")
Back to Journals » Journal of Blood Medicine » Volume 11
Current Perspectives on Diagnostic Assays and Anti-PF4 Antibodies for the Diagnosis of Heparin-Induced Thrombocytopenia
Authors Sahu KK , Jindal V , Anderson J, Siddiqui AD , Jaiyesimi IA
Received 28 March 2020
Accepted for publication 27 July 2020
Published 17 August 2020 Volume 2020:11 Pages 267—277
DOI https://doi.org/10.2147/JBM.S232648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Kamal K Sahu,1 Vishal Jindal,2 Joseph Anderson,2 Ahmad D Siddiqui,1 Ishmael A Jaiyesimi2
1Hemato-oncology Division, Department of Internal Medicine, Saint Vincent Hospital, Worcester, MA 01608, USA; 2Department of Hematology and Oncology, Oakland University William Beaumont School of Medicine, Royal Oak, MI 48073, USA
Correspondence: Kamal K Sahu
Hemato-Oncology Division, Department of Internal Medicine, Saint Vincent Hospital, 23 Summer Street, Worcester, Massachusetts 01608, USA
Email [email protected]
Vishal Jindal
Department of Hematology and Oncology, Oakland University-William Beaumont School of Medicine, 3601 W. 13 Mile Road, Royal Oak, MI 48073, USA
Tel +1 347 589-2541
Fax +1 248 551-2947
Email [email protected]
Abstract: Heparin-induced thrombocytopenia (HIT) is a recognized clinical entity in patients receiving unfractionated heparin and low–molecular weight heparin. Currently, diagnosing HIT includes the combination of a physician’s clinical suspicion based on a clinical scoring system and a series of laboratory tests. In the present article, we discuss challenges in suspecting and diagnosing HIT in consideration of the turnaround time of available tests and recent advances in techniques and methodologies of newer immunoassays and functional assays.
Keywords: heparin-induced thrombocytopenia, laboratory diagnosis, immunoassay, functional assay, platelets, platelet factor 4, serotonin-release assay, HIPA test
Introduction
Heparin-induced thrombocytopenia (HIT) is a complication following exposure to heparin. It is most commonly seen in patients receiving unfractionated heparin (UFH) and less often with low–molecular weight heparin (LMWH).1 The approximate proportion of patients developing HIT after receiving heparin for >4 days has been reported as up to 5%.2,3 Patients receiving UFH have a higher chance of developing HIT (~2.6%) than patients receiving LMWH (~0.2%).4 More importantly, higher mortality — up to 20% — has been reported with untreated HIT. However, with early recognition and treatment, mortality incidence has significantly reduced to <2%.
Although HIT occurs in a very small proportion of patients exposed to heparin and is unrelated to dosage, schedule, or route of administration, there is no predictor that can detect the predisposed population. Since most cases of thrombocytopenia in hospitalized patients on heparin are not caused by HIT, clinicians must be able to differentiate rare cases of HIT from the many without HIT.5,7 Similarly, overdiagnosing HIT contributes to an unnecessary burden on patient-care resources, in that alternative anticoagulants are frequently more expensive. As such, a detailed understanding of the pathophysiology, evaluation, and management of HIT facilitates appropriate and timely treatment.1
Functional assays such as the 14C serotonin–release assay (14C-SRA) and heparin-induced platelet activation (HIPA) test are considered gold-standard tests for detecting pathological HIT antibodies. However, the 14C-SRA is a time-consuming assay, expensive to perform, requires a specialized laboratory setup, and utilizes radioactive elements. A recent study has shown that absolute immature-platelet count could complement anti-PF4 heparin testing in evaluating patients suspected of HIT.8 Therefore, there is impetus for research focusing on alternative ways to diagnose HIT.
Review
Mechanisms of Heparin-Induced Thrombocytopenia
Heparin is a commonly used anticoagulant in various clinical settings, and can be given either intravenously or subcutaneously. In HIT, heparin interacts with PF4, a small cytokine belonging to the CXC chemokine family that is also known as CXCL4. This chemokine is released from α-granules of activated platelets during platelet aggregation, forming a complex with vascular endothelial heparin–like molecules and normally promoting platelet function at the site of vascular injury. With the administration of exogenous heparin, competitive binding results in the suppression of platelet function. HIT is caused by an autoimmune reaction, whereby the immune system identifies the heparin–PF4 complex as a threat and IgG antibodies are formed in response. Antibody–antigen complexes (IgG–heparin–PF4) bind to platelet FcγRIIA receptors, leading to platelet destruction. Platelet destruction in turn prompts the release of more PF4 molecules. This can trigger a cycle that results in a rapid and significant drop (eg, 50% or more) in platelet numbers, ultimately leading to thrombocytopenia.9 However, not all patients who receive heparin will develop autoimmune antibodies, and not all patients who develop antibodies will develop thrombocytopenia. Typically, thrombocytopenia results in bleeding, although up to 55% with HIT present with thrombotic events.9,16 It is also important to note that development and activity of these antibodies depends on the level of PF4 antigen in relation to heparin (or other GAGs). Studies have shown that PF4:heparin ratios of 3:1–0.7:1 have the potential to form ultralarge complexes.17,18 Similarly, extracellular GAG can mimic heparin and form a complex with PF4–GAG antibodies, which can indeed be pathogenic.19
Disease Burden of HIT
HIT is more commonly triggered by treatment with UFH (8%–17% of patients receiving heparin), followed by LMWH and fondaparinux (2%–8%).20,21 The production of antibodies against the PF4–heparin complex is an immunologic response that can be seen in variable percentages of patients (8%–50%).21 However, clinical sequelae of these antibodies are far less commonly noted (0.2%–3%).21 The destruction of platelets may generate cell fragments, which may predispose to the development of new or worsening thrombosis, known as HIT and thrombosis (HITT).22,23 If not detected in a timely fashion, HITT can be a catastrophic event, and mortality secondary to thrombus is generally cited as 5%–10%, with some reporting mortality as high as 20% among HIT patients.24 With advanced techniques of detection and early intervention, the mortality rate can be reduced to <2%.24 In the following sections, we discuss in detail newer investigations related to HIT diagnosis.
Clinical Diagnostic Scoring Systems
Diagnosis of HIT involves a stepwise process from suspicion to screening and finally making the diagnosis (Figure 1). The three most common clinical scoring systems studied are the 4 Ts, HEP score (HIT-expert probability) and HIT score for cardiopulmonary bypass.25,29 The 4T clinical scoring system is most commonly used in clinical practice and is based on typical clinical features and laboratory results of HIT. This scoring system is easy to apply and has a high negative predictive value, thereby meaning a low-probability 4T score serves as an excellent tool to rule out HIT. However, due to the low positive predictive value of this system for intermediate- and high-scoring groups, it is difficult clinically to rule out HIT in these subsets of patients.19 Therefore, laboratory testing constitutes an integral part of HIT diagnosis (Table 1).
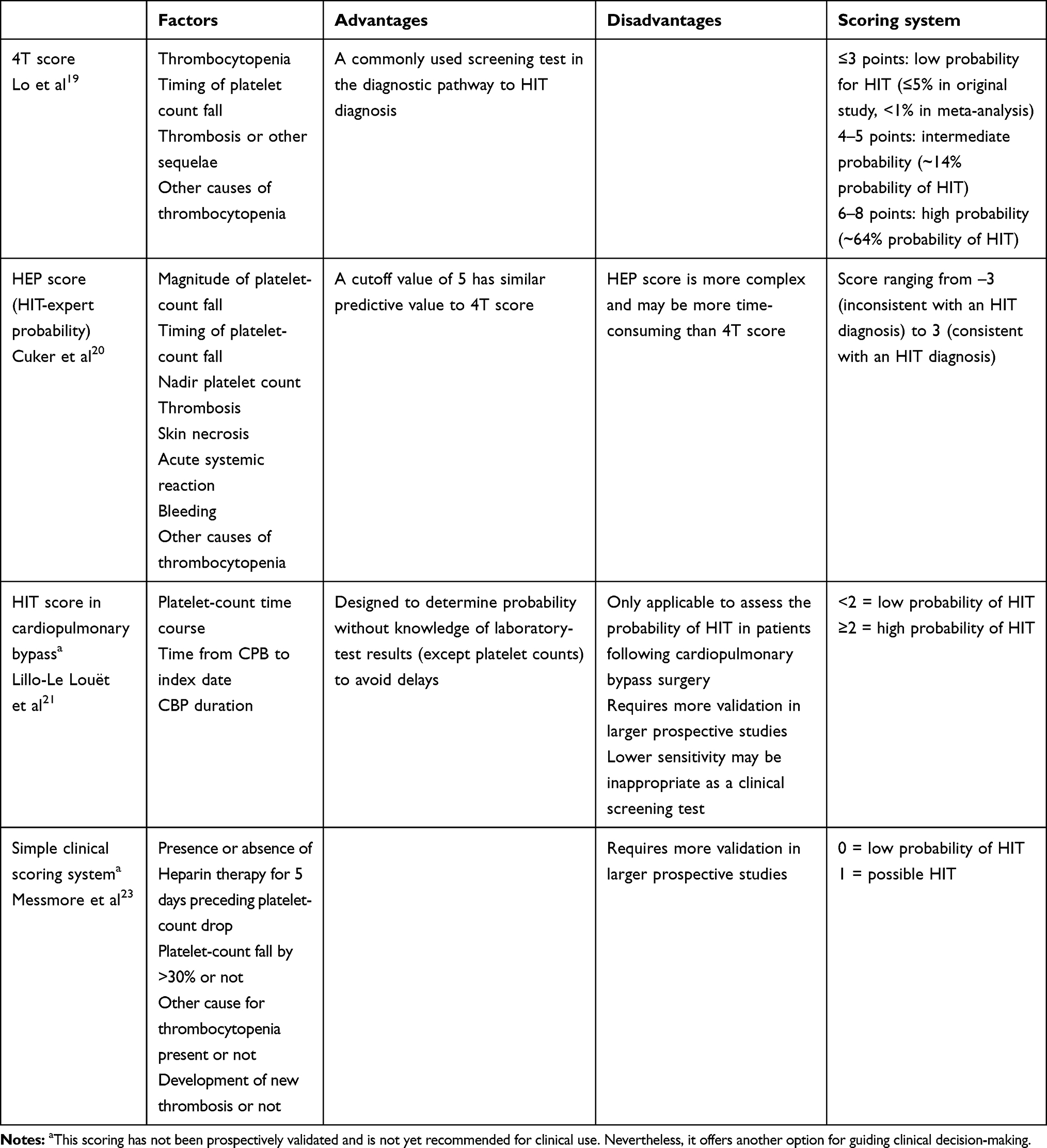 |
Table 1 Clinical scoring systems and variables used, with advantages and disadvantages |
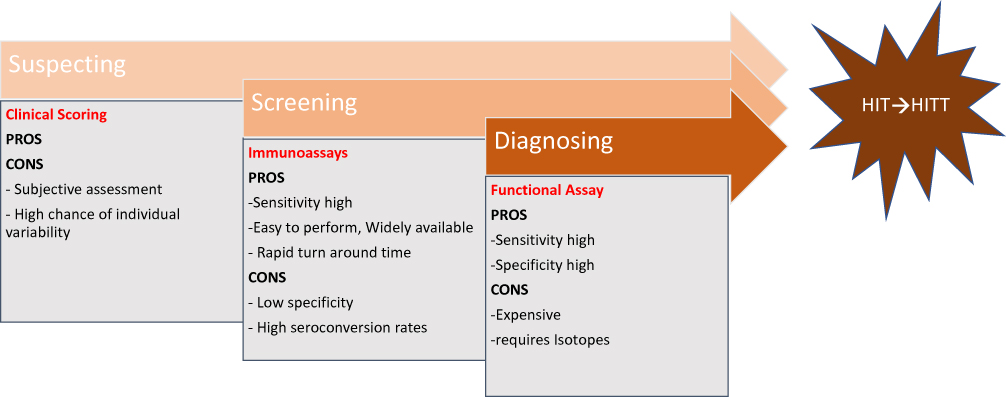 |
Figure 1 Guide to decision-making for HIT from suspicion to diagnosis, with advantages and disadvantages of the assays. |
Laboratory Diagnosis of HIT
Following clinical scoring, when a clinician suspects HIT the next step is evaluation of the patient with laboratory studies. Currently, two classes of assays are available: immunoassays (antigenic), which detect binding of anti-PF4–heparin antibodies, and functional assays (platelet-activation assays), which are more specific and detect if circulating antibodies have the capability to activate platelets. Each of these assays tests separate stages in the HIT pathway, with immunoassays capable of detecting initial immunoresponse and functional assays detecting specific pathogenic antibodies causing platelet activation in the presence of heparin.
The various phases of HIT as shown in Figure 2 are helpful to understand test results and subsequent interpretation. Suspected HIT is the first phase, when a patient is suspected of having HIT based on clinical scenarios. Acute HIT constitutes the phase when a laboratory confirmation is made, and this phase lasts till the point of platelet-count recovery. Following this, subacute HIT is divided into two subphases namely: A (both functional assay and immunoassay are positive), and B (functional assay is negative, immunoassay is still positive). The final phase is remote HIT, when immunoassays are no longer able to detect antibodies (Figure 2). It is important to remember that some patients who have clinical signs of HIT fail to have antibodies detected by current assays. For instance, in patients with HIT diagnosis clinically, up to 10% could have a negative H/PF4 ELISA result. Similarly, another subset of patients with positive tests may not necessarily have clinical HIT. This proportion is higher in surgical patients (up to 50%) than patients admitted for medical reasons (up to 20%).
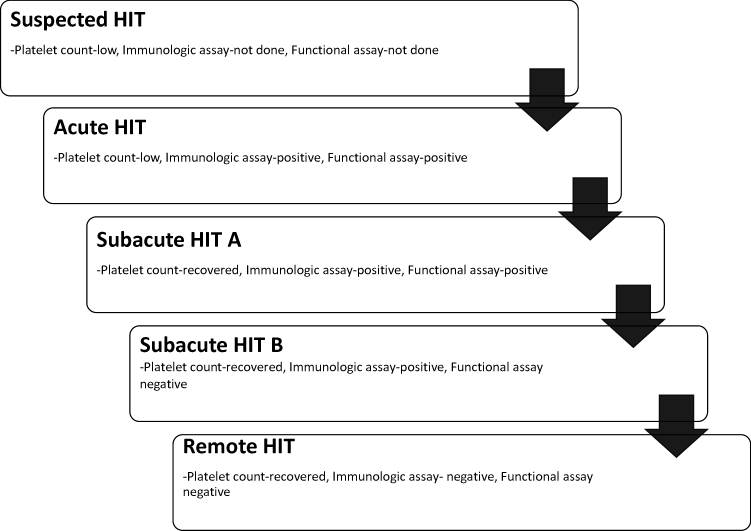 |
Figure 2 The five phases of HIT, ranging from cases of suspected HIT without confirmatory laboratory tests; acute HIT, a highly prothrombotic phase that persists until platelet-count recovery through subacute and remote HIT, where functional and immunologic assays return to negative. |
Immunoassays
Immunoassays measure the presence of anti-PF4–heparin antibodies using one of several antibody-capturing platforms (eg ELISA based, particle gel, turbidimetry). Immunoassays have high sensitivity (>99%), thereby making them ideal for use as an initial screening test for HIT. The drawback is low specificity (30%–70%) for a diagnosis of HIT, due to asymptomatic seroconversions.30,31 As such, they have a lower positive predictive value. In general, only 10%–50% of patients with positive immunoassay results have platelet-activating antibodies. Immunoassays have the advantage of being able to be performed by most laboratories routinely and a rapid response time.
Figure 3 shows the timeline from the discovery of platelets (1882) till the recent discovery of chimeric antibodies for diagnosis of HIT (2017). The first polyspecific ELISA to detect HIT antibodies was developed in 1992.21,32,45 In this test, suspected patients’ serum/plasma is added to PF4–polyanion complex–embedded microtiter plate wells. The semiquantitative nature of the test allows assessment of intensity of color change, and measures it as optical density (OD) at a specific wavelength (typically 405 nm), which is proportional to the concentration of bound antibodies. Since the development of ELISA for HIT, many studies have been done to assess the clinical performance of various in-house and commercially available ELISAs. It is important to note that there is no universally agreed-upon OD value for the interpretation of the results, as ELISA OD units vary based on the heparin–PF4 ELISA kit being used. Therefore, it is important for physicians to connect with local reporting laboratories for the OD cutoff set by the reporting laboratory, in order to interpret the ELISA results.46
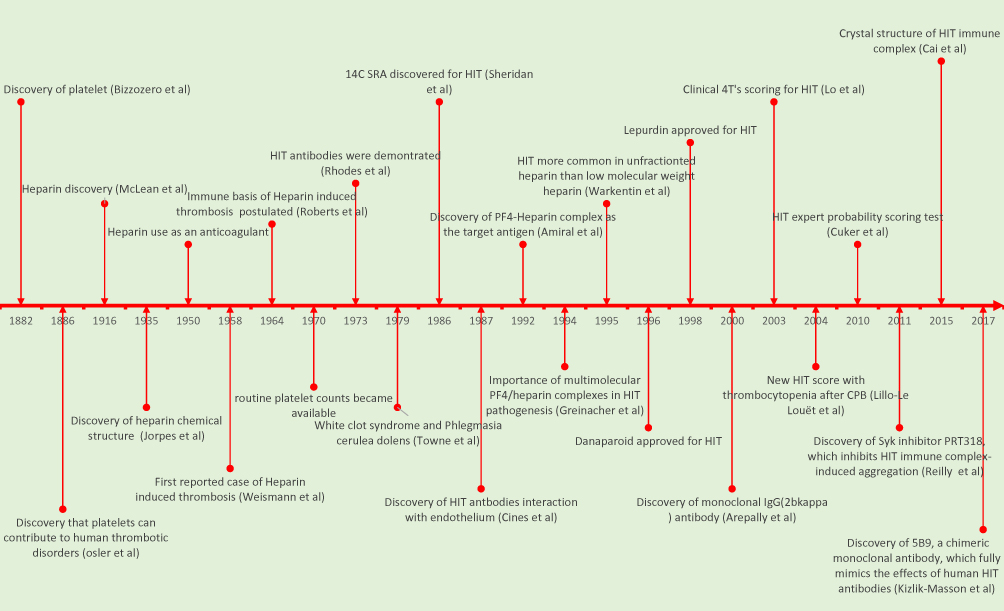 |
Figure 3 Timeline from discovery of platelets, HIT, and subsequent diagnostic milestones. |
Many laboratories report HIT ELISA results as positive or negative based on the prefixed OD cutoff set in their laboratories. However, an absolute value is more informative than nominal results, and is helpful in anticipating the test results of an SRA functional test. A study on 1,553 samples from suspected HIT patients was done to correlate OD results of HIT ELISA with functional SRAs.47 The study showed that a weakly positive OD value (0.4–<1) had a good correlation with a negative SRA in 95% or more of cases. On the other hand, an OD value of >2 correlated well with a positive SRA in 90% or more of cases. Many similar studies have evaluated the applicability and generalizability of various ELISA OD thresholds from special populations of patients, such as those on extracorporeal membrane oxygenation.48 Kataria et al studied 47 such patients. They identified an OD threshold of 1 achieved specificity and negative predictive value of 89% and 95%, respectively. They suggested that keeping the OD threshold at 1 could help clinicians in ruling out HIT.48 A study of 318 patients admitted with clinical suspicion of HIT attempted to correlate OD values of HIT ELISA with thrombosis. The authors reported that with every 1-unit rise in OD value, the odds of developing thrombosis doubled, with 19%, 32%, and 48% of patients developing clinically evident thrombosis at ODs of 1, 2, and 3, respectively.49
As discussed, once there is clinical suspicion of HIT, a screening immunoassay is the first step in a diagnostic workup for HIT. One of the major drawbacks of ELISA is that results may be available only once a day, considering the cumbersome procedure and requirement for a specialized laboratory with skilled technicians. Other drawbacks are that enzyme activity can be hampered by plasma constituents, the presence of heterophilic antibodies may generate false-positive results, and results may not be absolute. These drawbacks prompted researchers to look for newer alternatives that could have a rapid turnaround time without compromising the high sensitivity and cost-effectiveness of ELISA.50 Some other recently studied immunologic assays are particle-gel immunoassays (H–PF4 PaGIA), lateral-flow immunoassays (LFIAs; eg, STiC; Diagnostica Stago, Australia), chemiluminescent immunoassays (CLIA; eg, Hemosil AcuStar; Werfen, Australia) and latex-agglutination assays. These assays exploit alternative technologies and differ in methodology and end-point measurements (Table 2).
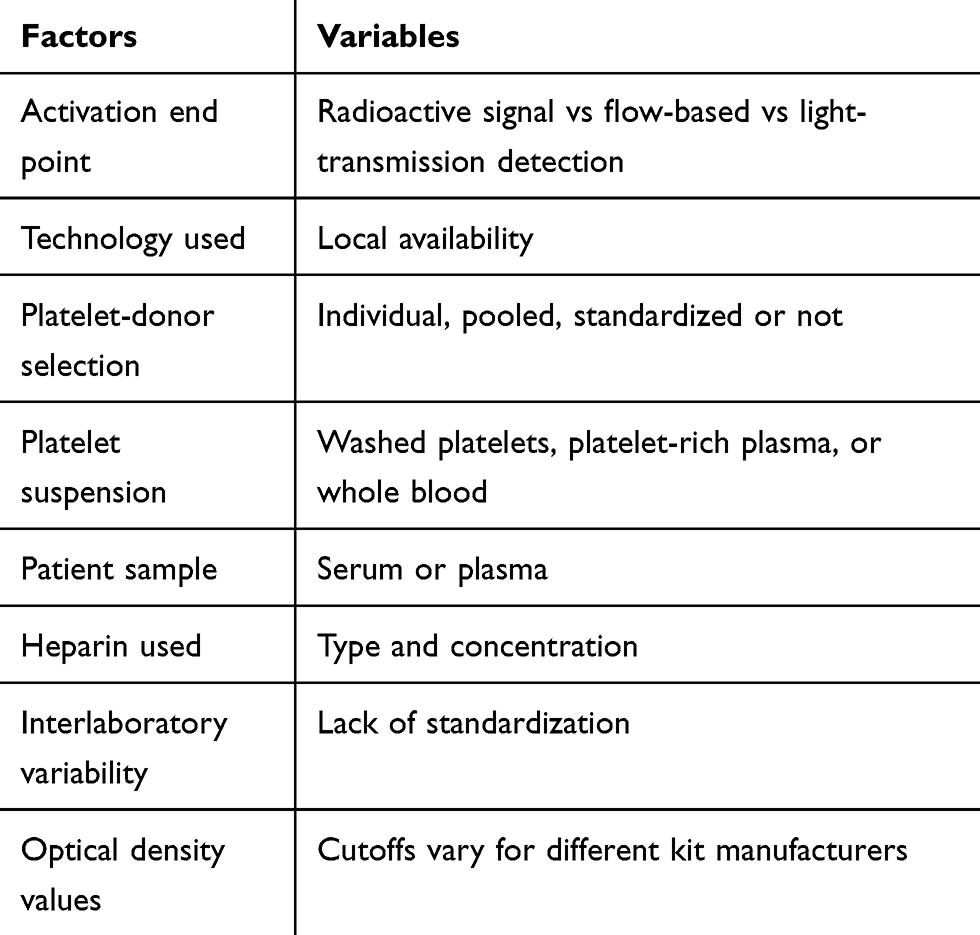 |
Table 2 Variables posing challenges while conducting functional assays and interpreting results |
Bankova et al compared ELISA with the newer immunologic assays AcuStar HIT-IgG and PaGIA for diagnostic accuracy, reproducibility, and analytical costs.50 Diagnostic accuracy was studied for three HIT ELISA OD-cutoff thresholds: low (0.4), intermediate (1.3), and high (2.0). The study showed that specificity increased with the rise in OD thresholds, and all three tests had specificity >90% in the intermediate- and high-threshold groups. For the low-threshold group, the specificity of AcuStar HIT-IgG (92.8%, p<0.05) was significantly higher than PaGIA (83.0%) and ELISA (81.8%). Total costs per test were US$120.10, $84.89, and $52.10 for PaGIA, AcuStar HIT-IgG, and ELISA, respectively. The study concluded that although the newer tests were 1.5- to twofold costlier than conventional HIT ELISA,when implemented in a 24-hour service they might improve patient care, considering their rapid turnaround, similar or better specificity, and adequate reproducibility.50
Recently, a meta-analysis by Nagler et al studied the diagnostic utility of various immunologic assays.30 The meta-analysis included 15,199 patients from 49 studies, and reported that the combination of high sensitivity (>95%) and high specificity (>90%) was found only in the following tests with specified thresholds: polyspecific ELISA with an intermediate threshold (Genetic Testing Institute, Asserachrom), PaGIA, LFIA, polyspecific CLIA with a high threshold, and IgG-specific CLIA with a low threshold. In addition, they found that diagnostic accuracy was inadequate in certain tests and specified thresholds: particle-immunofiltration assay, ELISA at high-dose heparin confirmation step, andIgG-specific CLIA with high and intermediate thresholds. The authors concluded that when evaluating a suspected HIT patient, physicians must utilize clinical scoring tools for pretest probabilities and estimate the posttest probabilities before running the test. Recent studies have also been conducted to look for additional pathological antibodies in patients detected negative for anti-PF4–heparin. Bashover et al recently conducted a study on anti-PF4-heparin–negative samples by using whole‐cell ELISA.51 They detected that 12.5% (eight of 64) of negative samples were found positive for antibodies against antigens on platelets or red blood cells.
Functional Assays/Platelet-Activation Assays
As immunoassays have low specificity and hence tend to overdiagnose HIT, they may erroneously lead to inappropriate discontinuation of heparin anticoagulants and compel the use of costlier alternative anticoagulants. These shortcomings mandate the performance of a functional assay in patients with a positive immunoassay to reduce overdiagnosis and erroneous discontinuation of UFH/LMWH in patients without HIT. In general, functional assays have high specificity (>95%) and high positive predictive values (89%–100%) but lower sensitivity (56%–100%). The sensitivity of this assay depends on the functional end point being studied (radioactive signal > flow-based > light-transmission detection).46 Technically, these studies are difficult to perform and hence not routinely available at many medical centers. Also, there are variables during the functional assay that need to be considered while conducting these tests and interpreting the results (Table 2). In clinical practice, when a functional assay is not available and if the patient has a high 4T score plus a strongly positive immunoassay, a functional assay may be excluded and the patient can be treated as a case of HIT. Similarly, a diagnosis of HIT can be entertained in conditions with a negative functional assay, a strongly positive immunoassay, and a high-probability 4T score.52
Functional assays detect platelet-conformation changes when a patient’s serum containing HIT antibodies is incubated with donor platelets and heparin. If HIT antibodies are pathologically significant, they are expected to form a heparin–antibody–PF4 complex, which in turn binds to FcRIIa receptors located on the donor’s platelet surfaces. This leads to donor-platelet activation with subsequent changes in platelet conformation and surface expression, including the release of dense granules, production of platelet microparticles, and translocation and surface expression of various glycoproteins (eg P-selectin or CD62P). It is the capture of these changes that constitutes the end point of various functional assays.52
The SRA end point is to detect the release of 14C serotonin from dense granules upon activation of donor platelets by HIT antibodies present in the patient’s serum. The test involves incubating radiolabeled donor platelets and the patient’s serum in the presence of therapeutic or excessive heparin concentrations. A positive test result is defined as the release of 14C serotonin with therapeutic concentrations of heparin (0.1 U/mL), but not with higher heparin concentrations (100 U/mL). Differential serotonin-release responses to two different heparin concentrates results, since the binding of HIT antibodies to antigens occurs only at a specific ratio of heparin to PF4.53
In the HIPA, the suspected patient’s serum or platelet-poor plasma is added to a healthy donor’s platelet-rich plasma with or without heparin. A positive test is indicated by visual recognition of strong platelet aggregation with low heparin concentrations (~0.1–0.3 U/mL), but no or minimal aggregation in the absence of heparin or presence of very high concentrations of heparin (~10–100 U/mL). The advantage of HIPA is visual measurement of platelet aggregation, unlike SRA, which utilizes radioactive agents.
In addition to SRA and HIPA, there are various other functional assays available in the commercial market or clinical trials (Tables 2 and 3). The goal of developing newer assays is to use simpler technologies (eg, flow cytometry, turbidometry, aggregometry, and luciferase activity) with rapid turnaround. Various chemical changes in platelet surface–antigen expression (eg, P-selectin or anionic phospholipids), release of granules (eg, ATP) or other platelet-derived microparticles, or physical changes like aggregation serve as end points in functional assays. The potential for wider acceptance of these alternate functional assays is encouraging, with generally good specificity (75%–>95%), but variable sensitivity (56%–100%), which could be due to multiple variables, as mentioned earlier (Table 3).
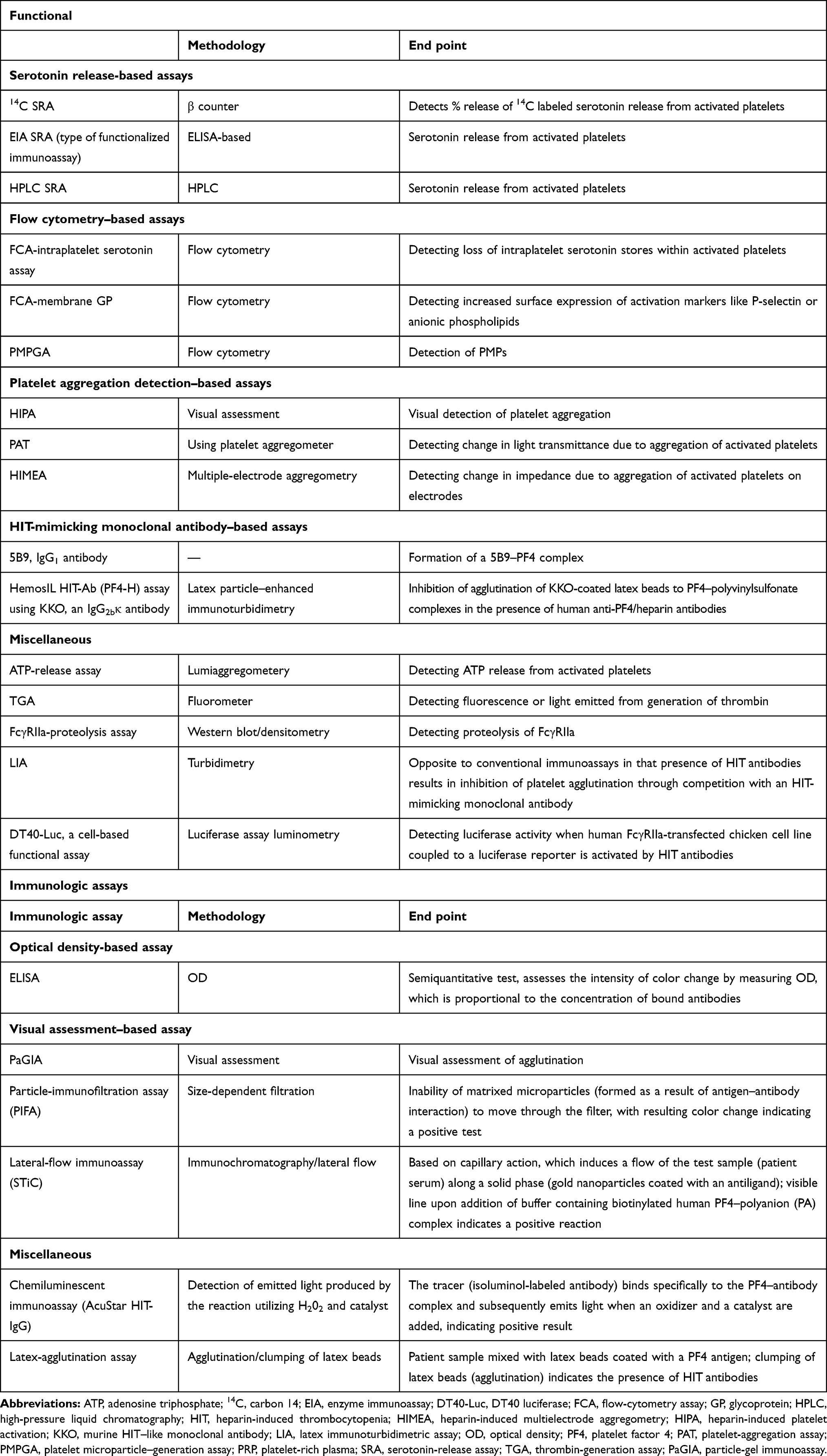 |
Table 3 Functional and immunologic assays: end points and methodological principles |
Initially, anti-PF4–heparin antibodies were considered solely responsible for the pathogenesis of HIT/HITT. However, with the discovery of the polyclonal and polyspecific nature of antibodies found in sera of HIT patients, researchers are now convinced that other antibodies contribute to HIT/HITT as well. Following that, researchers have studied many mouse and human models using HIT-mimicking monoclonal antibodies.44,45 Arepally et al developed a murine monoclonal IgG2bκ antibody against human PF4–heparin complexes.44 This antibody was used recently by Li et al to recognize a new second epitope named site 2, which was found adjacent to site 1 in the crystal structure of the PF4 tetramer.54 Similarly, Vayne et al developed 5B9, a monoclonal IgG fully mimicking human HIT antibodies, in their laboratory and used it to study the performance of alternative functional assays (HIMEA, LTA, and FC). They found good sensitivity for all the tested functional assays at a higher concentration of 5B9 (50 µg/mL).45 Similarly, Morel et al studied the murine IgG1 platelet–activating anti-CD9 antibody ALB6; however, unlike 5B9, ALB6 was aspecific for PF4–H complexes and had very poor affinity.55
Conclusion
Diagnosis of HIT requires a combined clinical and biochemical approach. Timely suspicion of HIT can essentially prevent fatal thrombotic events, but physicians should use the HIT panel judiciously in order to avoid overdiagnosis, unnecessary discontinuation of heparin treatment, and financial burden on patients. Since the time of the initial description of this disease almost six decades back, many novel, cheaper, and rapid confirmatory tests have been developed and shown promising results, but are not presently sensitive enough to replace SRA. Studies on rapid immunoassays, namely particle-gel immunoassay, LFIA, and CLIA, are encouraging. The sensitivity and specificity of various tests depends on antibody specificities and thresholds set for the analysis. As per the recent meta-analysis, pooled sensitivity of polyspecific ELISA improves only marginally from 96.7% at low threshold to 98.4% at intermediate threshold. However, specificity improves significantly from 86.8% to 94.9% by increasing the threshold from low to intermediate.30 Based on these observations, Bankova et al recommend applying intermediate thresholds to improve specificity of polyspecific assays.50 However, most studies have have not found AcuStar HIT-IgG to be sensitive enough to use at intermediate threshold.50,56,57
Cost-wise, rapid immunoassays are costlier when compared to the ELISA. However, a large-budget model study showed that availability of 24-hour in-hospital service for rapid immunoassays can reduce the time span of suspecion to diagnosis of HIT and would reduce the amount of anticoagulant-treatment costs.58 Therefore, an integrated diagnostic approach combining a clinical picture with immunoassays and functional assays serves as an ideal method for decision-making in suspected HIT. Also, clinicians should be aware of the type of tests used to diagnose HIT, their antibody specificities, and the thresholds considered before interpreting the results.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jindal V, Singh A, Siddiqui AD, Leb L. The appropriateness of testing platelet factor 4/heparin antibody in patients suspected of heparin-induced thrombocytopenia. Cureus. 2018;10(10):e3532. doi:10.7759/cureus.3532
2. Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood. 2005;106(8):2710–2715. doi:10.1182/blood-2005-04-1546
3. Girolami B, Prandoni P, Stefani PM, et al. The incidence of heparin-induced thrombocytopenia in hospitalized medical patients treated with subcutaneous unfractionated heparin: a prospective cohort study. Blood. 2003;101(8):2955–2959. doi:10.1182/blood-2002-07-2201
4. Warkentin TE, Sheppard J-AI, Sigouin CS, Kohlmann T, Eichler P, Greinacher A. Gender imbalance and risk factor interactions in heparin-induced thrombocytopenia. Blood. 2006;108(9):2937–2941. doi:10.1182/blood-2005-11-012450
5. Yanamandra U, Sahu KK, Malhotra P, Varma S. Thrombocytopaenia with absent radius (not radii). BMJ Case Rep. 2014;2014(jun02 1):bcr2014204844–bcr2014204844. doi:10.1136/bcr-2014-204844
6. Dhibar DP, Sahu KK, Dhir V, Singh S. Immune thrombocytopenia as a presenting manifestation of tuberculosis- challenge in resource constraint settings. J Clin Diagn Res. 2016;10(10):OD01–OD02. doi:10.7860/JCDR/2016/20911.8612
7. Sahu KK, Siddiqui AD, Rezaei N, Cerny J. Challenges for Management of Immune Thrombocytopenia during COVID-19 Pandemic. J Med Virol. 2020. doi:10.1002/jmv.26251
8. Chen W, Ha JP, Hong H, Maitta RW. Absolute immature platelet counts in the setting of suspected heparin-induced thrombocytopenia may predict anti-PF4-heparin immunoassay testing results. Transfus Apher Sci. 2018;57(4):507–511. doi:10.1016/j.transci.2018.04.001
9. Linkins L-A, Dans AL, Moores LK, et al. Treatment and prevention of heparin-induced thrombocytopenia: antithrombotic therapy and prevention of thrombosis, 9th ed: american college of chest physicians evidence-based clinical practice guidelines. Chest. 2012;141(2 Suppl):e495S–e530S. doi:10.1378/chest.11-2303
10. Sahu KK, Yanamandra U, Bhar V, Dhibar DP, Varma SC, Malhotra P. Dasatinib and dysfunction of platelets. Indian J Hematol Blood Transfus. 2016;32(Suppl S1):246–247. doi:10.1007/s12288-016-0659-x
11. Sahu KK, Mishra AK, George SV, Siddiqui AD. Managing retroperitoneal hematoma: associated complexities and its challenges. Am J Emerg Med. 2020. doi:10.1016/j.ajem.2020.02.003
12. Sahu KK, Mishra AK, Lal A. Update on retroperitoneal hematoma in children. World J Emerg Med. 2020;11(1):64. doi:10.5847/wjem.j.1920-8642.2020.01.010
13. Sahu KK, Lal A, Mishra AK. Additional insights regarding aortic intramural hematoma. Can J Anaesth. 2020;67(3):382–383. doi:10.1007/s12630-019-01490-w
14. Sahu KK, Mishra AK, Lal A, Davuluri V. An interesting case of gluteal haematoma. BMJ Case Rep. 2019;12(8):8. doi:10.1136/bcr-2019-230282
15. Sahu KK, Maradana S, Mishra A, Chastain I. A spontaneous rectus sheath hematoma. Intern Emerg Med. 2018;13(8):1341–1343. doi:10.1007/s11739-018-1932-9
16. Dhibar DP, Sahu KK, Deo P, Varma S. Haemophilia-A-related haematoma: management in resource constraint settings. BMJ Case Rep. 2016;2016. doi:10.1136/bcr-2016-215933.
17. Greinacher A, Alban S, Omer-Adam MA, Weitschies W, Warkentin TE. Heparin-induced thrombocytopenia: a stoichiometry-based model to explain the differing immunogenicities of unfractionated heparin, low-molecular-weight heparin, and fondaparinux in different clinical settings. Thromb Res. 2008;122(2):211–220. doi:10.1016/j.thromres.2007.11.007
18. Prechel MM, Walenga JM. Emphasis on the Role of PF4 in the Incidence, Pathophysiology and Treatment of Heparin Induced Thrombocytopenia. Thromb J. 2013;11(1):7. doi:10.1186/1477-9560-11-7
19. Poncz M, Rauova L, Cines DB. The role of surface PF4: glycosaminoglycan complexes in the pathogenesis of heparin-induced thrombocytopenia (HIT). Pathophysiol Haemost Thromb. 2006;35(1–2):46–49. doi:10.1159/000093543
20. Amiral J, Peynaud-Debayle E, Wolf M, Bridey F, Vissac AM, Meyer D. Generation of antibodies to heparin-PF4 complexes without thrombocytopenia in patients treated with unfractionated or low-molecular-weight heparin. Am J Hematol. 1996;52(2):90–95. doi:10.1002/(SICI)1096-8652(199606)52:2<90::AID-AJH4>3.0.CO;2-0
21. Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332(20):1330–1335. doi:10.1056/NEJM199505183322003
22. Dhibar DP, Sahu KK, Varma SC, et al. Intra-cardiac thrombus in antiphospholipid antibody syndrome: an unusual cause of fever of unknown origin with review of literature. J Cardiol Cases. 2016;14(5):153–156. doi:10.1016/j.jccase.2016.07.005
23. Sahu KK, Varma SC. Cortical vein thrombosis in a case of idiopathic thrombocytopenic purpura. Platelets. 2015;26(4):374–375. doi:10.3109/09537104.2014.898180
24. Fathi M. Heparin-induced thrombocytopenia (HIT): identification and treatment pathways. Glob Cardiol Sci Pract. 2018;2018(2):15. doi:10.21542/gcsp.2018.15
25. Lo GK, Juhl D, Warkentin TE, Sigouin CS, Eichler P, Greinacher A. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4(4):759–765. doi:10.1111/j.1538-7836.2006.01787.x
26. Cuker A, Arepally G, Crowther MA, et al. The HIT Expert Probability (HEP) Score: a novel pre-test probability model for heparin-induced thrombocytopenia based on broad expert opinion. J Thromb Haemost. 2010;8(12):2642–2650. doi:10.1111/j.1538-7836.2010.04059.x
27. Lillo-le Louët A, Boutouyrie P, Alhenc-Gelas M, et al. Diagnostic score for heparin-induced thrombocytopenia after cardiopulmonary bypass. J Thromb Haemost. 2004;2(11):1882–1888. doi:10.1111/j.1538-7836.2004.00949.x
28. Piednoir P, Allou N, Provenchère S, et al. Heparin-induced thrombocytopenia after cardiac surgery: an observational study of 1722 patients. J Cardiothorac Vasc Anesth. 2012;26(4):585–590. doi:10.1053/j.jvca.2011.11.009
29. Messmore HL, Fabbrini N, Bird ML, et al. Simple scoring system for early management of heparin-induced thrombocytopenia. Clin Appl Thromb Hemost. 2011;17(2):197–201. doi:10.1177/1076029610387126
30. Nagler M, Bachmann LM, Ten Cate H, Ten Cate-Hoek A. Diagnostic value of immunoassays for heparin-induced thrombocytopenia: a systematic review and meta-analysis. Blood. 2016;127(5):546–557. doi:10.1182/blood-2015-07-661215
31. Nagler M, Bakchoul T. Clinical and laboratory tests for the diagnosis of heparin-induced thrombocytopenia. Thromb Haemost. 2016;116(5):823–834. doi:10.1160/TH16-03-0240
32. Ribatti D, Crivellato E. Giulio Bizzozero and the discovery of platelets. Leuk Res. 2007;31(10):1339–1341. doi:10.1016/j.leukres.2007.02.008
33. McLEAN J. The discovery of heparin. Circulation. 1959;19(1):75–78. doi:10.1161/01.cir.19.1.75
34. Jorpes E. The chemistry of heparin. Biochem J. 1935;29(8):1817–1830. doi:10.1042/bj0291817
35. Weismann RE, Tobin RW. Arterial embolism occurring during systemic heparin therapy. AMA Arch Surg. 1958;76(2):219–225. doi:10.1001/archsurg.1958.01280200041005
36. Roberts B, Rosato FE, Rosato EF. HEPARIN–A CAUSE OF ARTERIAL EMBOLI? Surgery. 1964;55:803–808.
37. Rhodes GR, Dixon RH, Silver D. Heparin induced thrombocytopenia with thrombotic and hemorrhagic manifestations. Surg Gynecol Obstet. 1973;136(3):409–416.
38. Towne JB, Bernhard VM, Hussey C, Garancis JC. White clot syndrome. Peripheral vascular complications of heparin therapy. Arch Surg. 1979;114(4):372–377. doi:10.1001/archsurg.1979.01370280026004
39. Cines DB, Tomaski A, Tannenbaum S. Immune endothelial-cell injury in heparin-associated thrombocytopenia. N Engl J Med. 1987;316(10):581–589. doi:10.1056/NEJM198703053161004
40. Amiral J, Bridey F, Dreyfus M, et al. Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb Haemost. 1992;68(1):95–96. doi:10.1055/s-0038-1656329
41. Greinacher A, Pötzsch B, Amiral J, Dummel V, Eichner A, Mueller-Eckhardt C. Heparin-associated thrombocytopenia: isolation of the antibody and characterization of a multimolecular PF4-heparin complex as the major antigen. Thromb Haemost. 1994;71(2):247–251.
42. Reilly MP, Sinha U, André P, et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood. 2011;117(7):2241–2246. doi:10.1182/blood-2010-03-274969
43. Cai Z, Yarovoi SV, Zhu Z, et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat Commun. 2015;6(1):8277. doi:10.1038/ncomms9277
44. Arepally GM, Kamei S, Park KS, et al. Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood. 2000;95(5):1533–1540. doi:10.1182/blood.V95.5.1533.005k01_1533_1540
45. Kizlik-Masson C, Vayne C, McKenzie SE, et al. 5B9, a monoclonal antiplatelet factor 4/heparin IgG with a human Fc fragment that mimics heparin-induced thrombocytopenia antibodies. J Thromb Haemost. 2017;15(10):2065–2075. doi:10.1111/jth.13786
46. Pouplard C, Amiral J, Borg JY, Laporte-Simitsidis S, Delahousse B, Gruel Y. Decision analysis for use of platelet aggregation test, carbon 14-serotonin release assay, and heparin-platelet factor 4 enzyme-linked immunosorbent assay for diagnosis of heparin-induced thrombocytopenia. Am J Clin Pathol. 1999;111(5):700–706. doi:10.1093/ajcp/111.5.700
47. Warkentin TE, Sheppard JI, Moore JC, Sigouin CS, Kelton JG. Quantitative interpretation of optical density measurements using PF4-dependent enzyme-immunoassays. J Thromb Haemost. 2008;6(8):1304–1312. doi:10.1111/j.1538-7836.2008.03025.x
48. Kataria V, Moore L, Harrison S, Hernandez O, Vaughan N, Schwartz G. Evaluation of serotonin release assay and enzyme-linked immunosorbent assay optical density thresholds for heparin-induced thrombocytopenia in patients on extracorporeal membrane oxygenation. Crit Care Med. 2020;48(2):e82–e86. doi:10.1097/CCM.0000000000004090
49. Baroletti S, Hurwitz S, Conti NAS, Fanikos J, Piazza G, Goldhaber SZ. Thrombosis in suspected heparin-induced thrombocytopenia occurs more often with high antibody levels. Am J Med. 2012;125(1):44–49. doi:10.1016/j.amjmed.2011.06.025
50. Bankova A, Andres Y, Horn MP, Alberio L, Nagler M. Rapid immunoassays for diagnosis of heparin-induced thrombocytopenia: comparison of diagnostic accuracy, reproducibility, and costs in clinical practice. PLoS One. 2017;12(6):e0178289. doi:10.1371/journal.pone.0178289
51. Bashover EM, Stefaniuk CM, Harding CV, Maitta RW. Use of a whole-cell ELISA to detect additional antibodies in setting of suspected heparin-induced thrombocytopenia. Eur J Haematol. 2019;103(2):99–106. doi:10.1111/ejh.13263
52. Minet V, Dogné J-M, Mullier F. Functional Assays in the Diagnosis of Heparin-Induced Thrombocytopenia: A Review. Molecules. 2017;22:4. doi:10.3390/molecules22040617
53. Warkentin TE, Arnold DM, Nazi I, Kelton JG. The platelet serotonin-release assay. Am J Hematol. 2015;90(6):564–572. doi:10.1002/ajh.24006
54. Li ZQ, Liu W, Park KS, et al. Defining a second epitope for heparin-induced thrombocytopenia/thrombosis antibodies using KKO, a murine HIT-like monoclonal antibody. Blood. 2002;99(4):1230–1236. doi:10.1182/blood.v99.4.1230
55. Morel-Kopp M-C, Mullier F, Gkalea V, et al. Heparin-induced multi-electrode aggregometry method for heparin-induced thrombocytopenia testing: communication from the SSC of the ISTH. J Thromb Haemost. 2016;14(12):2548–2552. doi:10.1111/jth.13516
56. Althaus K, Hron G, Strobel U, et al. Evaluation of automated immunoassays in the diagnosis of heparin induced thrombocytopenia. Thromb Res. 2013;131(3):e85–e90. doi:10.1016/j.thromres.2013.01.005
57. Minet V, Baudar J, Bailly N, et al. Rapid exclusion of the diagnosis of immune HIT by AcuStar HIT and heparin-induced multiple electrode aggregometry. Thromb Res. 2014;133(6):1074–1078. doi:10.1016/j.thromres.2014.01.014
58. Caton S, O’Brien E, Pannelay AJ, Cook RG. Assessing the clinical and cost impact of on-demand immunoassay testing for the diagnosis of heparin induced thrombocytopenia. Thromb Res. 2016;140:155–162. doi:10.1016/j.thromres.2016.01.025
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.