")
Back to Archived Journals » International Journal of Clinical Transfusion Medicine » Volume 5
Current strategies and future directions for the prevention of transfusion-transmitted cytomegalovirus
Authors Harmon CM, Cooling LL
Received 21 January 2017
Accepted for publication 23 March 2017
Published 27 April 2017 Volume 2017:5 Pages 49—59
DOI https://doi.org/10.2147/IJCTM.S115669
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Cees Th. Smit Sibinga
Charles M Harmon, Laura L Cooling
Department of Pathology, University of Michigan, Ann Arbor, MI, USA
Abstract: Cytomegalovirus (CMV) is a pervasive DNA virus that infects a significant portion of individuals worldwide, and may be transmitted through the transfusion of blood products. Although CMV infection is of little consequence in immunocompetent individuals, patients with an impaired immune system are at risk of significant morbidity and mortality. Unlike other blood-borne infectious agents, it is impractical to defer all CMV-positive individuals from blood donation as this would exclude a substantial number of otherwise eligible donors. Other methods such as transfusion of CMV-seronegative and leukoreduced blood products must be employed to prevent the transmission of CMV to at-risk patients. In this study, the widespread use of current strategies for the prevention of transfusion-transmitted CMV (TT-CMV) infection and the evidence to support these methods in various at-risk groups were reviewed. In addition, emerging pathogen inactivation technologies that have the potential to eliminate TT-CMV were also discussed.
Keywords: blood transfusion, cytomegalovirus, leukoreduction, pathogen inactivation, hematopoietic stem cell transplantation, very low birth weight infants
Introduction
Human cytomegalovirus (CMV) is an ubiquitous, enveloped, double-stranded DNA virus belonging to the Herpesviridae family. Because CMV is limited to humans and does not survive long in the environment, primary infection requires close or intimate contact with the body fluids of a CMV-positive individual.1 Like other herpesviruses, primary CMV infection is followed by a lifelong latent infection of the host. For CMV, CD34+ hematopoietic progenitor cells serve as an important reservoir for latent virus.2 CMV is also present in an estimated 0.004%–0.12% of peripheral blood monocytes, with each infected cell containing 2–13 viral genomes.3–7 Reactivation of latent CMV infection can occur later in life as a result of immunosuppression due to disease, stem cell transplantation, or solid organ transplantation. In most populations, CMV has a high seroprevalence rate, ranging from ~30%–40% in Western populations to >90% in the Middle East, Asia, and Africa (Figure 1).8–11 In the USA, the CMV-seropositive rate after the age of 6 years is 58.9% and increases 1% per year, reaching 90% by the age of 80 years.12
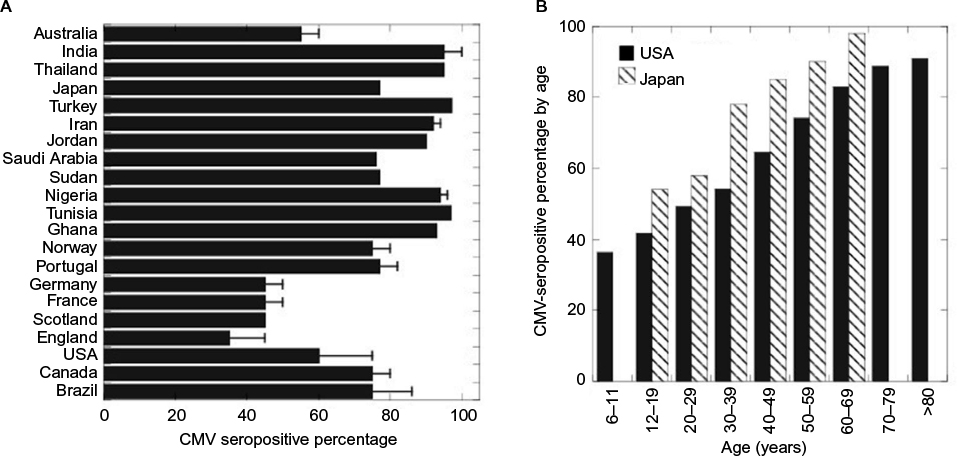 | Figure 1 Incidence of CMV seropositivity (A) by country and (B) by age. Notes: Data from Manicklal et al,8 Shaiegan et al,9 Das et al,10 Furui et al,11 and Staras et al.12 Abbreviation: CMV, cytomegalovirus. |
In immunocompetent hosts, CMV infection is usually asymptomatic, although some individuals experience an infectious mononucleosis-like syndrome. However, in immunocompromised individuals, this infection can lead to a life-threatening illness including pneumonia, enterocolitis, retinitis, encephalitis, and hepatitis.13–16 Groups at risk of severe disease from either primary CMV infection or reactivation of latent CMV infection include allogeneic hematopoietic stem cell transplant (HSCT) recipients, solid organ transplant recipients, patients with hematologic malignancies, fetuses, and premature infants with very low birth weight (VLBW).
Transmission of CMV infection through blood transfusion was first recognized >50 years ago in patients who had developed an infectious mononucleosis-like syndrome after transfusion.17–20 Historically, the incidence of transfusion-transmitted CMV (TT-CMV) infection has been reported to be as high as 13%–37% in immunocompromised patients.21 Since then, the prevention of TT-CMV infection has become an important priority, especially in the abovementioned high-risk patient groups. Unlike other viruses that can be transmitted by blood transfusion, it is impractical to defer all CMV-positive blood donors given the high rate of seroprevalence in nearly all populations. Therefore, other strategies to prevent TT-CMV infection must be considered which do not require permanent removal of a large portion of otherwise eligible blood donors from the donor pool.
CMV seroconversion in blood donors
Before discussing the strategies that are used to reduce the risk of TT-CMV infection, it is first important to review the natural course of CMV infection in immunocompetent healthy blood donors. Until recently, the window period between initial CMV infection and seroconversion was estimated to be 6–8 weeks.22 However, data by Ziemann et al indicated that only ~25% of donors have evidence of viremia, with low levels of CMV DNA (<30 IU/mL) detectable in their plasma during the window period (Figure 2).23 The counterintuitive reality is that the highest concentration of circulating CMV and the highest risk of TT-CMV infection occur shortly after seroconversion, but before neutralizing, high-avidity antibodies are formed. Seroconversion is initially characterized by the production of non-neutralizing IgM and low-avidity IgG antibodies directed against nonstructural immediate early CMV antigens and early CMV antigens. This is accompanied by an increase in the quantity of circulating cell-free CMV DNA in plasma (up to 1,600 IU/mL) in 80% of newly seroconverted donors.23 In another study, Ziemann et al detected CMV DNA in plasma for a median of 107 days, which was considerably longer than the leukocyte fraction (median =77 days).24 Clearance of detectable CMV DNA from both white blood cells (WBCs) and plasma required a median of 137 days (range, 0–269 days). In a related study, Gerna et al found detectable viral DNA in whole blood samples of immunocompetent subjects for a median of 85 days, whereas prolonged viremia (median =235 days) was observed in immunocompromised solid organ transplant recipients.25
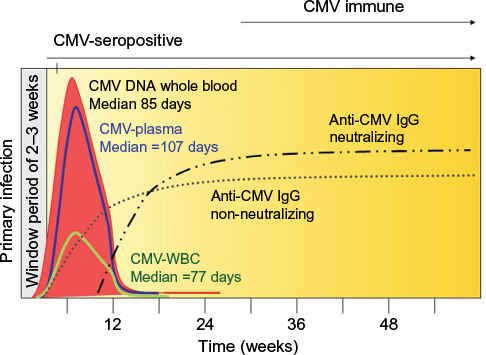 | Figure 2 Relative time course of primary CMV infection in immunocompetent individuals. After a window period of approximately 2–3 weeks, non-neutralizing IgM (not shown) and IgG antibodies to CMV are formed, accompanied by an increase in CMV DNA in peripheral blood. Subsequent clearance of CMV DNA coincides with development of neutralizing anti-CMV IgG. Data from Ziemann et al,23 Ziemann et al,24 and Gerna et al.25 Abbreviations: CMV, cytomegalovirus; WBC, white blood cell; IgG, immunoglobulin G; IgM, Immunoglobulin M. |
The clearance of CMV from blood coincides with the development of neutralizing antibodies. In general, these antibodies are directed against specific glycoproteins of the viral lipid envelope. The best studied of these are antibodies against glycoprotein B (gB), a viral envelope protein that is necessary for virus entry and cell-to-cell spread.24,26 On average, neutralizing antibodies to gB1 and gB2 are detected ~11 days and 62 days after seroconversion, respectively.24 With time, there is formation of high-avidity neutralizing antibodies (mean =141 days after seroconversion) and a loss of IgM antibodies (mean =220 days after seroconversion) with complete clearance of detectable CMV DNA (Figure 2).24
After resolution of primary infection and clearance of plasma-free virus, latent infection is established during which CMV in the peripheral blood is confined to WBCs, predominantly monocytes. However, the number of latently infected monocytes is usually at levels that are below the limit of reliable detection. In healthy long-term seropositive donors, it seems that there are rare episodes of reactivation during which CMV DNA is detectable in plasma. Of 7,303 long-term seropositive donors tested, only one donor (0.01%) was found to have detectable CMV DNA in plasma.27
Strategies for the reduction of TT-CMV infection
The most obvious strategy for the prevention of TT-CMV infection is transfusion of blood products from only CMV-seronegative donors to at-risk patients. Although this has proven to be an effective strategy since the 1980s, it carries a significant disadvantage of excluding a large portion of the donor population with the resultant difficulties in maintaining an adequate supply of CMV-seronegative blood products.28–33 In addition, the use of CMV-seronegative blood cannot completely eliminate the risk of TT-CMV infection, given the real possibility of window period donations.34 However, the risk of TT-CMV infection as a result of window period donations seems to be low, especially given the previously discussed finding that the peak levels of CMV DNA in primary infection occur only after seroconversion. Indeed, a recent study that included polymerase chain reaction (PCR)-based testing of plasma samples from 18,405 individual donors identified only one CMV DNA-positive window-period donor.35 In another study that included testing of 13,236 blood samples from CMV-seronegative donors, only two of them (0.02%) were positive for plasma CMV DNA.27
The other common method for decreasing the risk of TT-CMV infection is leukoreduction (LR) of blood products. As CMV infection is restricted to small numbers of WBCs during latent infection, the removal of these WBCs significantly decreases the risk of TT-CMV infection.36–38 Leukofiltration using filters composed of cellulose acetate, polyurethane, polycarbonate, or polyester is the most common method for leukodepletion of whole blood-derived components.39 The pore size of these filters is designed to impede the passage of leukocytes, but allow the passage of erythrocytes and platelets. Although bedside leukofiltration was commonly used in the past, quality control is extremely difficult with this method. In addition, delay in LR until the time of transfusion allows for the accumulation of inflammatory cytokines during storage, which are implicated in febrile nonhemolytic transfusion reactions.40 Hence, prestorage leukofiltration has become the widely accepted standard. For blood products collected by apheresis, various techniques are employed, including counterflow centrifugation, fluid particle bed separation, elutriation, and flow path geometry, in order to yield a collection product that is leukodepleted.39
The minimum viral load required for TT-CMV infection has not been well established in humans, as most estimates were based on mouse models. By extrapolating findings from experiments using murine CMV in mice, Roback et al estimated that WBC doses of <4×105 per kg would be unlikely to cause CMV infection in humans who are transfused with CMV-positive blood products.41 In a 70 kg adult, this amount would correspond to ~2.8×107 WBCs, which is a much greater quantity than the American Association of Blood Banks standard of <5×106 WBCs in leukoreduced units. Current methods of prestorage LR are highly reliable, with only rare (0.01%–0.11% of units) instances of failure.42,43 Because LR also decreases the incidence of febrile nonhemolytic transfusion reactions and human leukocyte antigen alloimmunization, nowadays, the vast majority of blood products are leukoreduced, and universal LR of blood products has been implemented in many countries.44,45
Although LR is very effective in removing leukocyte-associated CMV, it cannot remove CMV present in plasma.11 As a result, it is theoretically possible that a newly infected blood donor could transmit CMV despite effective LR. Although there is some evidence that cell-free CMV DNA in plasma may be infectious, this is not certain and a minimum infectious dose has not been characterized.42,46 This potential risk has led to proposals that LR-only blood products be collected from donors who have been seropositive for at least 1 year or that the donors also be screened for CMV by PCR.34,45,47 However, it remains to be determined whether the latter would be a cost-effective way to further decrease the risk of TT-CMV infection. For example, a recent study detected CMV DNA in the plasma of only 0.03% of blood donors.35 Similarly, Ziemann et al reported reproducibly detectable CMV DNA in the plasma of only 0.05% of unselected donors.27 Only a small minority of this small percentage of CMV DNA-positive blood products would be transfused to patients at risk of disease from TT-CMV infection.
Prevention of TT-CMV infection in specific patient groups
Over the past four decades, there have been many relatively small studies examining the prevention of TT-CMV infection in various at-risk patient groups. The most well-studied groups have been HSCT patients and VLBW infants, with fewer studies on other patient populations. The evidence supporting the various methods for the prevention of TT-CMV infection in each of these groups is discussed in the following sections (summarized in Table 1).
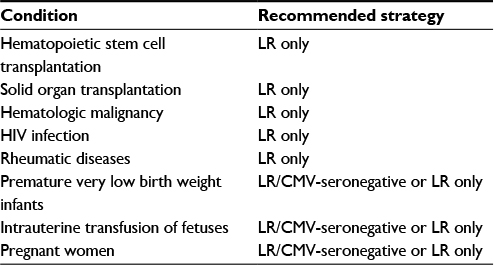 | Table 1 Recommendations for the prevention of TT-CMV infection in patient group Abbreviations: CMV, cytomegalovirus; LR/CMV, leukoreduction and CMV-seronegative; LR, leukoreduction; TT-CMV, transfusion-transmitted CMV. |
HSCT patients
CMV infection remains an important cause of morbidity and mortality in allogeneic HSCT recipients, with a much lower incidence of CMV disease in autologous HSCT patients.48 Historically, the mortality rate from CMV pneumonia in this patient population was as high as 85%.31 Although the mortality rate from CMV pneumonia has decreased with the introduction of ganciclovir and intravenous immunoglobulin therapy, it remains an important complication, with mortality rates remaining at >50%.48 Both allogeneic and autologous CMV-seropositive recipients (R+) are at the highest risk of CMV disease primarily due to the reactivation of a latent CMV infection.31 The risk of CMV reactivation and infection is even higher in CMV-seropositive allogeneic recipients transplanted with a CMV-seronegative donor (D−/R+).49,50
CMV-seronegative recipients transplanted with a CMV-seropositive donor (D+/R−) are also at high risk of CMV infection. However, the mechanism in these patients is primary infection from the CMV-seropositive donor. The estimated risk of CMV transmission in this scenario is ~20%–30%.13
CMV-seronegative recipients who receive a transplant from a CMV-seronegative donor (D−/R−) are at the lowest risk of CMV infection and disease. However, CMV disease may occur in D−/R− patients in the setting of primary CMV infection. TT-CMV was the historical major route of infection in D−/R− patients with a risk of CMV infection ranging from 28% to 57% with standard, unfiltered CMV random blood products.51 However, in recent decades, this rate of TT-CMV infection has been markedly decreased with changes in transfusion practice.
The effectiveness of blood products from CMV-seronegative donors in preventing TT-CMV infection in D−/R− HSCT patients was demonstrated by Bowden et al in a study that included 57 D−/R− patients who were randomized to receive either CMV-seronegative blood products or CMV-untested blood products.31 Only one of 32 (3.1%) patients who received CMV-seronegative blood products developed CMV infection compared with eight of 25 (32%) patients transfused with CMV-untested blood products (P<0.007). These results were confirmed in subsequent studies.32 However, in the late 1980s and early 1990s, several observational studies showed markedly reduced or absent TT-CMV infection in HSCT recipients who transfused with LR-only, CMV-untested blood products.52–54 These findings were followed by a seminal study conducted by Bowden et al that included 502 D−/R− HSCT patients who were prospectively randomized to receive CMV-seronegative or LR-only blood products.51 The primary endpoint of this study was chosen to be CMV infection and/or disease (clinical symptoms with tissue biopsy evidence of CMV infection) that developed >21 days after transplantation: CMV infections occurring <21 days after transplantation could arise from exposures other than transfusion and were excluded. No significant difference in the probability of CMV infection (1.3% versus 2.4%, P=1.0) or disease (0% versus 1.2%, P=0.25) was found at >21 days between patients who received CMV-seronegative and LR-only blood products, respectively. There was also no significant difference in CMV infection (1.4% versus 2.4%, P=0.5) between the two groups in a secondary analysis that included CMV infection that occurred at ≤21 days after transplantation. However, there was a small statistically significant lower probability of clinical CMV disease in the CMV-seronegative arm (0% versus 2.4%, P=0.03) than in the LR arm.
Although the overall results supported the equivalence of LR blood products and CMV-seronegative blood products in the prevention of TT-CMV infection in HSCT patients, some residual concern persisted about the safety of LR blood products, given the slightly higher probability of CMV disease in the LR arm of this study. These concerns persisted after a subsequent study by Nichols et al found a higher incidence of TT-CMV infection among HSCT patients who received any LR blood product than among those who received only CMV-seronegative products (5.6% versus 2.0%, P=0.05).55 However, this was a nonrandomized observational study in contrast to that of Bowden et al, with several confounding factors, including the fact that all the patients in the LR group of this study received both LR and CMV-seronegative blood products. In particular, the patients in the LR group only received LR/CMV random blood products as an alternative when CMV-seronegative blood products were unavailable. In addition, the blood product requirements of the patients in the LR group were significantly higher than that of the group that received only CMV-seronegative units (mean =35.1 units versus 12.4 units, P=<0.001), thereby resulting in more donor exposures. A recent meta-analysis of the combined data from the studies by Bowden et al and Nichols et al and one additional smaller study found no statistically significant difference in the risk of TT-CMV infection between LR-only blood products and CMV-seronegative blood products (relative risk [RR] =2.18 for LR blood products; 95% confidence interval [CI] =0.96–4.98).56
Although these older studies are helpful in supporting the equivalence of LR-only blood products to CMV-seronegative blood products, it is important to remember that many of these studies were carried out by using bedside leukofiltration as a means of LR, a method that is not nearly as consistent as the prestorage leukofiltration that is currently in widespread use. In addition, the LR filters used in many of the older studies were not as effective as the current-generation filters. Therefore, LR may be more effective now than in the past as a means of prevention of TT-CMV infection. Accordingly, two studies published within the past 6 years each reported no cases of TT-CMV infection among a combined group of 123 D−/R− HSCT patients with a combined transfusion of ~8,000 LR/CMV-untested (LR-only) blood products.33,57 Based on these data, the risk of TT-CMV infection with CMV-untested blood products that undergo prestorage leukodepletion with current-generation LR filters seems to be very low. These findings contrast sharply with those of Wu et al who reported three cases of possible TT-CMV infection among 46 previously CMV-seronegative patients transfused with leukoreduced blood products that had not been prospectively screened for CMV status.58 Although each of these three patients had received blood products from CMV-seropositive donors, none of the implicated donors’ available serum samples were positive for CMV DNA (no cellular samples were available for testing of WBC-associated CMV DNA). In addition, two of these three patients with presumed TT-CMV infection had reported risk factors for community-acquired CMV infection unrelated to transfusion, such as sick contacts and living with young children. Therefore, while TT-CMV infection seems to be a possibility, these cases of putative TT-CMV infection may be viewed with some degree of skepticism.
In an era when LR is the standard for nearly all blood products, the most pertinent question today is whether there is any clinical benefit to requiring both LR and CMV-seronegative products (“belt-and-suspenders” method) for patients at risk of TT-CMV infection. A relatively recent observational study published in 2013 by Kekre et al examined the risk of CMV viremia in 89 D−/R− HSCT patients who were transfused with LR/CMV-seronegative blood products compared with 77 patients who were transfused with LR-only blood products.59 There was no statistically significant difference in CMV infection between these two cohorts (P=0.6244). In fact, CMV viremia was detected in three patients (3.37%) in the LR/CMV-seronegative group compared with only one patient (1.30%) in the LR-only group. An older study by Ljungman et al that included blood products that were leukoreduced by bedside leukofiltration also found no statistically significant difference in the rate of CMV infection in HSCT patients who were transfused with LR/CMV-seronegative blood products (9.1% versus 12.2% LR/CMV-untested).60 Meta-analysis of the combined data from both of these studies similarly found no significant difference in the risk of TT-CMV infection in patients transfused using the “belt-and-suspenders” approach versus those transfused with LR-only blood products (RR =1.02; 95% CI =0.33–3.18).56 Although these studies are few in number with a high degree of uncertainty in the calculated RRs, there is no current evidence that LR/CMV-seronegative blood products are superior to LR-only blood products in the prevention of TT-CMV infection in HSCT patients. Until there is evidence to the contrary, the “belt-and-suspenders” approach in HSCT patients is not warranted, especially given the added expense and difficulty in maintaining an adequate CMV-seronegative blood supply.
Solid organ transplant patients
Like HSCT patients, recipients of solid organ transplants are at a substantial risk of CMV infection, reactivation, and disease that is unrelated to transfusion when either the donor or the recipient is CMV-seropositive. Therefore, the prevention of TT-CMV infection is most important in D−/R− solid organ transplant recipients. Unfortunately, very few studies have been conducted to assess the efficacy of strategies to prevent TT-CMV infection in solid organ transplant patients. An early paper published by Preiksaitis et al described no cases of TT-CMV infection in eight D−/R− heart transplant recipients who received only CMV-seronegative blood compared with a rate of CMV infection of 20% (1 of 5) in the group of D−/R− patients who received unscreened/non-LR blood.30 Although there has been a lack of any studies on the effectiveness of LR in preventing TT-CMV infection in the setting of solid organ transplantation, it is unlikely that solid organ transplant patients are at a higher risk of TT-CMV infection than HSCT patients. Therefore, data showing the effectiveness of an LR-only transfusion strategy in HSCT patients may be extrapolated to solid organ transplant patients.
Hematologic malignancy patients
Although patients with hematologic malignancies are less likely to develop severe CMV infection and its sequelae than HSCT patients, it is important to prevent TT-CMV infection in CMV-seronegative patients. In addition, many of these patients may subsequently undergo stem cell transplantation. LR has been shown to be an effective method in this patient population in the prevention of TT-CMV infection.36,38 Although head-to-head studies comparing CMV-seronegative versus LR-only versus LR/CMV-seronegative blood products in this patient population are lacking, it is unlikely that these patients would be at a higher risk of TT-CMV infection than HSCT patients. Therefore, in light of the evidence discussed above for HSCT patients, an LR-only approach for transfusion in patients with hematologic malignancies may be safely employed.
Human immunodeficiency virus (HIV) patients
Patients who are infected with HIV are at risk of CMV disease, through either primary infection or reactivation of latent CMV infection.61,62 CMV retinitis is by far the most frequent manifestation of CMV disease in HIV patients, but esophagitis, colitis, gastritis, encephalitis, hepatitis, and pneumonitis have also been described in this population.61,63 Although CMV infection through the transfusion of blood products is certainly plausible, the actual risk of TT-CMV infection in HIV patients is unknown.62,64 To the authors’ knowledge, there are no documented cases in the literature of TT-CMV infection as a cause of primary CMV infection and CMV disease in HIV patients. Not surprisingly, there have been no studies examining the most effective approach for the prevention of TT-CMV infection in this population. As in the case of solid organ transplant patients and hematologic malignancy patients, the authors’ institution employs an LR-only approach based on the extrapolation of data showing the effectiveness of this strategy in the HSCT population.
Patients with rheumatic diseases
Because patients with rheumatic diseases are often treated with immunosuppressive therapy, they are at risk of symptomatic CMV infection.65 However, there are no reported cases in the literature of TT-CMV infection in this population. Because it is unlikely that patients with rheumatic diseases would be at a greater risk of TT-CMV infection than HSCT patients, an LR-only transfusion strategy is reasonable in this group.
Premature VLBW infants, fetuses, and CMV-seronegative pregnant women
Congenital and postnatal CMV infections are a significant cause of morbidity and mortality among infants. Symptoms of congenital CMV infection in the neonatal period include hepatosplenomegaly, thrombocytopenia, petechiae, microcephaly, ventriculomegaly, and chorioretinitis.66,67 Although the majority of infants with congenital CMV infection are initially asymptomatic at birth, both symptomatic and asymptomatic infants are at risk of long-term sequelae including mental impairment, sensorineural hearing loss, visual loss, and motor deficits.66,68
The risk of congenital CMV infection is highest in CMV-seronegative women who acquire primary CMV infection during pregnancy with an overall rate of in utero transmission of ~20%–75%.69,70 The rate of congenital CMV infection varies according to the gestational age at which maternal CMV infection occurs, with the rates of 30%–42% in the first trimester, 34%–45% in the second trimester, and 40%–78% in the third trimester.69–72 Infants with congenital infection are more likely to be symptomatic if the maternal CMV infection occurs early in pregnancy, particularly during the first trimester.72 In contrast, congenital CMV infection due to the reactivation of latent virus in CMV-seropositive women is uncommon.
Postnatal CMV infection in otherwise healthy full-term infants is usually asymptomatic and of little consequence. However, preterm VLBW (<1500 g) infants are at risk of severe postnatal CMV infection including pneumonia, hepatitis, neutropenia, thrombocytopenia, and enterocolitis.73 These infants may also develop a sepsis-like syndrome (SLS) characterized by apnea, bradycardia, or respiratory deterioration.74
The most frequent source of postnatal CMV infection is breast milk. CMV DNA is detectable in breast milk of up to 96% of seropositive mothers.75 Through incompletely understood mechanisms, local CMV reactivation occurs in the mammary glands of CMV-positive mothers, which coincides with the beginning of lactation.73 A meta-analysis by Lanzieri et al demonstrated a CMV transmission rate of 19% (95% CI =11%–32%) in VLBW infants who were fed untreated breast milk from CMV-positive mothers.74 Moreover, 10% (95% CI =5%–17%) of infants fed untreated milk developed symptomatic CMV infection, and 4% (95% CI =2%–7%) of infants suffered from CMV-SLS.74 The methods used to prevent CMV transmission through breast milk include holder pasteurization and freezing. Holder pasteurization (heating at 63°C for 30 min with subsequent rapid cooling) completely inactivates CMV, whereas freezing reduces the risk of CMV transmission but does not completely eliminate it.73,76 Although the frequencies of CMV transmission (13%; 95% CI =7%–24%) and symptomatic CMV infection (7%; 95% CI =3%–14%) are lower among infants who are fed frozen milk, the percentage of infants who develop CMV-SLS (5%; 95% CI =2%–12%) is not different from that seen in infants fed untreated breast milk.74 Both pasteurization and freezing have been shown to alter the nutritional properties of breast milk, including a significant reduction in fat, lactose, and energy content.77
Because of the dire complications of CMV infection described above, the prevention of TT-CMV infection is essential when administering blood products to fetuses in utero, CMV-seronegative pregnant women, and premature VLBW infants. The effectiveness of CMV-seronegative blood products in the prevention of TT-CMV infection in neonates was demonstrated in an older landmark study by Yeager et al, which was conducted prior to the availability of LR products. In their study, infants were randomized to receive CMV-seronegative or CMV-untested blood products.29 In children born to CMV-seronegative mothers, there were no cases of CMV infection in infants transfused with CMV-seronegative blood products, whereas 13.5% of infants transfused with CMV-positive blood products developed CMV infection. In children born to CMV-seropositive mothers, CMV-seronegative blood provided no benefit (17.6% CMV-seronegative blood versus 15% CMV-seropositive), likely due to CMV transmission through breast milk. A subsequent randomized controlled trial performed by Gilbert et al found that LR was highly effective in reducing the rate of TT-CMV infection in infants of CMV-seronegative mothers (0% LR versus 21% non-LR; P=0.002).37 In the era of universal LR, Josephson et al showed that the transfusion of LR/CMV-seronegative (belt-and-suspenders) blood products completely prevented TT-CMV in a large cohort of 539 VLBW infants, of whom 310 infants were transfused.43 Nearly all (27 of 38; 96%) cases of postnatal CMV infection were linked to breast milk that was positive for CMV DNA. Because all the transfused infants in the study by Josephson et al received only LR/CMV-seronegative blood products with no comparison group, this study could not answer the question as to whether the belt-and-suspenders approach is superior to the transfusion of LR-only blood products.
Unfortunately, there has never been a head-to-head randomized controlled trial comparing the effectiveness of LR/CMV-seronegative blood products to LR-only blood products in the prevention of TT-CMV infection in VLBW infants. Since the rate of TT-CMV would be expected to be very low in both the groups, such a trial would require the enrollment of a large number of susceptible infants in order to achieve statistical power to detect a significant difference between the two approaches.78,79 This fact, combined with the expense of conducting such a study, makes it unlikely that a randomized controlled trial will ever be performed. However, Delaney et al recently published the findings of a pilot study of LR-only transfusion in VLBW infants using a similar cohort design to the previous study by Josephson et al so that the results of the two studies might be compared.79 This small study detected no cases of TT-CMV infection using the LR-only approach (0 of 8; 95% CI =0%–25.3%) although the 95% CI was very wide, given that only 20 VLBW infants were enrolled among whom only eight were transfused. However, the authors showed that if a study of similar size to that of Josephson et al (~300 transfused infants) could be conducted using the LR-only approach, it should be possible to demonstrate that the effectiveness of LR-only blood products is similar to that of LR/CMV-seronegative blood products in the prevention of TT-CMV infection.79 In the meantime, these and previous studies showed that both LR-only and LR/CMV-seronegative blood products are effective in the prevention of TT-CMV infection in VLBW infants and that either is an acceptable approach. However, it may be difficult to maintain a LR/CMV-seronegative blood supply depending on the CMV seroprevalence in the local donor population.
To the authors’ knowledge, there have not been any studies on the effectiveness of CMV-seronegative, LR-only, or LR/CMV-seronegative blood products in the prevention of TT-CMV infection in fetuses undergoing intrauterine transfusion or CMV-seronegative pregnant women. By extrapolating the data from other at-risk patient groups, it is reasonable to assume that each of these strategies is effective. However, as in VLBW infants, it is unknown whether LR/CMV-seronegative blood products are superior to LR-only products in the prevention of TT-CMV infection. Therefore, it is not surprising that transfusion practices vary widely across different countries and hospital settings.45,80
Future directions
Until recently, the approach for preventing the transmission of infectious agents through blood transfusion has been largely reactive. For example, when an emerging pathogen is recognized as being transmissible through blood, new testing modalities are implemented to either screen donors or blood products for infection. However, there is inevitably a period of time between the recognition of the pathogen and implementation of screening during which the blood supply is at risk.81 This cycle has recently played out once more during the ongoing Zika virus pandemic.82
Pathogen inactivation technologies (PITs) have the enormous potential of shifting this paradigm. Two types of PITs, amotosalen/ultraviolet (UV) light (INTERCEPT™, Cerus Corporation, Concord, CA, USA) and riboflavin/UV light (Mirasol PRT®, Terumo BCT, Lakewood, CO, USA), are relevant to the discussion of TT-CMV infection as these are the current technologies in widespread use for platelet products. Amotosalen is a psoralen compound that, when exposed to UV light, forms covalent bonds that result in cross-linking of nucleic acids, thereby rendering the pathogen incapable of carrying out transcription and replication.83 Riboflavin (vitamin B2) similarly attacks nucleic acids when activated by UV light but rather works through the oxidation of guanine residues, which leads to single-strand breaks in DNA and RNA.84 Both technologies have been shown to prevent the transmission of murine CMV by platelet transfusion in mouse models.85,86 In addition to the benefit of preventing TT-CMV, pathogen inactivation also prevents the transmission of bacteria and many other viruses. It also decreases the risk of transfusion-associated graft versus host disease through the inactivation of donor leukocytes, possibly obviating the need for irradiation of blood products in at-risk patients.87,88 Although a licensed pathogen inactivation system is not yet available for red blood cells, the development of such a technology is in progress that uses S-303, a frangible anchor linker effector compound that crosslinks nucleic acids, and glutathione as a quencher to minimize the interaction of S-303 with plasma proteins.89,90 Adaptation of the riboflavin/UV method of pathogen inactivation for packed red blood cells and whole blood is also underway.
As with all significant changes to the blood supply, there has been appropriate debate as to whether the benefits of PITs outweigh the costs. In particular, patients receiving pathogen-reduced platelet transfusion have been shown to have an inferior corrected count increment and require 7% more platelet transfusions than those patients who receive standard platelet transfusion.91 However, these studies also demonstrated no significant difference in mortality, clinically significant bleeding, or adverse events between these two groups of patients.91 Although these results support the equivalence of pathogen-reduced platelets and standard platelets in regard to outcomes, it is important to keep in mind that all such studies to date have been performed in the setting of prophylactic platelet transfusion, and the effectiveness of pathogen-reduced platelets in the context of massive transfusion, such as that occurs in trauma, remains to be prospectively studied by randomized controlled trials.92 The use of PITs will certainly increase the cost of blood products, but this increase in cost is expected to be partially offset by the elimination of the need for irradiation and several types of infectious disease testing (eg, CMV, West Nile virus, and bacterial culture) in pathogen-reduced blood products.88
Although important questions remain in regard to implementation, PITs will likely end the long-running debate as to whether the addition of CMV-seronegativity to blood products that are already leukoreduced is necessary for optimal prevention of TT-CMV infection.
Disclosure
The authors report no conflicts of interest in this work.
References
Forbes BA. Acquisition of cytomegalovirus infection: an update. Clin Microbiol Rev. 1989;2(2):204–216. | ||
Wills MR, Poole E, Lau B, Krishna B, Sinclair JH. The immunology of human cytomegalovirus latency: could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol Immunol. 2015;12(2):128–138. | ||
Slobedman B, Mocarski ES. Quantitative analysis of latent human cytomegalovirus. J Virol. 1999;73(6):4806–4812. | ||
Soderberg-Naucler C, Fish KN, Nelson JA. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell. 1997;91(1):119–126. | ||
Dumont LJ, Luka J, VandenBroeke T, Whitley P, Ambruso DR, Elfath MD. The effect of leukocyte-reduction method on the amount of human cytomegalovirus in blood products: a comparison of apheresis and filtration methods. Blood. 2001;97(11):3640–3647. | ||
Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair JH. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol. 1991;72(Pt 9):2059–2064. | ||
Ziemann M, Krueger S, Maier AB, Unmack A, Goerg S, Hennig H. High prevalence of cytomegalovirus DNA in plasma samples of blood donors in connection with seroconversion. Transfusion. 2007;47(11):1972–1983. | ||
Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev. 2013;26(1):86–102. | ||
Shaiegan M, Rasouli M, Zadsar M, Zolfaghari S. Meta-analysis of cytomegalovirus seroprevalence in volunteer blood donors and healthy subjects in Iran from 1992 to 2013. Iran J Basic Med Sci. 2015;18(7):627–634. | ||
Das B, Kaur G, Basu S. Seroprevalence of cytomegalovirus antibodies among blood donors and multitransfused recipients – a study from north India. Transfus Apher Sci. 2014;50(3):438–442. | ||
Furui Y, Satake M, Hoshi Y, Uchida S, Suzuki K, Tadokoro K. Cytomegalovirus (CMV) seroprevalence in Japanese blood donors and high detection frequency of CMV DNA in elderly donors. Transfusion. 2013;53(10):2190–2197. | ||
Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States, 1988-1994. Clin Infect Dis. 2006;43(9):1143–1151. | ||
Boeckh M, Ljungman P. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood. 2009;113(23):5711–5719. | ||
Adler SP, Chandrika T, Lawrence L, Baggett J. Cytomegalovirus infections in neonates acquired by blood transfusions. Pediatr Infect Dis. 1983;2(2):114–118. | ||
Kotton CN, Kumar D, Caliendo AM, et al. Updated international consensus guidelines on the management of cytomegalovirus in solid-organ transplantation. Transplantation. 2013;96(4):333–360. | ||
Whitley RJ, Jacobson MA, Friedberg DN, et al. Guidelines for the treatment of cytomegalovirus diseases in patients with AIDS in the era of potent antiretroviral therapy: recommendations of an international panel. International AIDS Society-USA. Arch Intern Med. 1998;158(9):957–969. | ||
Kaariainen L, Klemola E, Paloheimo J. Rise of cytomegalovirus antibodies in an infectious-mononucleosis-like syndrome after transfusion. Br Med J. 1966;1(5498):1270–1272. | ||
Smith DR. A syndrome resembling infectious mononucleosis after open-heart surgery. Br Med J. 1964;1(5388):945–948. | ||
Kreel I, Zaroff LI, Canter JW, Krasna I, Baronofsky ID. A syndrome following total body perfusion. Surg Gynecol Obstet. 1960;111:317–321. | ||
Anderson R, Larsson O. Fever, splenomegaly and atypical lymphocytes after open heart surgery. Lancet. 1963;2(7314):947. | ||
Roback JD, Drew WL, Laycock ME, Todd D, Hillyer CD, Busch MP. CMV DNA is rarely detected in healthy blood donors using validated PCR assays. Transfusion. 2003;43(3):314–321. | ||
Drew WL, Roback JD. Prevention of transfusion-transmitted cytomegalovirus: reactivation of the debate? Transfusion. 2007;47(11):1955–1958. | ||
Ziemann M, Heuft HG, Frank K, Kraas S, Gorg S, Hennig H. Window period donations during primary cytomegalovirus infection and risk of transfusion-transmitted infections. Transfusion. 2013;53(5):1088–1094. | ||
Ziemann M, Unmack A, Steppat D, Juhl D, Gorg S, Hennig H. The natural course of primary cytomegalovirus infection in blood donors. Vox Sang. 2010;99(1):24–33. | ||
Gerna G, Lilleri D, Fornara C, et al. Differential kinetics of human cytomegalovirus load and antibody responses in primary infection of the immunocompetent and immunocompromised host. J Gen Virol. 2015;96(Pt 2):360–369. | ||
Isaacson MK, Compton T. Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J Virol. 2009;83(8):3891–3903. | ||
Ziemann M, Juhl D, Gorg S, Hennig H. The impact of donor cytomegalovirus DNA on transfusion strategies for at-risk patients. Transfusion. 2013;53(10):2183–2189. | ||
Kumar A, Nankervis GA, Cooper AR, Gold E, Kumar ML. Acquisition of cytomegalovirus infection in infants following exchange transfusion: a prospective study. Transfusion. 1980;20(3):327–331. | ||
Yeager AS, Grumet FC, Hafleigh EB, Arvin AM, Bradley JS, Prober CG. Prevention of transfusion-acquired cytomegalovirus infections in newborn infants. J Pediatr. 1981;98(2):281–287. | ||
Preiksaitis JK, Rosno S, Grumet C, Merigan TC. Infections due to herpesviruses in cardiac transplant recipients: role of the donor heart and immunosuppressive therapy. J Infect Dis. 1983;147(6):974–981. | ||
Bowden RA, Sayers M, Flournoy N, et al. Cytomegalovirus immune globulin and seronegative blood products to prevent primary cytomegalovirus infection after marrow transplantation. N Engl J Med. 1986;314(16):1006–1010. | ||
Miller WJ, McCullough J, Balfour HH Jr, et al. Prevention of cytomegalovirus infection following bone marrow transplantation: a randomized trial of blood product screening. Bone Marrow Transplant. 1991;7(3):227–234. | ||
Nash T, Hoffmann S, Butch S, Davenport R, Cooling L. Safety of leukoreduced, cytomegalovirus (CMV)-untested components in CMV-negative allogeneic human progenitor cell transplant recipients. Transfusion. 2012;52(10):2270–2272. | ||
Roback JD, Josephson CD. New insights for preventing transfusion-transmitted cytomegalovirus and other white blood cell-associated viral infections. Transfusion. 2013;53(10):2112–2116. | ||
Vollmer T, Knabbe C, Dreier J. Systematic evaluation of different nucleic acid amplification assays for cytomegalovirus detection: feasibility of blood donor screening. J Clin Microbiol. 2015;53(10):3219–3225. | ||
Murphy MF, Grint PC, Hardiman AE, Lister TA, Waters AH. Use of leucocyte-poor blood components to prevent primary cytomegalovirus (CMV) infection in patients with acute leukaemia. Br J Haematol. 1988;70(2):253–254. | ||
Gilbert GL, Hayes K, Hudson IL, James J. Prevention of transfusion-acquired cytomegalovirus infection in infants by blood filtration to remove leucocytes. Neonatal Cytomegalovirus Infection Study Group. Lancet. 1989;1(8649):1228–1231. | ||
de Graan-Hentzen YC, Gratama JW, Mudde GC, et al. Prevention of primary cytomegalovirus infection in patients with hematologic malignancies by intensive white cell depletion of blood products. Transfusion. 1989;29(9):757–760. | ||
Hervig T, Seghatchian J. Leukocyte-reduced blood components: laboratory and clinical aspects. In: Simon TL, McCullough J, Snyder EL, Solheim BG, Strauss RG, editors. Rossi’s Principles of Transfusion Medicine. 5th ed. Chichester, West Sussex, Hoboken, NJ: John Wiley & Sons Inc.; 2016:278–285. | ||
Muylle L, Joos M, Wouters E, De Bock R, Peetermans ME. Increased tumor necrosis factor alpha (TNF alpha), interleukin 1, and interleukin 6 (IL-6) levels in the plasma of stored platelet concentrates: relationship between TNF alpha and IL-6 levels and febrile transfusion reactions. Transfusion. 1993;33(3):195–199. | ||
Roback JD, Su L, Zimring JC, Hillyer CD. Transfusion-transmitted cytomegalovirus: lessons from a murine model. Transfus Med Rev. 2007;21(1):26–36. | ||
Seed CR, Wong J, Polizzotto MN, Faddy H, Keller AJ, Pink J. The residual risk of transfusion-transmitted cytomegalovirus infection associated with leucodepleted blood components. Vox Sang. 2015;109(1):11–17. | ||
Josephson CD, Caliendo AM, Easley KA, et al. Blood transfusion and breast milk transmission of cytomegalovirus in very low-birth-weight infants: a prospective cohort study. JAMA Pediatr. 2014;168(11):1054–1062. | ||
Vamvakas EC, Blajchman MA. Universal WBC reduction: the case for and against. Transfusion. 2001;41(5):691–712. | ||
Lieberman L, Devine DV, Reesink HW, et al. Prevention of transfusion-transmitted cytomegalovirus (CMV) infection: standards of care. Vox Sang. 2014;107(3):276–311. | ||
James DJ, Sikotra S, Sivakumaran M, et al. The presence of free infectious cytomegalovirus (CMV) in the plasma of donated CMV-seropositive blood and platelets. Transfus Med. 1997;7(2):123–126. | ||
Ziemann M, Hennig H. Prevention of transfusion-transmitted cytomegalovirus infections: which is the optimal strategy? Transfus Med Hemother. 2014;41(1):40–44. | ||
Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol Oncol Clin North Am. 2011;25(1):151–169. | ||
Ozdemir E, Saliba RM, Champlin RE, et al. Risk factors associated with late cytomegalovirus reactivation after allogeneic stem cell transplantation for hematological malignancies. Bone Marrow Transplant. 2007;40(2):125–136. | ||
Patel SR, Ridwan RU, Ortin M. Cytomegalovirus reactivation in pediatric hemopoietic progenitors transplant: a retrospective study on the risk factors and the efficacy of treatment. J Pediatr Hematol Oncol. 2005;27(8):411–415. | ||
Bowden RA, Slichter SJ, Sayers M, et al. A comparison of filtered leukocyte-reduced and cytomegalovirus (CMV) seronegative blood products for the prevention of transfusion-associated CMV infection after marrow transplant. Blood. 1995;86(9):3598–3603. | ||
De Witte T, Schattenberg A, Van Dijk BA, et al. Prevention of primary cytomegalovirus infection after allogeneic bone marrow transplantation by using leukocyte-poor random blood products from cytomegalovirus-unscreened blood-bank donors. Transplantation. 1990;50(6):964–968. | ||
van Prooijen HC, Visser JJ, van Oostendorp WR, de Gast GC, Verdonck LF. Prevention of primary transfusion-associated cytomegalovirus infection in bone marrow transplant recipients by the removal of white cells from blood components with high-affinity filters. Br J Haematol. 1994;87(1):144–147. | ||
Narvios AB, Przepiorka D, Tarrand J, Chan KW, Champlin R, Lichtiger B. Transfusion support using filtered unscreened blood products for cytomegalovirus-negative allogeneic marrow transplant recipients. Bone Marrow Transplant. 1998;22(6):575–577. | ||
Nichols WG, Price TH, Gooley T, Corey L, Boeckh M. Transfusion-transmitted cytomegalovirus infection after receipt of leukoreduced blood products. Blood. 2003;101(10):4195–4200. | ||
Mainou M, Alahdab F, Tobian AA, et al. Reducing the risk of transfusion-transmitted cytomegalovirus infection: a systematic review and meta-analysis. Transfusion. 2016;56(6 Pt 2):1569–1580. | ||
Thiele T, Kruger W, Zimmermann K, et al. Transmission of cytomegalovirus (CMV) infection by leukoreduced blood products not tested for CMV antibodies: a single-center prospective study in high-risk patients undergoing allogeneic hematopoietic stem cell transplantation (CME). Transfusion. 2011;51(12):2620–2626. | ||
Wu Y, Zou S, Cable R, et al. Direct assessment of cytomegalovirus transfusion-transmitted risks after universal leukoreduction. Transfusion. 2010;50(4):776–786. | ||
Kekre N, Tokessy M, Mallick R, et al. Is cytomegalovirus testing of blood products still needed for hematopoietic stem cell transplant recipients in the era of universal leukoreduction? Biol Blood Marrow Transplant. 2013;19(12):1719–1724. | ||
Ljungman P, Larsson K, Kumlien G, et al. Leukocyte depleted, unscreened blood products give a low risk for CMV infection and disease in CMV seronegative allogeneic stem cell transplant recipients with seronegative stem cell donors. Scand J Infect Dis. 2002;34(5):347–350. | ||
Gronborg HL, Jespersen S, Honge BL, Jensen-Fangel S, Wejse C. Review of cytomegalovirus coinfection in HIV-infected individuals in Africa. Rev Med Virol. 2017;27(1). Epub 2016 Oct 7. | ||
Hillyer CD, Lankford KV, Roback JD, Gillespie TW, Silberstein LE. Transfusion of the HIV-seropositive patient: immunomodulation, viral reactivation, and limiting exposure to EBV (HHV-4), CMV (HHV-5), and HHV-6, 7, and 8. Transfus Med Rev. 1999;13(1):1–17. | ||
Gallant JE, Moore RD, Richman DD, Keruly J, Chaisson RE. Incidence and natural history of cytomegalovirus disease in patients with advanced human immunodeficiency virus disease treated with zidovudine. The Zidovudine Epidemiology Study Group. J Infect Dis. 1992;166(6):1223–1227. | ||
Preiksaitis JK. The cytomegalovirus-“safe” blood product: is leukoreduction equivalent to antibody screening? Transfus Med Rev. 2000;14(2):112–136. | ||
Takizawa Y, Inokuma S, Tanaka Y, et al. Clinical characteristics of cytomegalovirus infection in rheumatic diseases: multicentre survey in a large patient population. Rheumatology (Oxford). 2008;47(9):1373–1378. | ||
Kylat RI, Kelly EN, Ford-Jones EL. Clinical findings and adverse outcome in neonates with symptomatic congenital cytomegalovirus (SCCMV) infection. Eur J Pediatr. 2006;165(11):773–778. | ||
Boppana SB, Pass RF, Britt WJ, Stagno S, Alford CA. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11(2):93–99. | ||
Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326(10):663–667. | ||
Picone O, Vauloup-Fellous C, Cordier AG, et al. A series of 238 cytomegalovirus primary infections during pregnancy: description and outcome. Prenat Diagn. 2013;33(8):751–758. | ||
Bodeus M, Hubinont C, Goubau P. Increased risk of cytomegalovirus transmission in utero during late gestation. Obstet Gynecol. 1999;93(5 Pt 1):658–660. | ||
Revello MG, Fabbri E, Furione M, et al. Role of prenatal diagnosis and counseling in the management of 735 pregnancies complicated by primary human cytomegalovirus infection: a 20-year experience. J Clin Virol. 2011;50(4):303–307. | ||
Enders G, Daiminger A, Bader U, Exler S, Enders M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J Clin Virol. 2011;52(3):244–246. | ||
Gunkel J, Wolfs TF, de Vries LS, Nijman J. Predictors of severity for postnatal cytomegalovirus infection in preterm infants and implications for treatment. Expert Rev Anti Infect Ther. 2014;12(11):1345–1355. | ||
Lanzieri TM, Dollard SC, Josephson CD, Schmid DS, Bialek SR. Breast milk-acquired cytomegalovirus infection and disease in VLBW and premature infants. Pediatrics. 2013;131(6):e1937–e1945. | ||
Hamprecht K, Maschmann J, Vochem M, Dietz K, Speer CP, Jahn G. Epidemiology of transmission of cytomegalovirus from mother to preterm infant by breastfeeding. Lancet. 2001;357(9255):513–518. | ||
Lombardi G, Garofoli F, Manzoni P, Stronati M. Breast milk-acquired cytomegalovirus infection in very low birth weight infants. J Matern Fetal Neonatal Med. 2012;25 (Suppl 3):57–62. | ||
Garcia-Lara NR, Vieco DE, De la Cruz-Bertolo J, Lora-Pablos D, Velasco NU, Pallas-Alonso CR. Effect of Holder pasteurization and frozen storage on macronutrients and energy content of breast milk. J Pediatr Gastroenterol Nutr. 2013;57(3):377–382. | ||
Strauss RG. Optimal prevention of transfusion-transmitted cytomegalovirus (TTCMV) infection by modern leukocyte reduction alone: CMV sero/antibody-negative donors needed only for leukocyte products. Transfusion. 2016;56(8):1921–1924. | ||
Delaney M, Mayock D, Knezevic A, et al. Postnatal cytomegalovirus infection: a pilot comparative effectiveness study of transfusion safety using leukoreduced-only transfusion strategy. Transfusion. 2016;56(8):1945–1950. | ||
Smith D, Lu Q, Yuan S, Goldfinger D, Fernando LP, Ziman A. Survey of current practice for prevention of transfusion-transmitted cytomegalovirus in the United States: leucoreduction vs. cytomegalovirus-seronegative. Vox Sang. 2010;98(1):29–36. | ||
Snyder EL, Stramer SL, Benjamin RJ. The safety of the blood supply–time to raise the bar. N Engl J Med. 2015;372(20):1882–1885. | ||
Katz LM, Rossmann SN. Zika and the blood supply: a work in progress. Arch Pathol Lab Med. 2017;141(1):85–92. | ||
Lin L, Cook DN, Wiesehahn GP, et al. Photochemical inactivation of viruses and bacteria in platelet concentrates by use of a novel psoralen and long-wavelength ultraviolet light. Transfusion. 1997;37(4):423–435. | ||
Mundt JM, Rouse L, Van den Bossche J, Goodrich RP. Chemical and biological mechanisms of pathogen reduction technologies. Photochem Photobiol. 2014;90(5):957–964. | ||
Jordan CT, Saakadze N, Newman JL, et al. Photochemical treatment of platelet concentrates with amotosalen hydrochloride and ultraviolet A light inactivates free and latent cytomegalovirus in a murine transfusion model. Transfusion. 2004;44(8):1159–1165. | ||
Keil SD, Saakadze N, Bowen R, et al. Riboflavin and ultraviolet light for pathogen reduction of murine cytomegalovirus in blood products. Transfusion. 2015;55(4):858–863. | ||
Fast LD, Nevola M, Tavares J, Reddy HL, Goodrich RP, Marschner S. Treatment of whole blood with riboflavin plus ultraviolet light, an alternative to gamma irradiation in the prevention of transfusion-associated graft-versus-host disease? Transfusion. 2013;53(2):373–381. | ||
McCullough J, Goldfinger D, Gorlin J, et al. Cost implications of implementation of pathogen-inactivated platelets. Transfusion. 2015;55(10):2312–2320. | ||
Wiltshire M, Meli A, Schott MA, et al. Quality of red cells after combination of prion reduction and treatment with the intercept system for pathogen inactivation. Transfus Med. 2016;26(3):208–214. | ||
Salunkhe V, van der Meer PF, de Korte D, Seghatchian J, Gutierrez L. Development of blood transfusion product pathogen reduction treatments: a review of methods, current applications and demands. Transfus Apher Sci. 2015;52(1):19–34. | ||
Butler C, Doree C, Estcourt LJ, et al. Pathogen-reduced platelets for the prevention of bleeding. Cochrane Database Syst Rev. 2013;3:CD009072. | ||
Hess JR, Pagano MB, Barbeau JD, Johannson PI. Will pathogen reduction of blood components harm more people than it helps in developed countries? Transfusion. 2016;56(5):1236–1241. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.