")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 11
Current perspective on the role of insulin and glucagon in the pathogenesis and treatment of type 2 diabetes mellitus
Authors Ojha A , Ojha U, Mohammed R , Chandrashekar A , Ojha H
Received 23 January 2019
Accepted for publication 28 March 2019
Published 9 May 2019 Volume 2019:11 Pages 57—65
DOI https://doi.org/10.2147/CPAA.S202614
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Ashutosh Ojha,1 Utkarsh Ojha,2 Raihan Mohammed,3 Abhinaya Chandrashekar,2 Harsh Ojha4
1Shobhaben Pratapbhai Patel School Of Pharmacy & Technology Management, SVKM’s Narsee Monjee Institute of Management Studies, Mumbai, India; 2Faculty of Medicine, Imperial College London, London, UK; 3Department of Medicine, University of Cambridge, Cambridge, UK; 4Department of Life Sciences, University of Warwick, Coventry, UK
Abstract: According to the World Health Organization, 422 million adults worldwide live with diabetes mellitus (DM), a significant portion of whom have type 2 diabetes. The discovery of insulin as a key regulator of glucose metabolism has revolutionized our understanding of DM and provided several therapeutic avenues. Most studies have so far predominantly focused on the role of insulin in type 2 diabetes. However, the balance between insulin and glucagon is essential in ensuring glucose homeostasis. In this review, we begin by evaluating the principal differences between insulin and glucagon with regard to their mechanism and control of their secretion. Next, we discuss their mode of action and effects on metabolism. We further explore how the two hormones impact the natural history of type 2 diabetes. Finally, we outline how current and emerging pharmacological agents attempt to exploit the properties of insulin and glucagon to benefit patients with type 2 diabetes.
Keywords: type 2 diabetes mellitus, insulin, glucagon, glucose
Introduction
The discovery of insulin and glucagon as regulators of glucose metabolism has revolutionized our understanding of diabetes. Insulin is a 51-amino-acid peptide processed from proinsulin and secreted in pulses by the β-cells in the pancreatic islet of Langerhans.1 Insulin’s hypoglycaemic effect was first demonstrated by Banting and Best after successfully lowering blood glucose levels in a dog, following removal of the pancreas. Shortly after insulin’s discovery, Murlin et al2 suggested the effect of glucagon as a hyperglycaemic agent. In 1948, Sutherland and de Duve3 defined the α-cells of the islet of Langerhans as a source of this hormone. Glucagon is a 29-amino-acid peptide cleaved from proglucagon. These hormones respond to several nutritional,4 hormonal and neural5 factors. Their coordination is essential for glucose homeostasis.6
Diabetes mellitus (DM) is a metabolic disorder characterized by hyperglycemia. The global prevalence of DM has increased dramatically over the years, with 422 million affected adults7 in 2014, a figure that will almost double by 2040.8 Type 2 diabetes mellitus (T2DM) accounts for more than 90%9 of diabetes and typically features insulin resistance or insulin secretory defects.10 Understanding the role of insulin and glucagon in human physiology can provide novel therapeutic targets for the treatment of T2DM. This review will discuss the fundamental differences between insulin and glucagon and evaluate evidence supporting their impact on the natural history of T2DM.
The primary stimulant for insulin secretion from the pancreatic β-cell is hyperglycemia.11 Glucose is transported into the β-cell by the protein Glucose Transporter 2 (GLUT2).12 Following its entry, glucose is phosphorylated to glucose-6-phospahte (G6P) by the enzyme glucokinase. Further metabolism of G6P generates ATP.11 Studies13,14 show that K+ channels located on β-cells are sensitive to ATP and are responsible for the depolarization of the cell membrane. Closure of these K+ channels increases the influx of Ca2+ ions through L-type voltage-dependent calcium channels (L-VDCC). This entry raises cytosolic-free Ca2+ concentration which is considered to stimulate insulin exocytosis.15 This mechanism of insulin release is referred to as the “KATP channel-dependent” pathway (Figure 1). Evidence also indicates a different mechanism which is independent of the KATP channels.16,17 In these studies, closure of the K+ channel was prevented using K+ channel antagonists. The studies reported that glucose continued to augment Ca2+ influx resulting in insulin release. Taken together, the findings suggest that glucose controls insulin via two mechanisms.
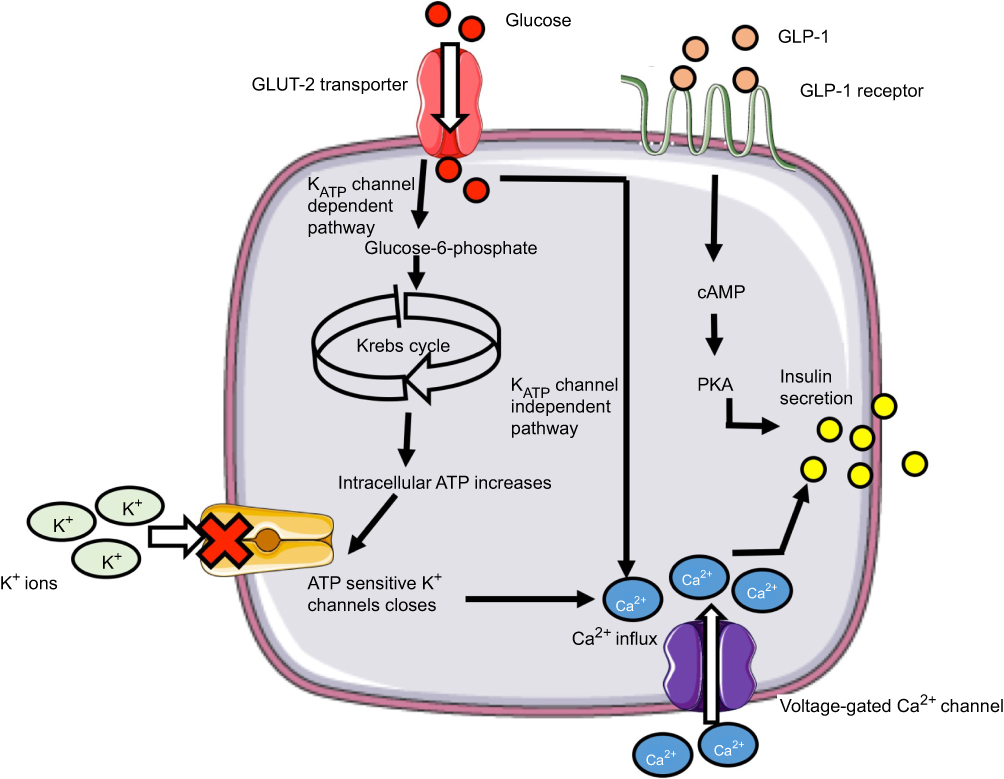 | Figure 1 Mechanism of insulin secretion from β-cells in response to glucose (left) and GLP-1 (right). |
Lang et al1 proposed that control of insulin secretion by glucose occurred in a pulsatile manner. The group measured plasma glucose and insulin concentration every minute for 2 hrs in ten subjects. The frequent sampling interval increases the accuracy of their results allowing identification of any anomalies. In five subjects, there was a regular cycle of basal plasma insulin concentration. A concurrent plasma glucose cycle was also demonstrated which began 2 mins in advance of the plasma insulin.
Furthermore, the incretin hormone Glucagon-like-peptide 1 (GLP-1) is considered to be an important regulator of insulin secretion. The GLP-1 receptor (GLP-1R) has been identified on β-cells.18 Gromada et al19 demonstrated GLP-1 mediates Ca2+-induced insulin secretion (Figure 1) Glucose and fatty acids are known to stimulate GLP-1 release from the distant ileum and colon. The enzyme dipeptidyl peptidase-4 is responsible for its degradation.20
The mechanism by which pancreatic α-cells release glucagon during hypoglycemia is widely debated. Several studies19,21,22 support the intrinsic model of glycemic control by α-cells. This model suggests activation of voltage-gated Na+ channels23 and VDCC drives glucagon exocytosis. Quesada and colleagues24 investigated the effects of glucose concentration on intracellular Ca2+ levels in both α- and β-cells of five human subjects. The group measured the intensity of intracellular Ca2+signalling at increasing glucose concentration using the Ca2+-sensitive dye Fluo-3. However, Fluo-3 is a non-ratiometric probe which is susceptible to external artifacts. This limitation could have been addressed by using a ratiometric probe for a more reliable measurement of Ca2+ signals. The study also conducted confocal imaging microscopy of cytosolic Ca2+ concentration. Confocal microscopes have a narrower depth of field than fluorescent and light microscopes and also eliminate background artifacts. This technique allows the authors to evaluate intracellular Ca2+ signals in individual cells and compare cell-to-cell characteristics. The results indicated that low glucose concentration electrical activity initiates pulsatile Ca2+ signals in α-cells. Conversely, at higher glucose levels, Ca2+ signaling was more potently stimulated in β-cells. Together, these results imply that α- and β-cells have opposite Ca2+ signaling patterns in response to glucose.
The intrinsic model also proposes that α-cell secretion of glucagon is mediated by the KATP-dependent pathway. MacDonald et al25 proposed that low glucose levels activate KATP channel creating a membrane potential around −60 mV. At this voltage, T- and N-type VDCC and VGNC on α-cells are open. Subsequent influx of Ca2+ via VDCC results in glucagon secretion. High influx of glucose via GLUT 1 in α-cells increases intracellular ATP which blocks KATP channel activity. As a result, the membrane potential of the α-cells falls within a range where the voltage-dependent channels are closed. Consequently, Ca2+ influx and glucagon secretion are inhibited. This study was conducted on intact islet cells in both rodents and humans. The results were replicated in both species. However, the authors neither specify their sample size nor provide a power calculation.
Alternatively, many studies suggest glucagon secretion is controlled by paracrine factors released by neighboring β- and δ-cells in response to glucose levels. Recent observation in isolated rat α-cells26 highlighted increasing glucose concentration continues to stimulate rather than inhibit glucagon release. This effect reverses following administration of somatostatin,26 GABA,27 insulin and Zn2+.28 Study by Franklin and colleagues28 demonstrated that Zn2+ and insulin secretion from β-cells suppress glucagon during hyperglycemia exocytosis by acting on α-cells. These findings are argued by Cheng-Xue et al26, who revealed that glucose can inhibit glucagon secretion independently of Zn2+. However, both of these experiments were conducted in pancreatic cells which were prepared extensively after isolation from the pancreas. Therefore, the property of the islet cells may have changed following extraction and not be representative of intact islet cells in vivo.
There have been several debates on whether there may be extra-pancreatic secretion of glucagon. Indeed, groups have shown that the most likely place that glucagon may be secreted from, apart from the α-cells in the pancreas, is the gastrointestinal tract. Whilst further studies are needed to evaluate the exact location of these extra-pancreatic secretions, they may provide a novel target for future drugs targeting glucagon secretion.
Mode of action and effect on metabolism
The effects of insulin are mediated by binding to its specific tyrosine kinase transmembrane receptor.29 The insulin receptor (IR) is a tetrameric protein with two extracellular α-subunits and two intracellular β-subunits. IR is present in both peripheral and central tissues. In peripheral tissues, insulin mediates metabolism via downstream activation of phosphatodylinostiol 3 kinase (P13K) and insulin receptor substrate (IRS). The IRS-P13K pathway results in activation of different kinases. Recent evidence suggests that insulin’s action on hepatocytes stimulates glycolysis and lipogenesis via the protein complex mTORC2.30 Insulin is also considered to reduce gluconeogenesis by downregulating the Forkhead box class O transcription factors.31 Further effects on metabolism also include glycogen and protein synthesis.
Moreover, activation of the IRS-P13K enzyme system has been shown to increase glucose uptake in peripheral tissues. Recent studies32–34 suggest insulin also signals via the IRS-P13K system in the hypothalamus to reduce hepatic glucose output. Obici et al33 assessed hepatic glucose production by administering intracerebroventricular (ICV) injections of insulin in male rats and comparing with vehicle-treated controls. The group also evaluated hepatic insulin sensitivity through systemic pancreatic-insulin clamp studies. The authors found glucose production significantly reduced during ICV infusion of insulin compared to the vehicle-treated mice. The study concluded that ICV infusion of insulin reduces hepatic glucose production by increasing hepatic insulin sensitivity. However, the authors do not specify whether the mice were pair-fed over the course of the experiment. As a result, it is not certain whether varying food intake amongst the mice impacted glucose production. Further study in mice by Pocai et al34 shows that ICV insulin infusion inhibits hepatic glucogenogensis by activating KATP channels in the mediobasal hypothalamus. The group also found that ICV administration of diazoxide downregulates mRNA expression of hepatic glucose-6-phosphatase (G6Pase) and phosphoenolypyruvate carboxykinase (PEPCK). These enzymes are drivers of gluconeogenesis in the liver. Moreover, hepatic vagotomy seemed to nullify this effect. Taken as a whole, these rodent studies suggest that insulin activity in the hypothalamus may signal to the liver via the vagus nerve to inhibit gluconeogenesis. Due to a difference between hypothalamic structure, it is not conclusive if insulin also mediates glucose metabolism in humans along this neuronal pathway.
Glucagon’s action is also initiated following binding to its receptor. Unlike IR, the glucagon receptor (GR) is a seven transmembrane protein and member of class B G-protein-coupled family receptor.35 The distribution pattern of these receptors also differs from IR. Dot blot analysis and RNase protection assays revealed that GR is most abundantly expressed in liver and kidneys.36 GR activation is coupled to a GTP-binding G protein, which leads to subsequent activation of cAMP and PKA. GR stimulation can also stimulate the phospholipase C-inositol phosphate pathway via Gq proteins resulting in intracellular Ca2+ store secretion.35 Glucagon increases hepatic glucose output by potentiating glycogenolysis and gluconeogenesis. Evidence shows37 glucagon upregulates transcription of the G6Pase and PEPCK gene through stimulation of the cAMP response element-binding protein and peroxisome proliferator-activated receptor γ-coactivator-1. Stimulation of these enzymes drives both gluconeogenesis and glycogenolysis. Studies38,39 have also suggested that glucagon stimulates breakdown of fatty acids. In a recent investigation, Arafat et al39 proposed glucagon potentiates lipolysis by acting on fibroblast growth factor 21 (FGF21). FGF21 has been previously shown to regulate lipolysis.40 This study reported that exogenous glucagon administration in isolated adipocytes induced lipolysis in both humans and rodents. The study also claimed glucagon increased circulating FGF21 in both species. However, the authors report these results were not replicated in vivo. The group also administered a relatively high dose of exogenous glucagon. Therefore, it is not conclusive if glucagon levels in humans will increase FGF21 and induce lipolysis. In addition to its role in metabolism, glucagon also plays a role in energy expenditure. Davidson et al41 demonstrated that infusing glucagon in rats increases their oxygen consumption. Further studies in rats42,43 have shown that glucagon administration leads to an increase in whole-body oxygen consumption, core body temperature, flood flow and mass of brown adipose tissue. This observation was also seen when conducted in human volunteers, in whom energy expenditure was increased following infusion of glucagon.44
Insulin, glucagon and the natural history of type 2 diabetes
Studies have widely examined the role insulin plays in developing T2DM. Martin et al45 followed 155 subjects from 86 families over 25 years. Although the sample size is relatively small, the length of the follow-up strengthens the study’s protocol. The group used intravenous glucose tolerance test with the minimum model assessment to estimate insulin sensitivity (Si). The authors reported subjects who developed T2DM had lower values of Si. This indicates that insulin resistance is an important factor in the pathophysiology of T2DM. Moreover, Godsland et al46 investigated β-cell insulin secretion at increasing fasting plasma glucose (FPG) concentrations. The study measured acute insulin response to glucose (AIRg) levels in 466 nondiabetic men. The authors reported a steep decline in AIRg levels after FPG levels of 5mmol/L. Taken together, these studies show that both insulin resistance and loss of β-cell function are implicated in the natural history of T2DM.
The impact of glucagon has also been recognized in diabetic patients.47 Normal response to a carbohydrate meal involves a rise in insulin levels and a reciprocal decrease in glucagon concentrations.48 In contrast, in patients with T2DM, postprandial insulin secretion is depressed, whereas glucagon levels are increased. Shah et al49 hypothesized that lack of α-cell suppression due to impaired insulin secretion in T2DM resulted in hyperglycemia. Their study evaluated the effect of glucagon suppression on glucose concentration in humans. The authors infused insulin and glucose into the systemic circulation at a rate which mimicked postprandial profiles in diabetic patients. The study reported that lack of glucagon suppression resulted in a significantly increased peak plasma glucose concentration (P<0.002) compared to glucagon-suppressed diabetic profile. From these results, the authors concluded failure of glucagon suppression in diabetic patients causes hyperglycemia. However, the study only assessed one postprandial insulin profile. In T2DM, insulin secretion varies greatly; therefore, the results of this study are limited only to this study. A recent rodent study has also suggested that β-cells dedifferentiate under stress to progenitor pluripotent cells which secrete glucagon.50 Whilst this study may highlight a further mechanism of increased glucagon secretion, further studies must investigate whether this process occurs in human β-cells and its involvement in the pathogenesis of T2DM.
Targeting insulin in the treatment of type 2 diabetes mellitus
The majority of current pharmacological treatments for type 2 diabetes target either increasing insulin secretion or potentiating its effect.
Metformin
Metformin is a drug in the biguanide class usually given as first-line pharmacological therapy to patients with T2DM. It is proposed that metformin may reduce serum glucose level by several different mechanisms. It potentiates the effects of insulin and is commonly thought of as an “insulin sensitizer”. Rodent51 and human52 studies have shown that metformin may act on IR and increase the activity of the IRS-PI3K pathway through the enzyme adenosine monophosphate kinase, thus resulting in the inhibition of key enzymes involved in gluconeogenesis and glycogen synthesis in the liver whilst stimulating insulin signaling and glucose transport in muscles. Furthermore, metformin increases the peripheral glucose disposal that arises largely through increased nonoxidative glucose disposal into skeletal muscle. In more recent years, there have been further studies to suggest that one of the main sites of actions of metformin may be the intestines.53
Significant adverse effects of metformin are yet to be reported. However, it can lead to lactic acidosis through inhibition of the cori cycle resulting in nausea, vomiting, abdominal pain and palpitations. Consequently, the drug is usually not advised in patients with end-stage renal failure.54–57
Sulfonylurea
Some drugs also aim to increase insulin secretion from β-cells. Sulfonylureas are a class of drug which binds to KATP channels on β-cells reducing K+ inflow. This initiates Ca+-induced insulin release. However, their mechanism of action requires the presence of residual pancreatic β-cells. Currently, sulfonylureas are used to control hyperglycemia in T2DM-affected patients who cannot achieve appropriate control with changes in diet alone. Known side effects of sulfonylyreas include hypoglycemia, weight gain, photosensitivity and in rare cases sulfonylyreas can cause hyponatremia by increasing the effect of anti-diuretic hormone on the collecting ducts. It can further cause skin reactions like erythema multiforme and exfoliative dermatitis.58–60
Thiazolidinediones
This class of drugs works via the perioxisome proliferator-activated receptor gamma (PPAR-γ) pathway. The PPAR-γ are nuclear receptors whose endogenous ligands are free fatty acids. Upon stimulation, the receptor binds DNA with the retinoid X receptor resulting in the transcription of specific genes. The up-regulation of these genes results in numerous responses including increase in insulin sensitivity, increase in adiponectin levels, decrease in leptin levels and decrease in certain interleukins such as IL-6. Rosiglitazone was one of the first classes of these drugs, marketed in 1999. Whilst the drug proved to be effective at reducing blood glucose levels, there were several reports of its adverse effects leading to heart failure, heart attacks, strokes, bone fractures and death. Currently, pioglitazone, is the only form of this drug available for medical use in the UK. It is recommended that the drug is started in combination with metformin when metformin alone has failed to achieve adequate blood glucose levels. Alternatively, pioglitazone may be given first line in circumstances where metformin is contraindicated. The blood glucose-lowering effects of thiazolidinedione can take time to be seen, with pioglitazone taking up to 4 months to achieve its maximal effects.61
Insulin analogs
Whilst insulin analogs are commonly used as therapeutic options for patients with type 1 diabetes mellitus (T1DM), the National Institute for Health and Care Excellence advocates the use of insulin in those who are not responding to a combination of the other pharmacological agents.62 Many small-scale studies have also suggested that commencing insulin in patients with T2DM can potentially induce remission for up to 2 years.63 One of the major adverse effects of insulin therapy is the risk of developing hypoglycemia, especially nocturnally. Therefore, patients must be reminded and educated on how to recognize the symptoms associated with hypoglycemia. Moreover, due to insulin’s lipogenic properties, patients often also complain of weight gain as a side effect. There are many models of insulin therapy, including basal, prandial and biphasic insulin. The American Diabetes Association and European Association for the Study of Diabetes advise starting on basal insulin, like glargine and detemir based on its relative efficacy and safety.64
Targeting glucagon in the treatment of type 2 diabetes mellitus
Targeting the incretin pathway has provided therapeutic benefits for patients with T2DM in reducing postprandial glucagon level.
GLP-1 analogs
GLP-1 analogs were first marketed in 2005. Exenatide, which shares approximately a 50% homology to the GLP-1 receptor,65 was derived from the saliva of the Glia monster and demonstrated considerable promise at reducing blood glucose levels. This drug work hs an incretin effect and reduces blood glucose by stimulating insulin release and preventing glucagon secretion. The levels of GLP-1 seem to increase postprandially. Therefore, it has been suggested that GLP-1 is a key regulator in inducing satiety. This is further supported by the observation that patients have an increase in GLP-1 secretion following gastric bypass.66 This property of GLP-1 has sparked further interest in targeting the GLP-1 receptor to manufacture effective antiobesity drugs which may prevent progression to T2DM. Currently, GLP-1 analogs such as exenatide and liraglutide are recommended when maximum oral hypoglycaemic therapy with metformin and sulfonylurea have failed to achieve the desired blood glucose levels.62 Indeed, due to their effect of satiety, they are also recommended for patients to lower their body mass index.62
DPP4 inhibitors
DPP-4 inhibitors are used to reduce GLP-1 degradation. Subsequently, raised levels of endogenous GLP-1 potentiates insulin secretion, inhibits glucagon secretion whilst also inducing satiety. Currently, it is recommended that DPP-4s such as the “gliptins” (e.g sitagliptins) are added when metformin alone has not achieved optimal glycemic control. Moreover, they are also recommended as first line in patients with T2DM in whom metformin is contraindicated as 1st line.62
Reported side effects of these classes of drugs include nasopharyngitis, headache, nausea and skin reactions.61 In addition, it may also cause hypoglycemia in those taking sulfonylurea alongside DPP-4 inhibitors.
Further developments in targeting glucagon in the treatment of type 2 diabetes mellitus
In recent years, there have been several more attempts to develop drugs that blunt glucagon secretion or impair its action. A variety of compounds have been identified as a potential target to achieve these results including glucagon-derived peptides, non-peptidic small molecules, monoclonal antibodies and antisense oligonucleotides. Early results in various animal models of diabetes were encouraging, and several pharmaceutical companies have since initiated phase I and phase II clinical trials.
The first drug which targeted the glucagon receptor was Bay 27-9955 in 2001.67 Healthy male volunteers were given exogenous glucagon which elevated plasma glucose concentration. During this hyperglucagonemic period, oral administration of Bay 27-9955 reduced glucose production in a dose-dependent manner. Although continued development of the drug was not pursued, it was the first demonstration that an effective competitive glucagon antagonist could work in humans.
Over 10 years later, another GRA, MK-0893, was developed by Merck and reached phase II clinical trials. Over a 12-week period, type 2 diabetes patients were given a daily, oral dose (2080 mg) of MK-0893. Their results showed a dose-dependent decrease in both fasting and in postprandial plasma glucose levels.68 However, in some patients, there was also significant elevation in plasma LDL, plasma cholesterol and liver transaminase levels, as well as increases in body weight and blood pressure, leading to MK-0983 being discontinued.
Recently, another GRA entered phase II clinical trials; 10–60 mg of Eli Lilly’s LY-2409021 was given to type 2 diabetes patients once daily for 24 weeks. It reduced fasting and postprandial plasma glucose and glycosylated HbA1c levels, with good tolerability and the incidence of hypoglycaemic episodes was not statistically significant from placebo.69 However, there was also a dose-dependent increase in mean serum aminotransferase levels. Several patients had reversible increases in alanine and aspartate (over three times normal) which exceeded the US Food and Drug Administration limits for drug-induced liver injury. Consequently, the overall risk-benefit profile did not support the clinical development of LY2409021.
Eli Lilly also had a monoclonal antibody drug in phase I/II clinical trials, LY-2786890, a humanized IgG4. It significantly reduced fasting plasma glucose in a dose-dependent manner, with infrequent, mild hypoglycaemic episodes. However, similar to LY-2409021, there were dose-dependent elevations in liver aminotransferase levels and further investigation was halted.70
Finally, Ionis Pharmaceuticals have an antisense oligonucleotide drug currently in phase II trials. ISIS-GCGRRx antagonizes glucagon by reducing glucagon receptor mRNA expression. A preliminary study in T2DM patients showed tighter glucose control (assessed by fasting plasma glucose and HbA1c), with no hypoglycemia but again mild increases in aminotransferases (similar to other GRAs).71 A further study gave T2DM patients once-weekly 50 and 75 mg IONIS-GCGRRx.72 A substantial number of these patients showed robust and sustained reductions in HbA1c after 26 weeks, with no clinically meaningful increases in liver enzymes. With this favorable safety and efficacy profile, IONIS are advancing this drug to phase II clinical trials. Furthermore, the high specificity of antisense technology causes no drug–drug interaction concerns, and thus, this class of drugs could be given in combination with other antidiabetic agents.
Thus, it appears that most GRAs are not close to being approved for use in clinical practice. Although the efficacy profile of these small molecules is promising, the side effects do not outweigh the beneficial glucose reduction. These include weight gain, increases in blood pressure, increases in LDL and cholesterol, increase in fatty liver and changes in liver enzymes73 which could further aggravate the cardiovascular risk in T2DM patients.
Conclusions
There have been significant efforts to understand the influence and actions of insulin and glucagon in controlling glucose metabolism. As a result, many different pharmacological sites have been identified for treating T2DM. Despite these advances, the global prevalence of diabetes is sharply increasing. The impact of insulin in the development of T2DM has been widely studied. However, there is still relatively limited knowledge about the dysregulation of α-cells in T2DM. Whilst rodent studies continue to provide insights into the mechanism by which glucagon regulates glucose homeostasis, further research in human subjects is required to establish the role of glucagon in T2DM and how its role may be exploited therapeutically.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lang D, Matthews D, Peto J, Turner R. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. N Engl J Med. 1979;301(19):1023–1027. doi:10.1056/NEJM197911083011903
2. Murlin JR, Clough HD, Gibbs CBF, Stokes AM. Aqueous extracts of the pancreas. Influence on the carbohydrate metabolism of depancreatized animals. J Biol Chem. 1923;56:253–296.
3. Sutherland EW, De Duve C. Origin and distribution of the hyperglycemic-glycogenolytic factor of the pancreas. J Biol Chem. 1948;175(2):663–674.
4. Leclercq-Meyer V, Marchand J, Malaisse WJ. Calcium dependency of glucagon release: its modulation by nutritional factors. Am J Physiol. 1979;236(2):98–104.
5. Quesada I, Tuduri E, Ripoll C, Nadal A. Physiology of the pancreatic-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol. 2008;199(1):5–19. doi:10.1677/JOE-08-0290
6. Unger R, Cherrington A. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Inves. 2012;122(1):4–12. doi:10.1172/JCI57313
7.
8.
9.
10. Ramlo-Halsted BA, Edelman SV. The natural history of type 2 diabetes. Implications for clinical practice. Prim Care. 1999;26(4):771–789. doi:10.1016/S0095-4543(05)70130-5
11. Fu Z, Gilbert RE, Liu D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr Diabetes Rev. 2013;9(1):25–53. doi:10.2174/157339913804143225
12. Wilcox G. Insulin and insulin resistance. Clin Biochem. 2005;26(2):19–39.
13. Cook D, Hales C. Intracellular ATP directly blocks K+ channels in pancreatic b-cells. Nature. 1984;311(5983):271–273.
14. Henquin J. ATP-sensitive K+ channels may control glucose-induced electrical activity in pancreatic b-cells. Biochem Biophys Res Commun. 1988;156(2):769–775.
15. Hoenig M, Sharp GW. Glucose induces insulin release and a rise in cytosolic calcium concentration in a transplantable rat insulinoma. Endocrinology. 1986;119(6):2502–2507. doi:10.1210/endo-119-6-2502
16. Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest. 1992;89(4):1288–1295. doi:10.1172/JCI115714
17. Best L, Yates A, Tomlinson S. Stimulation of insulin secretion by glucose in the absence of diminished potassium (86Rb+) permeability. Biochem Pharmacol. 1992;43(11):2483–2485. doi:10.1016/0006-2952(92)90330-L
18. Dillon J, Tanizawa Y, Wheeler M, et al. Cloning and functional expression of the human glucagon-like peptide-1 (GLP-1) receptor. Endocrinology. 1993;133(4):1907–1910. doi:10.1210/endo.133.4.8404634
19. Ronner P, Matschinsky F, Hang T, Epstein A, Buettger C. Sulfonylurea-binding sites and ATP-sensitive K+ channels in alpha-tC glucagonoma and beta-tC insulinoma cells. Diabetes. 1993;42(12):1760–1762.
20. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–2157. doi:10.1053/j.gastro.2007.03.054
21. Bokvist K, Olsen H, Høy M, et al. Characterisation of sulphonylurea and ATP-regulated K+ channels in rat pancreatic a-cells. Pflugers Arch. 1999;438(4):428–436.
22. Gromada J, Høy XM, Bokvist K, Salehi A, Berggren P, Rorsman P. ATP-sensitive K+ channel-dependent regulation of glucagon release and electrical activity by glucose in wild-type and SUR1-/- mouse alpha-cells. Diabetes. 2004;53(3):181–189.
23. Göpel SO, Kanno T, Barg S, Weng X-G, Gromada J, Rorsman P. Regulation of glucagon release in mouse α-cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J Physiol. 2000;528(3):509–520. doi:10.1111/tjp.2000.528.issue-3
24. Quesada I, Todorova MG, Alonso-Magdalena P, et al. Glucose induces opposite intracellular ca 2+ concentration oscillatory patterns in identified α- and β-cells within intact human islets of langerhans. Diabetes. 2006;55(9):2463–2469. doi:10.2337/db06-0272
25. MacDonald P, Marinis Y, Ramracheya R, et al. A KATP channel-dependent pathway within α cells regulates glucagon release from both rodent and human islets of langerhans. PLoS Biol. 2007;5(6):1236–1247. doi:10.1371/journal.pbio.0050143
26. Cheng-Xue R, Gomez-Ruiz A, Antoine N, et al. Tolbutamide controls glucagon release from mouse islets differently than glucose: involvement of KATP channels from both -cells and -cells. Diabetes. 2013;62(5):1612–1622. doi:10.2337/db12-0863
27. Wendt A, Birnir B, Buschard K, et al. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes. 2004;53(4):1038–1045.
28. Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim C. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes. 2005;54(6):1808–1815. doi:10.2337/diabetes.54.6.1808
29. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi:10.1038/nrm1837
30. Hagiwara A, Cornu M, Cybulski N, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15(5):725–738. doi:10.1016/j.cmet.2012.03.015
31. Tikhanovich I, Cox J, Weinman SA. Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol. 2013;15(28):125–131. doi:10.1111/jgh.12021
32. Niswender K, Morrison C, Clegg D, et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52(2):227–321. doi:10.2337/diabetes.52.2.227
33. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8(12):1376–1382. doi:10.1038/nm1202-798
34. Pocai A, Lam TKT, Gutierrez-Juarez R, et al. Hypothalamic KATP channels control hepatic glucose production. Nature. 2005;434(7036):1026–1031. doi:10.1038/nature03379
35. Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6(12):689–697. doi:10.1038/nrendo.2010.154
36. Svoboda M, Tastenoy M, Vertongen P, Robberecht P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol Cell Endocrinol. 1994;105(2):131–137. doi:10.1016/0303-7207(94)90162-7
37. Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413(6852):179–183. doi:10.1038/35093131
38. Carlson M, Snead W, Campbell P. Regulation of free fatty acid metabolism by glucagon. J Clin Endocrinol Metab. 1993;77(1):11–15.
39. Arafat AM, Kaczmarek P, Skrzypski M, et al. Glucagon increases circulating fibroblast growth factor 21 independently of endogenous insulin levels: A novel mechanism of glucagon-stimulated lipolysis? Diabetologia. 2012;56(3):588–597. doi:10.1007/s00125-012-2803-y
40. Hotta Y, Nakamura H, Konishi M, et al. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology. 2009;150(10):4625–4633. doi:10.1210/en.2009-0119
41. Davidson IW, Salter JM, Best CH. The effect of glucagon on the metabolic rate of rats. Am J Clin Nutr. 1960;8:540–546. doi:10.1093/ajcn/8.5.540
42. Yahata T, Habara Y, Kuroshima A. Effects of glucagon and noradrenaline on the blood flow through brown adipose tissue in temperature-acclimated rates. Jpn J Physiol. 1983;33(3):367–376. doi:10.2170/jjphysiol.33.367
43. Doi K, Kuroshima A. Modified metabolic responsiveness to glucagon in cold-acclimated and heat-acclimated rats. Life Sci. 1982;30(9):785–791.
44. Nair KS. Hyperglucagonemia increases resting metabolic rate in man during insulin deficiency. J Clin Endocrinol Metab. 1987;64:896–901. doi:10.1210/jcem-64-5-896
45. Martin B, Warram J, Krolewski A, Bergman R, Soeldner J, Kahn C. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet. 1992;340(8825):925–929. doi:10.1016/0140-6736(92)92814-V
46. Godsland IF, Jeffs JAR, Johnston DG. Loss of beta cell function as fasting glucose increases in the non-diabetic range. Diabetologia. 2004;47(7):1157–1166. doi:10.1007/s00125-004-1454-z
47. Unger RH, Aguilar-Parada E, Müller WA, Eisentraut AM. Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Inves. 1970;49(4):837–848. doi:10.1172/JCI106297
48. Sherwood LM, Parris EE, Unger RH. Glucagon physiology and Pathophysiology. N Engl J Med. 1971;285(8):443–449. doi:10.1056/NEJM197107152850301
49. Shah P, Basu A, Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol. 1999;277(2):283–290.
50. Talchai C, Xuan S, Lin H, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234. doi:10.1016/j.cell.2012.07.029
51. Kumar N, Dey CS. Metformin enhances insulin signalling in insulin-dependent and -independent pathways in insulin resistant muscle cells. Br J Pharmacol. 2002;137(3):329–336. doi:10.1038/sj.bjp.0704840
52. Gunton JE, Delhanty PJD, Takahashi S-I, Baxter RC. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J Clin Endocrinol Metab. 2003;88(3):1323–1332. doi:10.1210/jc.2002-021394
53. McCreight LJ, Bailey CJ, Pearson ER. Metformin and the gastrointestinal tract. Diabetologia. 2016;59(3):426–435. doi:10.1007/s00125-015-3844-9
54. Scheen AJ, Paquot N. Metformin revisited: a critical review of the benefit-risk balance in at-risk patients with type 2 diabetes. Diabetes Metab. 2013;39:179–190. doi:10.1016/j.diabet.2013.02.006
55. Baradaran A. Lipoprotein (a), type 2 diabetes and nephropathy; the mystery continues. J Nephropathol. 2012;1:126–129. doi:10.5812/nephropathol
56. Nasri H, Behradmanesh S, Maghsoudi AR, Ahmadi A, Nasri P, Rafieian-Kopaei M. Efficacy of supplementary vitamin D on improvement of glycemic parameters in patients with type 2 diabetes mellitus: A randomized double blind clinical trial. J Ren Inj Prev. 2014;3:31–34.
57. Gheissari A, Hemmatzadeh S, Merrikhi A, Fadaei Tehrani S, Madihi Y. Chronic kidney disease in children: A report from a tertiary care center over 11 years. J Nephropathol. 2012;1:177–182. doi:10.5812/nephropathol
58. Derosa G, Limas CP, Macías PC, Estrella A, Maffioli P. Dietary and nutraceutical approach to type 2 diabetes. Arch Med Sci. 2014;10:336–344. doi:10.5114/aoms.2014.42587
59. Groop L, Eriksson CJ, Huupponen R, Ylikahri R, Pelkonen R. Roles of chlorpropamide, alcohol and acetaldehyde in determining the chlorpropamide-alcohol flush. Diabetologia. 1984;26:34–38. doi:10.1007/BF00252260
60. Kadowaki T, Hagura R, Kajinuma H, Kuzuya N, Yoshida S. Chlorpropamide-induced hyponatremia: incidence and risk factors. Diabetes Care. 1983;6:468–471.
61. Chowdhury T, Grant P. Drug therapies in type 2 diabetes: an era of personalised medicine. Clin Med (Northfield Il). 2016;16(5):441–447. doi:10.7861/clinmedicine.16-5-441
62.
63. Chen HS, Wu TE, Jap TS, Hsiao LC, Lee SH, Lin HD. Beneficial effects of insulin on glycemic control and beta-cell function in newly diagnosed type 2 diabetes with severe hyperglycemia after short-term intensive insulin therapy. Diabetes Care. 2008;31(10):1927–1932. doi:10.2337/dc08-0075
64. Lovre D, Fonseca V. Benefits of timely basal insulin control in patients with type 2 diabetes. J Diabetes Complications. 2015;29(2):295–301. doi:10.1016/j.jdiacomp.2014.11.018
65. Prasad-Reddy L, Isaacs D. A clinical review of GLP-1 receptor agonists: efficacy and safety in diabetes and beyond. Drugs Context. 2015;4:212283. doi:10.7573/dic.212283
66. Dar MS, Chapman WH
67. Petersen KF, Sullivan JT. Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia. 2001;44:2018–2024. doi:10.1007/s001250100008
68. Xiong Y, Guo J, Candelore MR, et al. Discovery of a novel glucagon receptor antagonist N-[(4-{(1S)-1-[3-(3, 5-dichlorophenyl)-5-(6- methoxynaphthalen-2-yl)-1H-pyrazol-1-yl]ethyl}phenyl)carbo nyl]- beta-alanine (MK-0893) for the treatment of type II diabetes. J Med Chem. 2012;55:6137–6148. doi:10.1021/jm300579z
69. Kazda CM, Ding Y, Kelly RP, et al. Evaluation of efficacy and safety of the glucagon receptor antagonist LY2409021 in patients with type 2 diabetes: 12- and 24-week phase 2 studies. Diabetes Care. 2016;39:1241–1249. doi:10.2337/dc15-1643
70. Kelly RP, Garhyan P, Reynolds VL, et al. Glucagon receptor antibody LY2786890 reduced glucose levels in type 2 diabetes mellitus patients. Diabetes. 2015;64:
71. Morgan E, Bethune C, Watts L, et al. Reduction of hepatic glucagon receptor expression with an antisense drug (ISIS-GCGRRX) increases total GLP-1 levels without affecting cholesterol or BP in normal subjects. Diabetologia. 2013;56(Suppl1):S280,Abstract 691.
72. Morgan E, Tai L, Jung SB, et al. Low weekly doses of IONIS-GCGRRX, a second-generation antisense glucagon receptor antagonist, caused significant improvements in glycemic control in T2DM patients on stable metformin therapy. Diabetes. 2017;66:
73. Scheen AJ, Paquot N, Pierre J. Lefèbvre investigational glucagon receptor antagonists in phase I and II clinical trials for diabetes. Expert Opin Investig Drugs. 2017;26(12):1373–1389. doi:10.1080/13543784.2017.1395020
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.