")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Current Status and Perspectives of Protease Inhibitors and Their Combination with Nanosized Drug Delivery Systems for Targeted Cancer Therapy
Authors Rudzińska M , Daglioglu C , Savvateeva LV, Kaci FN , Antoine R , Zamyatnin Jr AA
Received 8 October 2020
Accepted for publication 15 December 2020
Published 6 January 2021 Volume 2021:15 Pages 9—20
DOI https://doi.org/10.2147/DDDT.S285852
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tuo Deng
Magdalena Rudzińska,1 Cenk Daglioglu,2 Lyudmila V Savvateeva,1 Fatma Necmiye Kaci,3 Rodolphe Antoine,4 Andrey A Zamyatnin Jr1,5,6
1Institute of Molecular Medicine, Sechenov First Moscow State Medical University, Moscow 119991, Russia; 2Biotechnology and Bioengineering Application and Research Center, Integrated Research Centers, Izmir Institute of Technology, Urla, Izmir 35430, Turkey; 3Department of Molecular Biology and Genetics, Faculty of Science, Erzurum Technical University, Yakutiye, Erzurum 25050, Turkey; 4CNRS, Institut Lumière Matière, Univ Lyon, Université Claude Bernard Lyon 1, Lyon F-69622, France; 5Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119991, Russia; 6Department of Biotechnology, Sirius University of Science and Technology, Sochi 354340, Russia
Correspondence: Magdalena Rudzińska; Andrey A Zamyatnin Jr
Institute of Molecular Medicine, Sechenov First Moscow State Medical University, Moscow 119991, Russia
Tel +74956229843
Email [email protected]; [email protected]
Abstract: In cancer treatments, many natural and synthetic products have been examined; among them, protease inhibitors are promising candidates for anti-cancer agents. Since dysregulated proteolytic activities can contribute to tumor development and metastasis, antagonization of proteases with tailored inhibitors is an encouraging approach. Although adverse effects of early designs of these inhibitors disappeared after the introduction of next-generation agents, most of the proposed inhibitors did not pass the early stages of clinical trials due to their nonspecific toxicity and lack of pharmacological effects. Therefore, new applications that modulate proteases more specifically and serve their programmed way of administration are highly appreciated. In this context, nanosized drug delivery systems have attracted much attention because preliminary studies have demonstrated that the therapeutic capacity of inhibitors has been improved significantly with encapsulated formulation as compared to their free forms. Here, we address this issue and discuss the current application and future clinical prospects of this potential combination towards targeted protease-based cancer therapy.
Keywords: proteases, inhibitors, nanoscale drug-delivery system, combination therapy, cancer treatment
Plain Language Summary
Proteases include widely distributed enzymes that are crucial for protein homeostasis and regulate many cellular processes, such as gene expression, differentiation, immunological defence, migration and cell death. Recent studies have indicated that the balance between production, activation and inhibition of proteases is often disturbed in malignant tumors, leading to tumor progression and dissemination. Numerous proteases are therefore under investigation for therapeutic purposes and their inhibition properties may serve potential for anti-cancer strategies. However, since protease inhibitors have mostly been developed in the form of small molecules or peptides, until recently their insufficient metabolic stability has remained an obstacle. Consequently, formulating protease inhibitors with a nanoparticle-based delivery system offers a new perspective on protease-targeted therapies. Protease inhibitors loaded in nanocarriers may provide improved bioavailability, increased circulation time and accumulate specifically in the tumoral tissues. This novel concept for the administration of protease inhibitors expands their potential in biomedical applications and gives the oportunity to achieve more effective treatment.
Proteases and Their Inhibitors
Proteases are a complex of enzymes that hydrolyze peptide bonds and are responsible for the breakdown of proteins into their individual components.1 Based on their amino acid core in the active site and the mechanism of peptide bond cleavage, proteases can be classified into six groups: cysteine, serine, threonine, glutamic acid, aspartate proteases and metalloproteases.1,2 Their control over protein synthesis, turnover and function enables them to regulate many biological processes, such as protein digestion, fertilization, immunological response, wound healing, cellular growth, differentiation, migration and apoptosis.3 Their enhanced activity and changes in expression have frequently been connected with disease propagation. Therefore, antagonization of these enzymes with designed inhibitors is a promising strategy against a wide range of protease-associated diseases such as parasitic,4 fungal5 and various infectious (eg, malaria,6 human immunodeficiency virus [HIV],7 hepatitis,8 herpes9); inflammatory,10 respiratory,11 cardiovascular,12 neurodegenerative disorders13 as well as cancer disease.14
In the context of cancerogenesis, most extracellular proteases are involved in various processes associated with tumor development and progression by different mechanisms. Thus, they can act as the base for membrane and matrix degradation, inactivation of natural protease inhibitors and chemotherapeutics, cell viability regulation, immune response modulation and inflammatory cell recruitment.15–18 Moreover, proteases localized within the intracellular compartments, including in the cytosol, nucleus, membrane and mitochondria are associated with many signaling pathways through which they can promote: adhesion, proliferation, migration, de-differentiation and epithelial to mesenchymal transition of cancer cells.18–20 The release of proteases into various cellular compartments and their altered expression can result from different factors, including the activation of membrane receptors (such as the tumor necrosis factor receptor21) or the generation of reactive oxygen species.22,23
These diverse tumor-promoting roles of proteases has emphasized their potential and drawn much attention to them as molecular targets.18,24 The inhibition of (1) matrix metalloproteases (MMPs), (2) the serine protease – urokinase-type plasminogen activator (uPA) system and (3) several cathepsins (Cts): CtsB, L, S, K, Z/X and D have all been recognized as promising approaches for anti-cancer therapy.2,25
Structural information on proteases and their active sites is vital to fit, design and introduce the inhibitor molecules to the biological systems. Proteolytic blocking of these enzymes by inhibitors can naturally occur through two different mechanisms that base on their interaction with the target protease: (i) irreversible trapping interaction, which initiates by cleaving an internal peptide bond in the inhibitor and causes its permanent conformational change (Figure 1A) and (ii) reversible tight-binding reactions, which directly bind to the active site of the protease1,26,27 (Figure 1B).
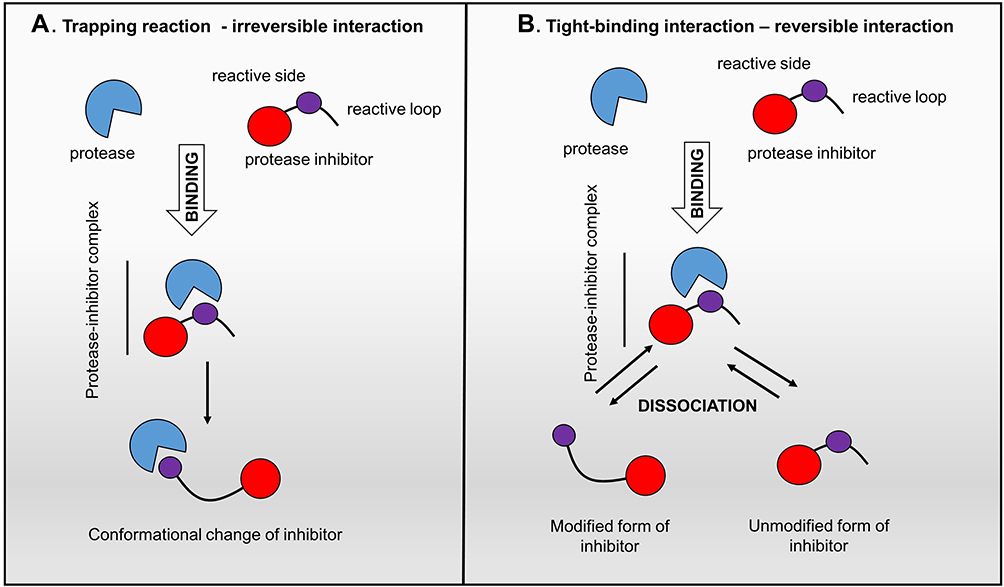 |
Figure 1 Protease inhibitor action mechanisms. Left panel: “trapping reaction” – the binding of inhibitor with protease activates the cleavage of an internal peptide bond, triggering an irreversible conformational change of the inhibitor; Right panel: “tight-binding interaction” – the reversible interplay of the inhibitor with the active site of protease is similar to the enzyme-substrate reaction.27,134 |
Apart from exogenous inhibiting molecules, the activity of proteases is also regulated by interactions with endogenous inhibitors. Among them, tissue inhibitors of metalloproteinases (TIMPs) are key regulators for targeting metalloproteases,28 while serpins are directed against serine proteases29 and cystatins are predominantly inhibiting cysteine proteases.30 However, it is important to note that the majority of protease inhibitors may interact with more than one type of protease, eg, serpins act with serine protease but can also inhibit the cysteine proteases.26
Matrix Metalloproteinase Inhibitors
Two general classes of MMP inhibitors, which consist of endogenous (TIMPs) and synthetic agents, are mostly non-selective. A group of non-peptidic inhibitors, including MMI270, prinomastat, marimastat and batimastat was designed with a zinc-binding hydroxamate group.31 Investigations to eliminate the significant limitations of early agents and decrease their side effects, low lability and specificity prompted the development of less toxic and more potent inhibitors.31 The second generation of non-hydroxamate MMP inhibitors was designed with different peptidomimetic and non-peptidomimetic structures, such as carboxylic acids (tanomastat), thiols (rebimastat) and others.31,32 Although the early inhibitors were improved, both the hydroxamate and non-hydroxamate groups failed to pass the early stages of human clinical trials.33 Despite their enhanced inhibitory effects, the broad-spectrum synthetic MMP inhibitors did not meet the relevant criteria when tested in humans.34 Moreover, there were several MMP inhibitor limitations leading to these failures including, metabolic instability, poor oral bioavailability and dose-limiting toxicities. Prolonged treatment with broad-spectrum MMP inhibitors was associated with unwanted side effects and this resulted in the use of lower, often inadequate, MMP inhibitor doses in trials.35 After the development of more potent selective antibodies, high affinity to MMPs was achieved without side effects such as musculoskeletal syndrome.36 One of the selective antibodies Andecaliximab (GS-5745; anti-MMP-9), has undergone clinical trials and been approved for its beneficial control over tumor growth and metastasis in a colorectal carcinoma model.37 Nevertheless, the potential of anti-MMP antibodies has yet to be fully realized, because they undergo proteolytic degradation during circulation, limiting systemic therapeutic application and synthesizing the agent at a reasonable cost is difficult and relatively slow.38,39
Importantly, the failure of MMP inhibitor treatments was also partly due to the inhibition of MMP anti-targets that are critical in host protection.40 It is tightly connected with the role of MMPs in the homeostatic regulation of the extracellular environment and controlling innate immunity.41 Thus, the knock-down of MMPs, such as MMP-3, −8 and −9 enhanced tumorigenesis and metastasis in some animal models of cancer.42,43 For a promising cancer therapy by MMP inhibition, the treatment must be selective against validated MMP targets.
Urokinase-Type Plasminogen Activator Inhibitors
Flavonoids are a group of secondary plant metabolites with a polyphenolic structure, which have been demonstrated to inhibit trypsin-like serine proteases such as uPA.44 The natural flavonoid quercetin was found to inhibit serine proteases and show anti-metastatic effects against a broad range of cancer models such as prostate, cervical, lung, breast and colorectal cancers.44,45 Research with uPA inhibitors developed based on aryl guanidine, aryl amidine or acyl guanidine backbones has shown their modest potency and poor selectivity.46 One uPA inhibitor – mesupron has been studied in clinical trials and entered Phase II, presenting a suppressor effect on breast cancer metastasis and reduction of primary tumor growth.47 However, its specificity is too low due to it targeting the S1 pockets, which are highly conserved among the serine protease family.48 In contrast, peptide-based inhibitors (cyclic peptides) showed higher selectivity for contacting multiple other regions outside the S1 pockets.49 Their unique structure provided by cyclization makes them more resistant to proteolytic degradation and better absorbed after oral administration.48,50 Cyclic peptides are of considerable interest as uPA modulators because they exhibit much higher specificity than small-molecule inhibitors.51 For example, a peptide-based uPA inhibitor CJ-463 (benzylsulfonyl-D-Ser-Ser-4- amidinobenzylamide) with Ki (inhibitory constant) value of 20 nM51 experimentally demonstrated good anti-tumor efficacy in a lung cancer animal mode.51,52
Cathepsin Inhibitors
Cts are expressed as pro-enzymes with inactive form, which on cleavage of the pro-domain, are usually dissociated from mature enzymes, thereby activating their catalytic function, whereas endogenous and exogenous protease inhibitors regulate their activities.53 The endogenic inhibitors including cystatins, thyropins, alpha2-macroglobulin, cytotoxic antigen 2β and members of the serpin family protect Cts from lysosomes that may be released, but they also possess other specialized roles.53 Therefore, misbalanced ratio of inhibitor family members to Cts can promote tumor growth, invasion and metastasis.18 Exogenous inhibitors can be divided into molecules isolated from animals, microorganisms, plants and fungi, monoclonal antibodies and small synthetic molecules. Among the natural inhibitors, the best-known is E-64 (1-[L-N-(trans-epoxysuccinyl)leucyl] amino-4-guanidinobutane), a wide-range peptidase inhibitor, isolated from Aspergillus japonicus, which serves as the base for more specific derivatives.54 Owing to the low cell permeability, off-target binding and the irreversible nature of inhibition, development has been directed towards covalent and non-covalent reversible inhibitors.55 The difference is based on the mechanistic approaches to enzyme inactivation. Generally, irreversible covalent inhibitors, permanently modify the protein of interest via the formation of a stable bond; its an off-rate that is slow relative to the rate of re-synthesis of the target protein.52,56 Reversible inhibitors bind to the target protein using non-covalent or covalent interactions, depending on the binding strength; reversible covalent bonds increases the binding affinity to proteases by 10–1000 times.57 While both reversible and irreversible inhibitors have their pros and cons, there has been a tendency to avoid irreversible, covalent inhibitors due to the risk of the immune response,58,59 unpredictable side effects (such as the generation of allergenic modified proteins; haptens), non-specific, irreversible modification of off-target proteins; and the difficulty in tracking metabolites when covalently bound to proteins.55
Irreversible specific CtsB (CA-074 and CA-03060) and CtsL (CLIK-148 and CLIK-19561) inhibitors have been developed from the structure of epoxysuccinate derivatives using X-ray crystallography and in vivo test results have shown that they reduced breast cancer metastasis.62
Oral therapy using the reversible CtsK inhibitor (L-235) can protect against tumor-induced osteolytic lesions and cortical disruption. The administration of L-235 in an experimental model of breast cancer bone metastasis reduced skeletal tumor burden.63
Furthermore, several small molecule inhibitors of cathepsins have been tested in clinical trials with good outcomes observed. Following, a selected anti-CtsS antibody, Fsn0503, significantly blocked the invasion of different tumor cell lines with the most significant result for colorectal carcinoma cells (HCT116). Moreover, a better anti-tumoral effect of Fsn0503 was shown in vivo where it retarded the tumor vascularization.64,65
Future Aims for Protease Inhibitors Designing
Most protease inhibitors (eg marimastat, prinomastat, non-hydroxamates neovastat, rebimastat, and tanomasta), which have been tested in pre-clinical and clinical trials for cancer treatments, have failed due to poor outcomes and pharmacological effects.33,34,66 Only a few, such as proteasome (multicatalytic protease complex) inhibitors: bortezomib, carfilzomib, ixazomib and MMPs inhibitor (doxycycline) have passed the Food and Drug Administration (FDA)-approval process. Using them with other therapies, including a combination of proteasome inhibitors with DNA-damaging drugs (eg doxorubicin and melphalan), has shown promise in the treatment of multiple myeloma and demonstrated the potential benefits for the development of antitumor therapy.67,68
Taken together, the in-depth exploration of natural inhibitors and synthesis of peptidomimetic antagonists, which control proteases, has provided many promising approaches for cancer studies.46,69 However, therapy with protease inhibitor is complicated in practical applications since different types of cancers use various proteases at different stages of tumor.3 As a result, no single inhibitor can be used on all classes of proteases. To develop suitable protease inhibitors successfully, the small molecule inhibitors need to be designed and tested against dedicated diseases and tissue.70,71 In this context, transcriptomic and proteomic analyses are crucial to validate inhibitors as specific targets.72,73 Additionally, comprehensive molecular experiments need to be performed to investigate the cross-talk between signal transduction pathways and protease activation cascades,70,74 which will be a base for maximizing their specificity and approving their repressor potential in vivo. Moreover, as an artificial class of drugs, protease inhibitors need to be designed with proper conformation to provide an extended interaction with specific proteases, while at the same time avoiding random degradation by the digestive tract, in plasma and inside cells.46,75
Furthermore, protease inhibitor delivery can incur problems due to (1) low membrane permeability,59 (2) rapid elimination from plasma,76 (3) high first-pass metabolism,77 (4) very low oral bioavailability,78 (5) fast renal clearance79 and (5) short in vivo retention times.80 To address these obstacles, finally, nanotechnology has been deployed to ensure the bioavailability of these inhibitors at the target site, thereby rendering improved pharmacokinetic and drug performance properties. In recent years, novel nanoparticle (NP) formulations such as non-traditional drug distribution platforms have been studied extensively for selective targeting and neutralizing proteases.81–85
Nanosized Drug Delivery Systems
Nanosized drug delivery systems are rapidly being developed in science and medicine where materials (liposomes, solid lipid NPs, dendrimers, polymers, silicon or carbon materials and magnetic NPs) in the nanoscale (10 nm to < 1000 nm) are used to serve as a scaffold for both diagnostic and therapeutic moieties.86 NP-based drug delivery systems enable the encapsulation of a variety of therapeutic agents, such as protein-based drugs, nucleic acids, peptides and small-molecule inhibitors, thereby increasing their therapeutic index and clinical significance (Figure 2).87 Considerable efforts have been devoted to improving novel nano-platforms toward the development of next-generation NP-based delivery systems.88–91 Among nanomaterials, ultra-small fluorescent metal gold nanoclusters92 (size < 2 nm) have gained increased attention for fluorescence imaging,93–95 diagnosis96 and cancer therapy.97,98 Protein protected gold nanoclusters (GNC) as a dual-functional nanoplatform for drug delivery and fluorescence imaging of tumors have recently been developed. The GNC were conjugated with a reduction-sensitive cisplatin prodrug and then functionalized with folic acid target ligand, targeting breast tumors.99 GNC could efficiently inhibit the growth of the primary tumor and suppress the metastasis of cancer cells. Insights for designing new GNC-based drug delivery systems are reported by Wu and coworkers who developed gold nanocage/cluster hybrid structures displaying high antitumor efficacy owing to the combined actions of epidermal growth factor receptor signaling pathway blockage and photothermal therapy.100 These have been developed as an all-in-one biomedical platform with proposed configurations with (i) significantly improved stability and solubility of treatments, (ii) prevention of degradation or premature interactions, (iii) improved drug absorption, (iv) targeted delivery of the drug to specific tissues, cells or subcellular compartments, (v) controlled drug distribution and pharmacokinetics.101,102 Furthermore, by engineering the NP formulations, more properties could be incorporated into the NPs according to requirements of disease states, such as multi-drug co-administration, programmed drug-delivery or triggered drug release, thereby reducing adverse effects of inhibitor/drug combinations to the healthy tissues or organs and improving the quality of life for patients.101,102
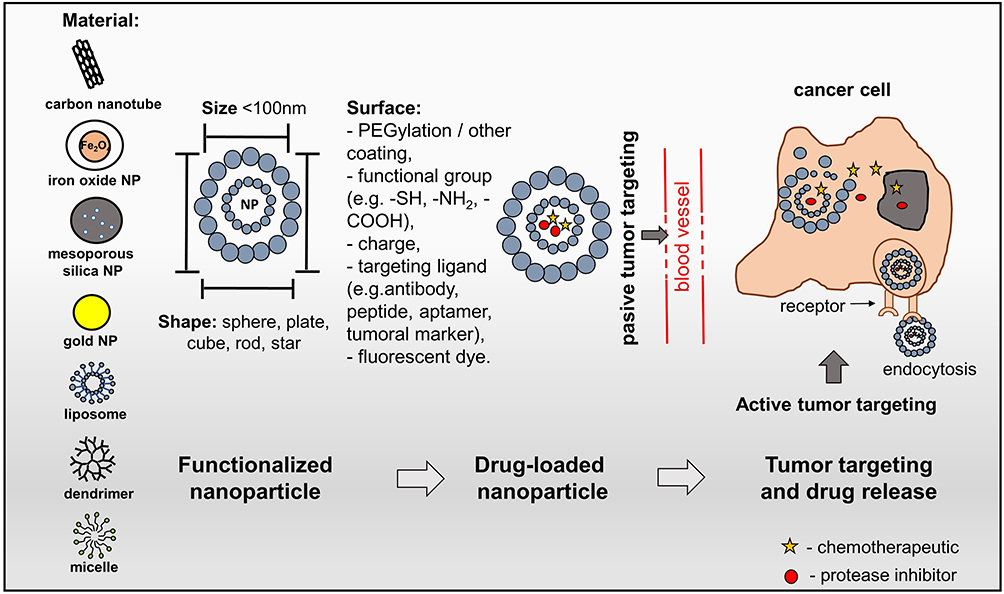 |
Figure 2 Nanostructured drug delivery systems. Schematic representation of the nanocarriers used in smart drug delivery systems showing their structure: size, material, shape and surface. A smart drug delivery system uses passive targeting and active targeting. Passive targeting employs the enhanced permeability and retention to locate cancer sites. The accumulation rate of nanoparticles (NPs) in a tumor is higher than in healthy tissue due to the leaky endothelium of the tumor vasculature. Furthermore, a deficiency of the lymphatic system leads to the retention of NPs in the tumor. Active targeting uses the ligand-receptor interaction to locate cancer cells; drug-loaded NPs are conjugated with targeting ligands.135,136 |
Consequently, in this context, formulating protease inhibitors with nanostructured carriers presents new perspectives on protease-targeted cancer therapies. Currently, there are several promising studies in this field. The proteasome inhibitors, bortezomib- and carfilzomib-loaded different NP systems (such as PEGylated lipid micelle, liposomes, gold NPs, mesoporous silica NPs) have demonstrated superior therapeutic effects such as biocompatibility, higher efficacy, increased circulation time and decreased systemic toxicity as compared to free drug formulations in breast cancer models, non-small cell lung and colon carcinomas.103,104
Combination of Matrix Metalloprotease Inhibitors with Nanosized Drug Delivery Systems
Wide-range MMP inhibitor (marimastat), loaded into lysolipid-containing thermosensitive liposomes (LTSLs), has exhibited higher accumulation in tumor tissue accompanied by decreased cancer angiogenesis and metastatic potential. Compared to saline control, the treatment with marimastat-LTSLs of mice-bearing the human breast tumor presented a 20-fold decrease in tumor growth and lower metastatic potential. Moreover, the MMP-2 and MMP-9 expression and activity were remarkably reduced.105 To conclude this effect, the encapsulation of active compounds into the bilayer of the liposomes facilitates the drug solubilization in aqueous media and also provides additional protection and control against drug degradation.106
Doxorubicin and Fab fragments (Fab’222–1D8) co-conjugated PEGylated liposomes directed against membrane type-1 matrix metalloproteinase led to reduced tumor growth and side effects, while prolonging the survival time of tested mice.107 PEGylated liposomes contain a hydrophilic polymer polyethylene glycol (PEG) moiety onto the surface of the vesicle.108 Herein the Fab′ fragments of antibody were conjugated at a distal end of PEG for sterically stabilization of doxorubicin-encapsulating liposomes. This modification significantly enhanced the cellular uptake of carriers into the HT1080 fibrosarcoma cells in comparison with the non-targeted liposomes. Systemic in vivo administration of Fab-doxorubicin structure into the tumor-bearing mice showed the notable suppression of tumor growth compared to control nanocarriers without Fab fragment.
Another strategy, which combines a monoclonal nucleosome-specific antibody (mAb 2C5) expressed on carcinogenic cells with an MMP-2 cleavable peptide, provided additional selectivity for tumor cells and shielded a carried cell-penetrating peptide.109 This system represents a liposomal nanocarrier in which two-sensitive bond between PEG and lipid undergoes cleavage in the tumor by the highly expressed extracellular MMP2. The conjugates combined with mAb 2C5 allow for the specific targeting of tumor cells. Additionally, cell-penetrating peptide triggering the enhanced intracellular delivery of the system after long-chain PEG removal and exposure. As a result, paclitaxel loaded PEG-poly-caprolactone NPs, which formulated with cell-penetrating peptides, attached through an MMP-2/9 cleavable sequence and showed longer survival in mice with glioblastoma than in controls.110
Combination of Urokinase-Type Plasminogen Activator Inhibitors with Nanosized Drug Delivery Systems
The novel formulation strategy, which encapsulated a high concentration of arsenic (As) in a stable precipitate with nickel acetate, was presented as a treatment that inhibited tumor cell growth in an orthotopictriple-negative human breast cancer xenograft model.111 However, in further experiments, this composition showed a poor pharmacokinetic profile, including a short plasma half-life and dose-limiting toxicity, therefore As2O3 formulation served as a solution for these disadvantages.112 To improve the anti-tumor properties of arsenic nanobins, then the anti-uPA antibody (ATN-291) was conjugated.113 In experiments with ovarian cancer cells, uPA-targeted nanobins were internalized about four-fold higher when compared with untargeted nanobins. The in vivo treatment with ATN-293-As2O3 nanobins decreased 47% of tumor burden in tested mice bearing orthotopic ovarian tumors.
Besides, several groups have demonstrated that NPs with different scaffolds (like liposomes, iron oxide, or gold NPs) conjugate with an inactive amino-terminal fragment (ATF) or 11-amino-acid sequence (U11) to target the expression of uPA in cancer cells.114–116
Iron oxide nanoparticles conjugated with ATF can work as the therapeutic system internalized via endocytosis pathway117 and also provide probes for monitoring the drug delivery by using MRI.118 Following, it was shown that ATF-conjugated iron oxide nanoparticles loaded with doxorubicin,118 noscapine119 or gemicitabine120 efficiently inhibited the breast, prostate and pancreatic tumor growth, respectively. Furthermore, the retained MRI contrasting effect making them as useful for the theranostic approach.
In another technique, PEGylated polylactide-based NPs, activated by uPA-specific sequence, have been designed to hinder nonspecific internalization of cell-penetrating peptides (CPP) and improve the therapeutic potential of drug treatment with paclitaxel.121 CPPs are a class of peptides with the ability to cross the cellular membrane without incurring significant cytotoxicity, therefore they became a good vectors for carrier systems.122 Due to the ectopic expression of uPA in tumor tissues, the uPA specific sequence was added to the CPP-conjugated nanoparticles and protected them from nonspecific internalization. Next, the uPA cleavage allowed the CPP-conjugated nanoparticles to internalize. The uPA-activated CPP-conjugated nanoparticles loaded with paclitaxel showed greater therapeutic efficacy in an in vivo glioma mouse model.121
Combination of Cysteine Cathepsin Inhibitors with Nanosized Drug Delivery Systems
Encapsulated cystatin into PLGA (Poly(lactic-co-glycolic acid)) has been shown to be more stable compared to its free form, while strongly absorbed by human breast epithelial cell line, MCF-10A neoT and presenting significantly decreased CtsB activity. The created complex was able to pass from cellular membrane to the lysosomal compartments that are rich in proteases enzymes after five minutes through endocytosis.123 At the other work, the same carriers were loaded with cystatin and labeled with the monoclonal antibody anty- cytokeratins, which are overexpressed in breast tumor cells. This selective uptake of tested treatment was presented in co-cultures of invasive breast cells, enterocytes (Caco-2) and monocytes/macrophages. Additionally, the provided drug delivery system significantly inhibited the proteolytic activity and reduced the invasive and metastatic potential of tumor cells.124 On the other hand, the broad-spectrum inhibitor JPM-565 has been successfully loaded in ~95 nm lipidated magnetic nanocarrier (ferri-liposomes) and tested in in vivo orthotopic murine models of primary mammary cancer (MMTV-PyMT). The results showed that the liposome-based application markedly decreased the cysteine Cts activity and, consequently, the tumor growth. Importantly, these nanocarriers possess contrast properties and were able to target the tumoral part and its microenvironment – stroma cells.125 Next, the lipidated CtsB inhibitor (NS-629) was designed to target extracellular CtsB exclusively. NS-629 was incorporated through a lipid linker into the preformed sterically stabilized PEG-coated nanosized stealth liposomes, thereby forming a lipidated nanocarrier capable of specific CtsB targeting (LNC-NS-629). Encapsulated doxorubicin in LNC-NS-629 has demonstrated a 22-fold improvement in inhibitory activity as compared to its free form against mammary tumor cells.126
Future Aims for Nanosized Drug Delivery Development
Although remarkable efforts have been made in recent years toward the development of next-generation NPs to improve the pharmacological and therapeutic properties of traditional medicine, there are still rate-limiting steps that need to be addressed. NP-based targeting systems can be subjected to aggregation with their small size and large surface area, or be phagocytosed by macrophages, which may cause cytotoxic effects through their intracellular degradation.127 Other important issues, including (1) low drug loading capacity,128 (2) lack of control during the distribution of carriers,129 (3) lack of biocompatibility,130 (4) controlling the interaction with biological barriers,131 (5) detecting and monitoring the exposure level132 and (6) assessing the impact on the environment,133 are also seen as significant drawbacks. In this context, novel techniques for developing and implementing nano-devices with approved quality and severe toxicity are needed. A real therapeutic breakthrough can be achieved solely by carrying out rigorous studies in the field of nano-therapy. As a result of these, nanosized systems combining multifunctional properties with pharmacological targets may improve the curative effect of antagonist agents and thus contribute to making an effective cancer treatment tool.
Conclusions
Conventional chemotherapy uses individual anti-cancer agents or combinations for the treatment of tumor cells, which intervenes primarily with the macromolecular synthesis processes, interfering with DNA synthesis and mitosis and leading to the death of proliferating cancer cells. However, tumor cells often present challenges to chemotherapeutic agents and endow high levels of chemoresistance. Therefore, new therapeutic approaches are urgently needed. The protease inhibitors are promising candidates for anti-cancer agents. By combining them with NP-based delivery systems, they can be exploited as a powerful medical tool, which may help to overcome the chemoresistance. This novel therapy form may substantially improve drug bioavailability, increase circulation time and achieve specific accumulation of treatment in the tumor tissues. Eventually, the decreased doses will protect healthy tissues, reduce side effects and ultimately lead to better outcomes and a higher quality of life for patients.
Abbreviations
HIV, human immunodeficiency virus; MMP, matrix metalloprotease; uPA, urokinase-type plasminogen activator; Cts, cathepsin; Ki, inhibitory constant; TIMP, tissue inhibitors of metalloproteinase; FDA, Food and Drug Administration; NP, nanoparticle; GNC, gold nanocluster; LTSL, lysolipid-containing thermosensitive liposome; PEG, polyethylene glycol; As, arsenic; U11, 11-amino-acid sequence; ATF, amino-terminal fragment; CPP, cell-penetrating peptide; PLGA, poly(lactic-co-glycolic acid).
Acknowledgments
We thank Alexandr V. Bazhin for fruitful discussions in relation to this paper.
Funding
This research was funded by the Russian Science Foundation (grant # 16-15-10410).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Berg JM, Tymoczko JL, Stryer L. Proteases: Facilitating a Difficult Reaction.
2. Eatemadi A, Aiyelabegan HT, Negahdari B, et al. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed Pharmacother. 2017;86:221–231. doi:10.1016/j.biopha.2016.12.021
3. López-Otín C, Bond JS. Proteases: multifunctional Enzymes in Life and Disease. J Biol Chem. 2008;283(45):30433–30437.
4. Brindley PJ, Kalinna BH, Wong JY, et al. Proteolysis of human hemoglobin by schistosome cathepsin D. Mol Biochem Parasitol. 2001;112(1):103–112.
5. Abad-Zapatero C, Goldman R, Muchmore SW, et al. Structure of a secreted aspartic protease from C. albicans complexed with a potent inhibitor: implications for the design of antifungal agents. Protein Sci. 1996;5(4):640–652.
6. Li Z, Chen X, Davidson E, et al. Anti-malarial drug development using models of enzyme structure. Chem Biol. 1994;1(1):31–37.
7. Wlodawer A, Erickson JW. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–585.
8. Love RA, Parge HE, Wickersham JA, et al. The Crystal Structure of Hepatitis C Virus NS3 Proteinase Reveals a Trypsin-like Fold and a Structural Zinc Binding Site. Cell. 1996;87(2):331–342.
9. Gibson W, Hall MR. Assemblin, an essential herpesvirus proteinase. Drug Des Discov. 1997;15(1):39–47.
10. Bernstein PR, Edwards PD, Williams JC. Inhibitors of human leukocyte elastase. Prog Med Chem. 1994;31:59–120.
11. Fath MA, Wu X, Hileman RE, et al. Interaction of Secretory Leukocyte Protease Inhibitor with Heparin Inhibits Proteases Involved in Asthma. J Biol Chem. 1998;273(22):13563–13569. doi:10.1074/jbc.273.22.13563
12. Stubbs MT, Bode W. A player of many parts: the spotlight falls on thrombin’s structure. Thromb Res. 1993;69(1):1–58. doi:10.1016/0049-3848(93)90002-6
13. Vassar R, et al. Beta-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science. 1999;286(5440):735–741. doi:10.1126/science.286.5440.735
14. Yan S, Sameni M, Sloane BF. Cathepsin B and human tumor progression.. Biol Chem. 1998;379(2):113–123.
15. Sevenich L, Joyce JA. Pericellular proteolysis in cancer. Genes Dev. 2014;28(21):2331–2347. doi:10.1101/gad.250647.114
16. Petushkova AI, Savvateeva LV, Korolev DO, Zamyatnin AA. Cysteine Cathepsins: potential Applications in Diagnostics and Therapy of Malignant Tumors. Biochemistry (Mosc). 2019;84(7):746–761. doi:10.1134/S000629791907006X
17. Soond SM, Kozhevnikova MV, Zamyatnin JAA. ‘Patchiness’ and basic cancer research: unravelling the proteases. Cell Cycle. 2019;18(15):1687–1701. doi:10.1080/15384101.2019.1632639
18. Rudzińska M, Parodi A, Soond SM, et al. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int J Mol Sci. 2019;20(14):3602. doi:10.3390/ijms20143602
19. Zhivotovsky B, Gahm A, Orrenius S. Two different proteases are involved in the proteolysis of lamin during apoptosis. Biochem Biophys Res Commun. 1997;233(1):96–101. doi:10.1006/bbrc.1997.6411
20. Glynn SE. Multifunctional Mitochondrial AAA Proteases. Front Mol Biosci. 2017;4:34.
21. Guicciardi ME, Deussing J, Miyoshi H, et al. Cathepsin B contributes to TNF-α–mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106(9):1127–1137.
22. Zdolsek J, Zhang H, Roberg K. Brunk U. H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic Res Commun. 1993;18(2):71–85.
23. Petushkova AI, Zamyatnin AAJ. Redox-Mediated Post-Translational Modifications of Proteolytic Enzymes and Their Role in Protease Functioning. Biomolecules. 2020;10(4):650.
24. Besingi RN, Clark PL. Extracellular Protease Digestion to Evaluate Membrane Protein Cell Surface Localization. Nat Protoc. 2015;10(12):2074–2080.
25. Rudzińska M, Parodi A, Maslova VD, et al. Cysteine Cathepsins Inhibition Affects Their Expression and Human Renal Cancer Cell Phenotype. Cancers. 2020;12(5):1310.
26. Farady CJ, Craik CS. Mechanisms Of Macromolecular Protease Inhibitors. Chembiochem. 2010;11(17):2341–2346.
27. Rawlings ND, Tolle DP, Barrett AJ. Evolutionary families of peptidase inhibitors. Biochem J. 2004;378(Pt 3):705–716.
28. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803(1):55–71.
29. Law RH, Zhang Q, McGowan S, et al. An overview of the serpin superfamily. Genome Biol. 2006;7(5):216.
30. The K-JN. Role of Cysteine Proteinases and their Inhibitors in the Host-Pathogen Cross Talk. Curr Protein Pept Sci. 2012;13(8):767–775.
31. Laronha H, Carpinteiro I, Portugal J, et al. Challenges in Matrix Metalloproteinases Inhibition. Biomolecules. 2020;10(5):717.
32. Liu J, Khalil RA. Matrix Metalloproteinase Inhibitors as Investigational and Therapeutic Tools in Unrestrained Tissue Remodeling and Pathological Disorders. Prog Mol Biol Transl Sci. 2017;148:355–420.
33. Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5(9):785–799.
34. Winer A, Adams S, Mignatti P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: turning Past Failures Into Future Successes. Mol Cancer Ther. 2018;17(6):1147–1155.
35. Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2020;13(12):904–927.
36. Fields GB. The Rebirth of Matrix Metalloproteinase Inhibitors: moving Beyond the Dogma. Cells. 2019;8(9):984.
37. Marshall DC, Lyman SK, McCauley S, et al. Selective Allosteric Inhibition of MMP9 Is Efficacious in Preclinical Models of Ulcerative Colitis and Colorectal Cancer. PLoS One. 2015;10(5):e0127063.
38. Fischer T, Riedl R. Inhibitory Antibodies Designed for Matrix Metalloproteinase Modulation. Molecules. 2019;24(12):2265.
39. Tauro M, McGuire J, Lynch CC. New approaches to selectively target cancer-associated matrix metalloproteinase activity. Cancer Metastasis Rev. 2014;33(4):1043–1057.
40. Overall CM. Dilating the degradome: matrix metalloproteinase 2 (MMP-2) cuts to the heart of the matter. Biochem J. 2004;383(Pt. 3):e5–e7.
41. Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4(8):617–629.
42. Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6(3):227–239.
43. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174.
44. Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci. 2016;5:e47.
45. Shin SA, Moon SY, Kim WY. Structure-Based Classification and Anti-Cancer Effects of Plant Metabolites. Int J Mol Sci. 2018;19(9):2651.
46. Fear G, Komarnytsky S, Raskin I. Protease inhibitors and their peptidomimetic derivatives as potential drugs. Pharmacol Ther. 2017;113(2):354–368.
47. Schmitt M, Harbeck N, Brünner N, et al. Cancer therapy trials employing level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev Mol Diagn. 2011;11(6):617–634.
48. Xu P, Andreasen PA, Huang M. Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors. Int J Biol Sci. 2017;13(10):1222–1233.
49. Duffy FJ, Devocelle M, Shields DC. Computational approaches to developing short cyclic peptide modulators of protein-protein interactions. Methods Mol Biol. 2015;1268:241–271.
50. Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81(1):136–147.
51. Schweinitz A, Steinmetzer T, Banke IJ, et al. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J Biol Chem. 2004;279(32):33613–33622.
52. Henneke I, Greschus S, Savai R, et al. Inhibition of urokinase activity reduces primary tumor growth and metastasis formation in a murine lung carcinoma model. Am J Respir Crit Care Med. 2010;181(6):611–619.
53. Verma S, Dixit R, Pandey KC. Cysteine Proteases: modes of Activation and Future Prospects as Pharmacological Targets. Front Pharmacol. 2016;25(7):107.
54. Hanada K, Tamai M, Yamagishi M, Ohmura S, Sawada J, Isolation TI. Characterization of E–64, a New Thiol Protease Inhibitor. Agric Biol Chem. 2014;9:0002–1369.
55. Siklos M, BenAissa M, Thatcher GRJ. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B. 2015;5(6):506–519.
56. Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2020;10(4):307–317.
57. Leung D, Abbenante G, Fairlie DP. Protease inhibitors: current status and future prospects. J Med Chem. 2000;43(3):305–341.
58. Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med Chem. 2010;2(6):949–964.
59. Dana D, Pathak SKA. Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules. 2020;25(3):698.
60. Murata M, Miyashita S, Yokoo C, et al. Novel epoxysuccinyl peptides. Selective inhibitors of cathepsin B, in vitro. FEBS Lett. 1991;280:2.
61. Katunuma N, Murata E, Kakegawa H, et al. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 1999;458(1):6–10.
62. Katunuma N. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(2):29–39.
63. Duong LT, Wesolowski GA, Leung P, et al. Efficacy of a cathepsin K inhibitor in a preclinical model for prevention and treatment of breast cancer bone metastasis. Mol Cancer Ther. 2014;13(12):2898–2909.
64. Burden RE, Gormley JA, Jaquin TJ, et al. Antibody-mediated inhibition of cathepsin S blocks colorectal tumor invasion and angiogenesis. Clin Cancer Res. 2009;15(19):6042–6051.
65. Ward C, Kuehn D, Burden RE, et al. Antibody targeting of cathepsin S inhibits angiogenesis and synergistically enhances anti-VEGF. PLoS One. 2010;5(9):e12543.
66. O’Halloran TV, Ahn R, Hankins P, Swindell E, Mazar AP. The Many Spaces of uPAR: delivery of Theranostic Agents and Nanobins to Multiple Tumor Compartments through a Single Target. Theranostics. 2013;3(7):496–506.
67. Hideshima T, Bradner JE, Wong J, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Nat Acad Sci U S A. 2005;102(24):8567–8572.
68. Mitsiades N, Mitsiades CS, Richardson PG, et al. The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003;101(6):2377–2380.
69. Mabonga L, Kappo AP. Peptidomimetics: A Synthetic Tool for Inhibiting Protein–Protein Interactions in Cancer. Int J Pept Res Ther. 2019;26(1):225–241.
70. Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011;21(4):228–237.
71. Verhamme IM, Leonard SE, Perkins RC. Proteases: pivot Points in Functional Proteomics. Methods Mol Biol. 2019;1871:313–392.
72. Kappelhoff R, Puente XS, Wilson CH, Seth A, López-Otín C, Overall CM. Overview of transcriptomic analysis of all human proteases, non-proteolytic homologs and inhibitors: organ, tissue and ovarian cancer cell line expression profiling of the human protease degradome by the CLIP-CHIP™ DNA microarray. Biochim Biophys Acta Mol Cell Res. 2017;1864(11 Pt B):2210–2219.
73. Verbovsek U, Motaln H, Rotter A, et al. Expression analysis of all protease genes reveals cathepsin K to be overexpressed in glioblastoma. PLoS One. 2014;9(10):e111819.
74. López-Otín C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer. 2010;10(4):278–292.
75. Kim EE, Baker CT, Dwyer MD, et al. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J Am Chem Soc. 1995;117(3):1181–1182.
76. Mueller BU, Anderson BD, Farley MQ, et al. Pharmacokinetics of the protease inhibitor KNI-272 in plasma and cerebrospinal fluid in nonhuman primates after intravenous dosing and in human immunodeficiency virus-infected children after intravenous and oral dosing. Antimicrob Agents Chemother. 1998;42(7):1815–1818.
77. Barry M, Gibbons S, Back D, Mulcahy F. Protease inhibitors in patients with HIV disease. Clin Important Pharm Considerations Clin Pharm. 1997;32(3):194–209.
78. Williams GC, Sinko PJ. Oral absorption of the HIV protease inhibitors: a current update. Adv Drug Deliv Rev. 1999;39(1–3):211–238.
79. Yeh KC, Deutsch PJ, Haddix H, et al. Single-dose pharmacokinetics of indinavir and the effect of food. Antimicrob Agents and Chemother. 1998;42:2.
80. Fitzsimmons ME, Collins JM. Selective biotransformation of the human immunodeficiency virus protease inhibitor saquinavir by human small-intestinal cytochrome P4503A4: potential contribution to high first-pass metabolism. Drug Metab Dispos. 1997;25(2):256–266.
81. Tatham LM, Rannard SP, Owen A. Nanoformulation strategies for the enhanced oral bioavailability of antiretroviral therapeutics. Ther Deliv. 2015;6(4):469–490.
82. Kigen G, Edwards G. Enhancement of saquinavir absorption and accumulation through the formation of solid drug nanoparticles. BMC Pharmacol Toxicol. 2018;19(1):79.
83. Imperiale JC, Nejamkin P, Del Sole MJ, Lanusse CE, Sosnik A. Novel protease inhibitor-loaded Nanoparticle-in-Microparticle Delivery System leads to a dramatic improvement of the oral pharmacokinetics in dogs. Biomaterials. 2015;37:383–394.
84. Varshosaz J, Fard MM, Mirian M, Hassanzadeh F. Targeted Nanoparticles for Co-delivery of 5-FU and Nitroxoline, a Cathepsin B Inhibitor, in HepG2 Cells of Hepatocellular Carcinoma. Anticancer Agents Med Chem. 2020;20(3):346–358.
85. Dheer D, Nicolas J, Shankar R. Cathepsin-sensitive nanoscale drug delivery systems for cancer therapy and other diseases. Adv Drug Deliv Rev. 2019;151–152:130–151.
86. Rizvi SAA, Saleh AM. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm J. 2018;26(1):64–70.
87. Patra JK, Das G, Fraceto LF, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnology. 2018;16(1):71.
88. Parodi A, Miao J, Soond SM, et al. Albumin Nanovectors in Cancer Therapy and Imaging. Biomolecules. 2019;9(6):218.
89. Maysinger D, Gran ER, Bertorelle F, et al. Gold nanoclusters elicit homeostatic perturbations in glioblastoma cells and adaptive changes of lysosomes. Theranostics. 2020;10(4):1633–1648.
90. Parodi A, Rudzińska M, Deviatkin AA, et al. Established and Emerging Strategies for Drug Delivery Across the Blood-Brain Barrier in Brain Cancer. Pharmaceutics. 2019;11(5):245.
91. Zou Y, Zhang L, Yang L, et al. “Click” chemistry in polymeric scaffolds: bioactive materials for tissue engineering. J Control Release. 2018;273:160–179.
92. Chen LY, Wang CW, Yuan Z, et al. Fluorescent gold nanoclusters: recent advances in sensing and imaging. Anal Chem. 2015;87(1):216–229.
93. Shen D, Henry M, Trouillet V, et al. Zwitterion functionalized gold nanoclusters for multimodal near infrared fluorescence and photoacoustic imaging. APL Materials. 2017;5(5):053404.
94. Zhang C, Li C, Liu Y, et al. Gold Nanoclusters-Based Nanoprobes for Simultaneous Fluorescence Imaging and Targeted Photodynamic Therapy with Superior Penetration and Retention Behavior in Tumors. Adv Funct Mater. 2015;25(8):1314–1325.
95. Yang P, Zhang S, Chen X, Liu X, Wang Z, Li Y. Recent developments in polydopamine fluorescent nanomaterials. Mater Horiz. 2020;7:746–761.
96. Wang Y, Wang Y, Zhou F, Kim P, Protein-Protected XY. Au Clusters as a New Class of Nanoscale Biosensor for Label-Free Fluorescence Detection of Proteases. Small. 2012;8(24):3769–3773.
97. Colombé C, Le Guével X, Martin-Serrano A, et al. Gold nanoclusters as a contrast agent for image-guided surgery of head and neck tumors. Nanomedicine. 2019;20:102011.
98. Zhang X-D, Luo Z, Chen J, et al. Ultrasmall Au10− 12(SG)10−12Nanomolecules for High Tumor Specificity and Cancer Radiotherapy. Adv Mater. 2014;26(26):4565–4568.
99. Zhou F, Feng B, Yu H, et al. Cisplatin Prodrug-Conjugated Gold Nanocluster for Fluorescence Imaging and Targeted Therapy of the Breast Cancer. Theranostics. 2016;6(5):679–687.
100. Zhan Z, Huang Y, Lin G, Huang S, Zeng F, Wu S. C, Huang Y, Lin G, Huang S, Zeng F, Wu S. A Gold Nanocage/Cluster Hybrid Structure for Whole-Body Multispectral Optoacoustic Tomography Imaging, EGFR Inhibitor Delivery, and Photothermal Therapy. Small. 2019;15(33):1900309. doi:10.1002/smll.201900309
101. Daglioglu C, Kaci FN. Cascade therapy with doxorubicin and survivin-targeted tailored nanoparticles: an effective alternative for sensitization of cancer cells to chemotherapy. Int J Pharm. 2019;561:74–81. doi:10.1016/j.ijpharm.2019.02.036
102. Daglioglu C, Synthesis OB. Characterization of AICAR and DOX Conjugated Multifunctional Nanoparticles as a Platform for Synergistic Inhibition of Cancer Cell Growth. Bioconjug Chem. 2016;27(4):1098–1111. doi:10.1021/acs.bioconjchem.6b00080
103. Shen S, Xiao-Jiao D, Liu J. Delivery of bortezomib with nanoparticles for basal-like triple-negative breast cancer therapy. J Control Release. 2015;208:14–24. doi:10.1016/j.jconrel.2014.12.043
104. Park JE, Park J, Jun Y, et al. Expanding Therapeutic Utility of Carfilzomib for Breast Cancer Therapy by Novel Albumin-coated Nanocrystal Formulation. J Control Release. 2019;302:148–159. doi:10.1016/j.jconrel.2019.04.006
105. Lyu Y, Xiao Q, Yin L, Yang L, He W. Potent delivery of an MMP inhibitor to the tumor microenvironment with thermosensitive liposomes for the suppression of metastasis and angiogenesis. Signal Transduct Target Ther. 2019;4(1):26. doi:10.1038/s41392-019-0054-9
106. Lombardo D, Kiselev MA, Caccamo MT. Smart Nanoparticles for Drug Delivery Application: development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. J Nanomater. 2009;2(9):1–26.
107. Hatakeyama H, Akita H, Ishida E, et al. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int J Pharm. 2007;342(1–2):194–200. doi:10.1016/j.ijpharm.2007.04.037
108. Nunes SS, Fernandes RS, Cavalcante CH, et al. Influence of PEG coating on the biodistribution and tumor accumulation of pH-sensitive liposomes. Drug Deliv Transl Res. 2019;9(1):123–130. doi:10.1007/s13346-018-0583-8
109. Zhu L, Kate P, Torchilin VP, Metalloprotease M. 2-Responsive Multifunctional Liposomal Nanocarrier for Enhanced Tumor Targeting. ACS Nano. 2012;6(4):3491–3498. doi:10.1021/nn300524f
110. Gu G, Xia H, Hu Q, et al. PEG-co-PCL nanoparticles modified with MMP-2/9 activatable low molecular weight protamine for enhanced targeted glioblastoma therapy. Biomaterials. 2013;34(1):196–208. doi:10.1016/j.biomaterials.2012.09.044
111. Ahn RW, Chen F, Chen H, et al. A novel nanoparticulate formulation of arsenic trioxide with enhanced therapeutic efficacy in a murine model of breast cancer. Clin Cancer Res. 2010;16(14):3607–3617. doi:10.1158/1078-0432.CCR-10-0068
112. Uslu R, Sanli UA, Sezgin C, et al. Arsenic trioxide-mediated cytotoxicity and apoptosis in prostate and ovarian carcinoma cell lines.. Clin Cancer Res. 2000;6(12):4957–4964.
113. Zhang Y, Kenny HA, Swindell EP, et al. Urokinase Plasminogen Activator System-Targeted Delivery of Nanobins as a Novel Ovarian Cancer Therapy. Mol Cancer Ther. 2013;12(12):2628–2639. doi:10.1158/1535-7163.MCT-13-0204
114. Ming Wang M, Löwik DWPM, Miller AD, Thanou M. Targeting the urokinase plasminogen activator receptor with synthetic self-assembly nanoparticles. Bioconjug Chem. 2009;20(1):32–40. doi:10.1021/bc8001908
115. Galbiati E, Cassani M, Verderio P, et al. Peptide-Nanoparticle Ligation Mediated by Cutinase Fusion for the Development of Cancer Cell-Targeted Nanoconjugates. Bioconjug Chem. 2015;26(4):680–689. doi:10.1021/acs.bioconjchem.5b00005
116. Huang X, Peng X, Wang Y, Wang Y, Shin DM. A Reexamination of Active and Passive Tumor Targeting by Using Rod-Shaped Gold Nanocrystals and Covalently Conjugated Peptide Ligands. ACS Nano. 2010;4(10):5887–5896. doi:10.1021/nn102055s
117. Sun X-H, Tan L, Li C-Y, et al. A novel gene delivery system targeting urokinase receptor. Acta Biochim Biophys Sin (Shanghai). 2004;36(7):485–491. doi:10.1093/abbs/36.7.485
118. Yang L, Cao Z, Sajja HK, et al. Development of Receptor Targeted Magnetic Iron Oxide Nanoparticles for Efficient Drug Delivery and Tumor Imaging. J Biomed Nanotechnol. 2008;4(4):439–449. doi:10.1166/jbn.2008.007
119. Abdalla MO, Karna P, Sajja HK, et al. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J Control Release. 2011;149(3):314–322. doi:10.1016/j.jconrel.2010.10.030
120. Lee GY, Qian WP, Wang L, et al. Theranostic nanoparticles with controlled release of gemcitabine for targeted therapy and MRI of pancreatic cancer. ACS Nano. 2013;7(3):2078–2089. doi:10.1021/nn3043463
121. Zhang B, Zhang Y, Liao Z, et al. UPA-sensitive ACPP-conjugated nanoparticles for multi-targeting therapy of brain glioma. Biomaterials. 2015;36:98–109. doi:10.1016/j.biomaterials.2014.09.008
122. Copolovici DM, Langel K, Eriste E, Langel Ü. Cell-penetrating peptides: design, synthesis, and applications. ACS Nano. 2014;8(3):1972–1994. doi:10.1021/nn4057269
123. Cegnar M, Kos J, Kristl J. Intracellular delivery of cysteine protease inhibitor cystatin by polymeric nanoparticles. J Nanosci Nanotechnol. 2006;6(9):3087–3094. doi:10.1166/jnn.2006.401
124. Obermajer N, Kocbek P, Repnik U, et al. Immunonanoparticles − an effective tool to impair harmful proteolysis in invasive breast tumor cells. FEBS Journal. 2007;274(17):4416–4427. doi:10.1111/j.1742-4658.2007.05971.x
125. Mikhaylov G, Mikac U, Magaeva AA, et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat Nanotechnol. 2011;6(9):594–602. doi:10.1038/nnano.2011.112
126. Mikhaylov G, Klimpel D, Schaschke N, et al. Selective Targeting of Tumor and Stromal Cells By a Nanocarrier System Displaying Lipidated Cathepsin B Inhibitor. Angew Chem Int Ed Engl. 2014;53(38):10077–10081. doi:10.1002/anie.201402305
127. De J. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. doi:10.2147/IJN.S596
128. Shen S, Wu Y, Liu Y, Wu D. High drug-loading nanomedicines: progress, current status, and prospects. Int J Nanomedicine. 2017;12:4085–4109. doi:10.2147/IJN.S132780
129. Gusić N, Ivković A, VaFaye J, et al. Nanobiotechnology and bone regeneration: a mini-review. Int Orthop. 2014;38(9):1877–1884. doi:10.1007/s00264-014-2412-0
130. Mokhtarzadeh A, Alibakhshi A, Yaghoobi H, Hashemi M, Hejazi M, Ramezani M. Recent advances on biocompatible and biodegradable nanoparticles as gene carriers. Expert Opin Biol Ther. 2016;16(6):771–785. doi:10.1517/14712598.2016.1169269
131. Tsai H, Imae T. Fabrication of dendrimers toward biological application. Prog Mol Biol Transl Sci. 2011;104:101–140.
132. Karimi M, Ghasemi A, Zangabad PS, et al. Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems. Chem Soc Rev. 2016;45(5):1457–1501.
133. Jahangirian H, Lemraski EG, Webster TJ, Rafiee-Moghaddam R, Abdollahi Y. A review of drug delivery systems based on nanotechnology and green chemistry: green nanomedicine. Int J Nanomedicine. 2017;12:2957–2978. doi:10.2147/IJN.S127683
134. Clemente M, Corigliano MG, Pariani SA, Sánchez-López EF, Sander VA, Ramos-Duarte VA. Plant Serine Protease Inhibitors: biotechnology Application in Agriculture and Molecular Farming. Int J Mol Sci. 2019;20(6):1345. doi:10.3390/ijms20061345
135. Hossen S, Hossain MK, Basher MK, Mia MNH, Rahman MT, Uddin MJ. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J Adv Res. 2019;15:1–18. doi:10.1016/j.jare.2018.06.005
136. Masood F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy. Mater Sci Eng C Mater Biol Appl. 2016;60:569–578. doi:10.1016/j.msec.2015.11.067
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.