test. (D) Intra-TG injection of AKT inhibitor IV in Cxcr3−/- mice did not affect the mechanical allodynia. Two-way RM ANOVA followed by the Bonferroni test. |
Discussion
In the present study, we found that global deletion or specific inhibition of Cxcr3 in the TG alleviated pIONL-induced mechanical allodynia in the orofacial area. In addition, pIONL persistently increased CXCR3 and CXCL10 expression in TG neurons. Intra-TG injection of CXCL10 induced pain hypersensitivity and activated ERK and AKT via CXCR3. Finally, pIONL-induced activation of ERK and AKT was reduced in Cxcr3−/− mice, and AKT inhibition attenuated pIONL-induced orofacial mechanical allodynia. Thus, our data revealed an important role of CXCL10/CXCR3 in the TG in regulating intracellular kinase activation and trigeminal neuropathic pain.
The Involvement of CXCR3 in the Pathogenesis of Trigeminal Neuropathic Pain
CXCR3 is constitutively expressed in neurons of the neocortex, hippocampus, striatum, and spinal cord, where it plays a vital role in the pathogenesis of a variety of neuroinflammatory and neurodegenerative diseases, including Alzheimer’s disease, bipolar disorder, multiple sclerosis, and chronic itch.32–35 Recent studies have shown that CXCR3 expression is increased in neurons, microglia, and astrocytes in the spinal cord in animal models of neuropathic pain and cancer pain.8,18 CXCR3 is also increased in DRG neurons in a bone cancer pain model in rats or a chronic constriction injury (CCI) model in mice.18,20 Here, low CXCR3 expression levels were observed in TG neurons of naïve mice, and these levels were persistently increased after pIONL. Although the mechanisms underlying CXCR3 upregulation were not investigated in this study, a previous report showed that decreased methylation of the Cxcr3 promoter and increased binding of the transcriptional factor C/EBPα with Cxcr3 caused upregulation of Cxcr3 in the spinal dorsal horn after SNL,8 indicating epigenetic regulation of CXCR3 under neuropathic pain conditions.
Previous studies have shown that intrathecal injection of the CXCR3-specific antagonist NBI-74330 alleviates chronic pain symptoms induced by SNL, CCI, or bone cancer.8,18,20 Cxcr3−/− mice show normal basal pain thresholds and motor function,8 but neuropathic pain induced by SNL is alleviated from day 3 to day 28.8 Intraspinal injection of LV-Cxcr3 shRNA to specifically inhibit CXCR3 in the spinal cord attenuates SNL-induced mechanical allodynia and heat hyperalgesia,8 indicating the role of spinal CXCR3 in the maintenance of neuropathic pain. In the present study, pIONL-induced mechanical allodynia was attenuated in Cxcr3−/− mice in the first 2 weeks, but not on day 21, suggesting that CXCR3 may not contribute to the maintenance of the late phase of trigeminal neuropathic pain. Lentivirus-expressing shRNA can rapidly and persistently reduce mRNA expression.6,7 Our data also showed that specific knockdown of Cxcr3 in the TG attenuated pIONL-induced mechanical allodynia from 7 to 14 days, supporting the role of CXCR3 in the TG in the maintenance of neuropathic pain. Given that CXCR3 in the spinal dorsal horn plays an important role in mediating neuropathic pain, and the medullary dorsal horn (MDH) is important in transmitting and mediating noxious signals from the orofacial area to the brain, we cannot exclude the possible role of CXCR3 in the MDH in mediating trigeminal neuropathic pain.
CXCL10/CXCR3 Induces Trigeminal Neuropathic Pain via Activation of ERK and AKT in the TG
A microarray assay showed that CXCL10 is a highly upregulated chemokine in the spinal cord after SNL.8 In addition, CXCL10 is constitutively expressed in dorsal horn neurons but induced in astrocytes after SNL; however, CXCR3 is expressed and upregulated in neurons. Thus, CXCL10 and CXCR3 mediate neuropathic pain via astrocyte-neuron interactions in the spinal cord.8 In contrast, CXCL10 and CXCR3 were both expressed in TG neurons after pIONL, suggesting that CXCL10/CXCR3 signaling may have an autocrine/paracrine function within the TG. Recent studies have demonstrated that chemokine pairs such as CCL2/CCR2 and CXCL13/CXCR5 are also expressed in TG neurons and contribute to the development and maintenance of trigeminal neuropathic pain.26,36 Notably, deletion of Cxcr5 attenuated pIONL-induced mechanical allodynia for more than 21 days,26 suggesting that different chemokines may have distinct roles in various phases of neuropathic pain.
CXCL10/CXCR3 induced ERK activation in dorsal horn neurons.8 Ligation of the ION has been shown to induce activation of ERK and p38, but not JNK, in the TG.26,27 ERK activation in TG neurons is also induced by migraine or lingual nerve crush.26,37,38 Our results showed that intra-TG injection of CXCL10 induced CXCR3-dependent activation of ERK, but not p38 or JNK, in WT mice, suggesting that ERK is a downstream kinase of CXCL10/CXCR3. Inhibition of ERK activation by PD98059 (MEK inhibitor) has been shown to attenuate pIONL-induced mechanical allodynia and lingual nerve crush-induced pain hypersensitivity.26,38 PD98059 also decreases pIONL-induced upregulation of TNF-α and IL-1β in the TG.26 Furthermore, intra-TG injection of the TNF-α inhibitor etanercept or the IL-1β inhibitor diacerein attenuates pIONL-induced mechanical allodynia for up to 6 h.26 In addition, activated ERK increases expression of the NaV1.8 sodium channel and regulates TRPV1 activity in peripheral sensory neurons.39,40 Proinflammatory cytokines upregulate NaV1.7 and NaV1.6 expression, further contributing to peripheral sensitization and neuropathic pain.41,42 Therefore, activation of CXCL10/CXCR3 may increase neuroinflammation and further regulate the excitability of TG neurons.
AKT is an important downstream target of PI3K and regulates multifarious cellular processes, including cell survival and death.43 Evidence has shown that the PI3K/AKT signaling pathway is required for central spinal sensitization after nerve injury.44 Inhibition of PI3K/AKT signaling pathway activation exerts analgesic effects in neuropathic pain models.45,46 Our present results showed that CXCL10/CXCR3 induced AKT activation, and inhibition of AKT attenuated pIONL-induced mechanical allodynia, indicating that CXCL10/CXCR3 contributes to trigeminal neuropathic pain via activation of ERK and AKT.
In summary, we explored the role of CXCR3 signaling in the TG in trigeminal neuropathic pain. Our results demonstrated that CXCR3 is activated by CXCL10 and induces downstream ERK/AKT activation, which may increase neuroinflammation and neuronal excitability and further contribute to the maintenance of neuropathic pain. Because of the important role of CXCL10/CXCR3 in the spinal cord in central sensitization, the CXCL10/CXCR3 pathway may be a promising target for neuropathic pain treatment.
Conclusion
CXCL10 acts on CXCR3 to induce ERK and AKT activation in TG neurons and contributes to the maintenance of trigeminal neuropathic pain.
Acknowledgment
We thank Lisa Kreiner, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This study was supported by funds from the Scientific Research Project of “226 Engineering” of Nantong Municipality (2020) to H. Wu and the Science and Technology Planning Project of Nantong Municipality, China (JC2020038) to L.-J. Ma.
Disclosure
All authors report no conflicts of interest for this work.
References
1. Al-Quliti KW. Update on neuropathic pain treatment for trigeminal neuralgia: the pharmacological and surgical options. Neurosciences. 2015;20(2):107. doi:10.17712/nsj.2015.2.20140501
2. Bista P, Imlach WL. Pathological mechanisms and therapeutic targets for trigeminal neuropathic pain. Medicines. 2019;6(3):91. doi:10.3390/medicines6030091
3. Zhang Z-J, Jiang B-C, Gao Y-J. Chemokines in neuron–glial cell interaction and pathogenesis of neuropathic pain. Cell Mol Life Sci. 2017;74(18):3275–3291. doi:10.1007/s00018-017-2513-1
4. Xie R-G, Gao Y-J, Park C-K, et al. Spinal CCL2 promotes central sensitization, long-term potentiation, and inflammatory pain via CCR2: further insights into molecular, synaptic, and cellular mechanisms. Neurosci Bull. 2018;34(1):13–21. doi:10.1007/s12264-017-0106-5
5. Gao Y-J, Ji -R-R. Chemokines, neuronal–glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126(1):56–68. doi:10.1016/j.pharmthera.2010.01.002
6. Jiang B-C, Cao D-L, Zhang X, et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 2016;126(2):745–761. doi:10.1172/JCI81950
7. Zhang Z-J, Cao D-L, Zhang X, Ji -R-R, Gao Y-J. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. PAIN®. 2013;154(10):2185–2197. doi:10.1016/j.pain.2013.07.002
8. Jiang B-C, He L-N, Wu X-B, et al. Promoted interaction of C/EBPα with demethylated Cxcr3 gene promoter contributes to neuropathic pain in mice. J Neurosci. 2017;37(3):685–700. doi:10.1523/JNEUROSCI.2262-16.2016
9. Gao Y-J, Zhang L, Samad OA, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29(13):4096–4108. doi:10.1523/JNEUROSCI.3623-08.2009
10. Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. 2005;6(7):434–438. doi:10.1016/j.jpain.2005.02.001
11. Kiguchi N, Kobayashi Y, Saika F, Sakaguchi H, Maeda T, Kishioka S. Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain. 2015;156(4):684–693. doi:10.1097/j.pain.0000000000000097
12. Saika F, Kiguchi N, Kobayashi Y, Fukazawa Y, Kishioka S. CC‐chemokine ligand 4/macrophage inflammatory protein‐1β participates in the induction of neuropathic pain after peripheral nerve injury. Eur j Pain. 2012;16(9):1271–1280. doi:10.1002/j.1532-2149.2012.00146.x
13. Jeon S-M, Lee K-M, Cho H-J. Expression of monocyte chemoattractant protein-1 in rat dorsal root ganglia and spinal cord in experimental models of neuropathic pain. Brain Res. 2009;1251:103–111. doi:10.1016/j.brainres.2008.11.046
14. Yu Y, Huang X, Di Y, Qu L, Fan N. Effect of CXCL12/CXCR4 signaling on neuropathic pain after chronic compression of dorsal root ganglion. Sci Rep. 2017;7(1):1–11. doi:10.1038/s41598-016-0028-x
15. Holmes FE, Arnott N, Vanderplank P, et al. Intra‐neural administration of fractalkine attenuates neuropathic pain‐related behaviour. J Neurochem. 2008;106(2):640–649. doi:10.1111/j.1471-4159.2008.05419.x
16. Koper OM, Kamińska J, Sawicki K, Kemona H. CXCL9, CXCL10, CXCL11, and their receptor (CXCR3) in neuroinflammation and neurodegeneration. Advan Clin Exp Med. 2018;27(6):849–856. doi:10.17219/acem/68846
17. Tokunaga R, Zhang W, Naseem M, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation–a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47. doi:10.1016/j.ctrv.2017.11.007
18. Guan X-H, Fu Q-C, Shi D, et al. Activation of spinal chemokine receptor CXCR3 mediates bone cancer pain through an Akt-ERK crosstalk pathway in rats. Exp Neurol. 2015;263:39–49. doi:10.1016/j.expneurol.2014.09.019
19. Wu X-B, He L-N, Jiang B-C, et al. Spinal CXCL9 and CXCL11 are not involved in neuropathic pain despite an upregulation in the spinal cord following spinal nerve injury. Mol Pain. 2018;14:1744806918777401. doi:10.1177/1744806918777401
20. Chen Y, Yin D, Fan B, et al. Chemokine CXCL10/CXCR3 signaling contributes to neuropathic pain in spinal cord and dorsal root ganglia after chronic constriction injury in rats. Neurosci Lett. 2019;694:20–28. doi:10.1016/j.neulet.2018.11.021
21. Ji -R-R, Gereau RW
22. Chen S-P, Zhou Y-Q, Liu D-Q, et al. PI3K/Akt pathway: a potential therapeutic target for chronic pain. Curr Pharm Des. 2017;23(12):1860–1868. doi:10.2174/1381612823666170210150147
23. Xu J-T, Tu H-Y, Xin W-J, Liu X-G, Zhang G-H, Zhai C-H. Activation of phosphatidylinositol 3-kinase and protein kinase B/Akt in dorsal root ganglia and spinal cord contributes to the neuropathic pain induced by spinal nerve ligation in rats. Exp Neurol. 2007;206(2):269–279. doi:10.1016/j.expneurol.2007.05.029
24. Zhang C, Li Z, Xu L, et al. CXCL9/10/11, a regulator of PD-L1 expression in gastric cancer. BMC Cancer. 2018;18(1):462. doi:10.1186/s12885-018-4384-8
25. Bonacchi A, Romagnani P, Romanelli RG, et al. Signal transduction by the chemokine receptor CXCR3 activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276(13):9945–9954. doi:10.1074/jbc.M010303200
26. Zhang Q, Cao D-L, Zhang Z-J, Jiang B-C, Gao Y-J. Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J Neuroinflammation. 2016;13(1):1–13. doi:10.1186/s12974-016-0652-1
27. Zhang Q, Zhu M-D, Cao D-L, Bai X-Q, Gao Y-J, Wu X-B. Chemokine CXCL13 activates p38 MAPK in the trigeminal ganglion after infraorbital nerve injury. Inflammation. 2017;40(3):762–769. doi:10.1007/s10753-017-0520-x
28. Neubert JK, Mannes AJ, Keller J, Wexel M, Iadarola MJ, Caudle RM. Peripheral targeting of the trigeminal ganglion via the infraorbital foramen as a therapeutic strategy. Brain Res Protocols. 2005;15(3):119–126. doi:10.1016/j.brainresprot.2005.05.003
29. Zhang Y, Chen Y, Liedtke W, Wang F. Lack of evidence for ectopic sprouting of genetically labeled Aβ touch afferents in inflammatory and neuropathic trigeminal pain. Mol Pain. 2015;11:
30. Kernisant M, Gear RW, Jasmin L, Vit J-P, Ohara PT. Chronic constriction injury of the infraorbital nerve in the rat using modified syringe needle. J Neurosci Methods. 2008;172(1):43–47. doi:10.1016/j.jneumeth.2008.04.013
31. Zhang Z-J, Guo J-S, Li -S-S, et al. TLR8 and its endogenous ligand miR-21 contribute to neuropathic pain in murine DRG. J Exp Med. 2018;215(12):3019–3037. doi:10.1084/jem.20180800
32. Jing P-B, Cao D-L, Li -S-S, et al. Chemokine receptor CXCR3 in the spinal cord contributes to chronic itch in mice. Neurosci Bull. 2018;34(1):54–63. doi:10.1007/s12264-017-0128-z
33. Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1α and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci, 96 12 6873–6878 doi: 10.1073/pnas.96.12.6873.
34. Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. J Neuroimmunol. 2000;108(1–2):227–235. doi:10.1016/S0165-5728(00)00285-X
35. Israelsson C, Bengtsson H, Lobell A, et al. Appearance of Cxcl10‐expressing cell clusters is common for traumatic brain injury and neurodegenerative disorders. Eur J Neurosci. 2010;31(5):852–863. doi:10.1111/j.1460-9568.2010.07105.x
36. Zhang Z-J, Dong Y-L, Lu Y, Cao S, Zhao Z-Q, Gao Y-J. Chemokine CCL2 and its receptor CCR2 in the medullary dorsal horn are involved in trigeminal neuropathic pain. J Neuroinflammation. 2012;9(1):1–13. doi:10.1186/1742-2094-9-136
37. Hawkins JL, Moore N, Miley D, Durham P. Secondary traumatic stress increases expression of proteins implicated in peripheral and central sensitization of trigeminal neurons. Brain Res. 2018;1687:162–172. doi:10.1016/j.brainres.2018.03.003
38. Mikuzuki L, Saito H, Katagiri A, et al. Phenotypic change in trigeminal ganglion neurons associated with satellite cell activation via extracellular signal‐regulated kinase phosphorylation is involved in lingual neuropathic pain. Eur J Neurosci. 2017;46(6):2190–2202. doi:10.1111/ejn.13667
39. Hudmon A, Choi J-S, Tyrrell L, et al. Phosphorylation of sodium channel Nav1. 8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28(12):3190–3201. doi:10.1523/JNEUROSCI.4403-07.2008
40. Zhuang Z-Y, Xu H, Clapham DE, Ji -R-R. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24(38):8300–8309. doi:10.1523/JNEUROSCI.2893-04.2004
41. Ding -H-H, Zhang S-B, Lv -Y-Y, et al. TNF-α/STAT3 pathway epigenetically upregulates Nav1. 6 expression in DRG and contributes to neuropathic pain induced by L5-VRT. J Neuroinflammation. 2019;16(1):29. doi:10.1186/s12974-019-1421-8
42. Tamura R, Nemoto T, Maruta T, et al. Up-regulation of NaV1. 7 sodium channels expression by tumor necrosis factor-α in cultured bovine adrenal chromaffin cells and rat dorsal root ganglion neurons. Anesth Analg. 2014;118(2):318–324. doi:10.1213/ANE.0000000000000085
43. Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114(2):368–378. doi:10.1161/CIRCRESAHA.113.300536
44. Liu W, Lv Y, Ren F. PI3K/Akt pathway is required for spinal central sensitization in neuropathic pain. Cell Mol Neurobiol. 2018;38(3):747–755. doi:10.1007/s10571-017-0541-x
45. Jiang S-P, Zhang Z-D, Kang L-M, Wang Q-H, Zhang L, Chen H-P. Celecoxib reverts oxaliplatin-induced neuropathic pain through inhibiting PI3K/Akt2 pathway in the mouse dorsal root ganglion. Exp Neurol. 2016;275:11–16. doi:10.1016/j.expneurol.2015.11.001
46. Zhang W, Suo M, Yu G, Zhang M. Antinociceptive and anti-inflammatory effects of cryptotanshinone through PI3K/Akt signaling pathway in a rat model of neuropathic pain. Chem Biol Interact. 2019;305:127–133. doi:10.1016/j.cbi.2019.03.016
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.