")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Current Opinions on the Clinical Utility of Ravulizumab for the Treatment of Paroxysmal Nocturnal Hemoglobinuria
Authors Gurnari C , Nautiyal I, Pagliuca S
Received 19 October 2021
Accepted for publication 28 November 2021
Published 14 December 2021 Volume 2021:17 Pages 1343—1351
DOI https://doi.org/10.2147/TCRM.S273360
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Carmelo Gurnari,1,2 Ishani Nautiyal,1 Simona Pagliuca1,3
1Department of Translational Hematology and Oncology Research, Taussig Cancer Center, Cleveland Clinic Foundation, Cleveland, OH, USA; 2Department of Biomedicine and Prevention, PhD in Immunology, Molecular Medicine and Applied Biotechnology, University of Rome Tor Vergata, Rome, Italy; 3ED561 Hematology, Oncogenesis and Biotherapies, University of Paris, Paris, France
Correspondence: Carmelo Gurnari
Translational Hematology and Oncology Research, Cleveland Clinic Foundation, 9620 Carnegie Ave, Building NE6-250, Cleveland, OH, 44106, USA
Email [email protected]
Abstract: Paroxysmal nocturnal hemoglobinuria (PNH) is a rare disorder of hematopoietic stem cells genetically defined by the acquisition of somatic mutations in the X-linked phosphatidylinositol glycan anchor biosynthesis, class A (PIGA) gene. PIGA is essential for the synthesis of glycosyl phosphatidylinositol (GPI) anchor proteins and its mutations result in a deficiency of such molecules on the membrane of blood cells derived from the mutant clone. In particular, the lack of the GPI-linked complement regulatory proteins CD55 and CD59 is responsible for the increased sensitivity of PNH erythrocytes to complement-mediated destruction. Indeed, the classical clinical picture of PNH includes signs and symptoms of intravascular hemolysis along with variable degrees of cytopenia and a strong tendency to thrombosis, hallmarks of the disease. Before the introduction of anti-complement inhibitors, PNH was characterized by a high mortality primarily due to thrombotic events. The approval of the terminal anti-complement inhibitor eculizumab in 2007 introduced a paradigm shift in the treatment of the disease with improvement of the chronic hemolytic process and dramatic reduction of the thrombotic rate. However, eculizumab has a relatively short half-life when considering a life-long treatment, with obvious consequences as to the quality of life of treated patients necessitating relatively frequent drug administrations. Moreover, up to 30% of PNH patients undergoing eculizumab therapy show a suboptimal response, continuing to require red cell transfusions because of extravascular hemolysis or breakthrough hemolytic episodes. In 2019, the FDA approved the second-generation C5 inhibitor ravulizumab, a long-lasting agent with a better control of disease manifestations. Herein, we discuss the use of ravulizumab in PNH, its differences with first-generation C5 inhibitors, the research evidence supporting the safety and efficacy of this drug and its impact on costs for health systems and quality of life of PNH patients.
Keywords: paroxysmal nocturnal hemoglobinuria, ravulizumab, anti-complement therapy
Paroxysmal Nocturnal Hemoglobinuria: A Curious Disease
Paroxysmal nocturnal hemoglobinuria (PNH) is a clonal stem cell disorder genetically defined by loss-of-function somatic mutations of phosphatidylinositol glycan anchor biosynthesis, class A (PIGA) gene (Xq22.2) belonging to the nosological spectrum of bone marrow failure syndromes.1,2 The picturesque moniker of the disease derives from the classical observation of Coca-Cola colored urine (“hemoglobinuria”) during particular changes in bloodstream acid-base equilibrium (i.e., not continuous, “paroxysmal”), typically occurring at night (“nocturnal”).3 PNH blood cells (erythrocytes, leukocytes and platelets) deriving from the PIGA-mutant clone lack glycosylphosphatidylinositol (GPI)-anchored proteins (in particular CD55 and CD59), which, among others, regulate complement functions.4 In particular, CD55 inhibits the formation of the C3 and C5 convertases while CD59 prevents the terminal polymerization of the membrane attack complex (MAC) via interaction with C9. Furthermore, the degree of CD55/CD59 deficiency based on immunofluorescent staining distinguishes PNH erythrocytes in type-1 characterized by normal expression, type-2 with partial reduction, and type-3 with their total absence.4,5
The PNH cells (especially erythrocytes) lacking these GPI-linked moieties are particularly vulnerable to complement-mediated destruction, from which the classical intravascular anemia arises, a hallmark of the disease.3 Along with anemia, the chronic hemolysis generates a variety of pathophysiological consequences responsible for a thrombophilic state, the other cardinal manifestation of PNH causing high morbidity and mortality when the disease is left untreated.6 Indeed, the free hemoglobin released after erythrocytes lysis causes a depletion in nitric oxide (NO) into the serum, neutralizing a natural defense that in normal conditions prevents platelets aggregation and adhesion.7 Besides, other processes are at play in generating the hypercoagulative status of patients with PNH and complement activation also seems to have a major role, probably mediated by inflammatory cytokines generation.8 Complement is part of the innate immune system and encompasses three different pathways (classic, alternative, and lectin binding) leading to C3 and C5 convertases activation and formation of the MAC with lysis of the target cell (Figure 1). Given its pivotal role in PNH pathogenesis, all the efforts in the last decades have tried to find feasible therapeutic targets within the intricate complement system pathway.9 Its crucial role in the pathogenesis of the disease manifestations explains the results in terms of reduction of hemolysis and thrombotic rate obtained after treatment with eculizumab, a first-in-class C5 inhibitor FDA-approved for PNH in 2007.10 This drug completely changed the natural history of the disease with improvement of hemolysis, survival and quality of life of PNH patients across the globe.11,12 However, the experience derived from clinical practice showed that a suboptimal response is registered in up to 30% of patients undergoing eculizumab treatment and is mainly due to the occurrence of breakthrough hemolytic events.2 These are defined as the return of PNH symptoms along with a new increase in lactate dehydrogenase (LDH) levels and are caused by changes in either pharmacokinetic or pharmacodynamic conditions. The first are chiefly due to insufficient drug dosing (with free C5 levels above the complete inhibition threshold of 0.5 μg/mL) and typically occur right before a new eculizumab administration (biweekly according to the label dosing) as the average drug half-life is 10 days, if excluding variations due to individual characteristics (e.g., weight).13 The latter are instead derived from conformational changes of C5, which becomes active as a result of the excess of C3b during complement-amplifying conditions (CAC) such as pregnancy, surgery and infections.14,15 Indeed, these conditions are characterized by a strong complement activation, which leads to C3b opsonization of erythrocytes and decreased binding affinity between C5 and its inhibitors without an actual increase in free C5 levels.2
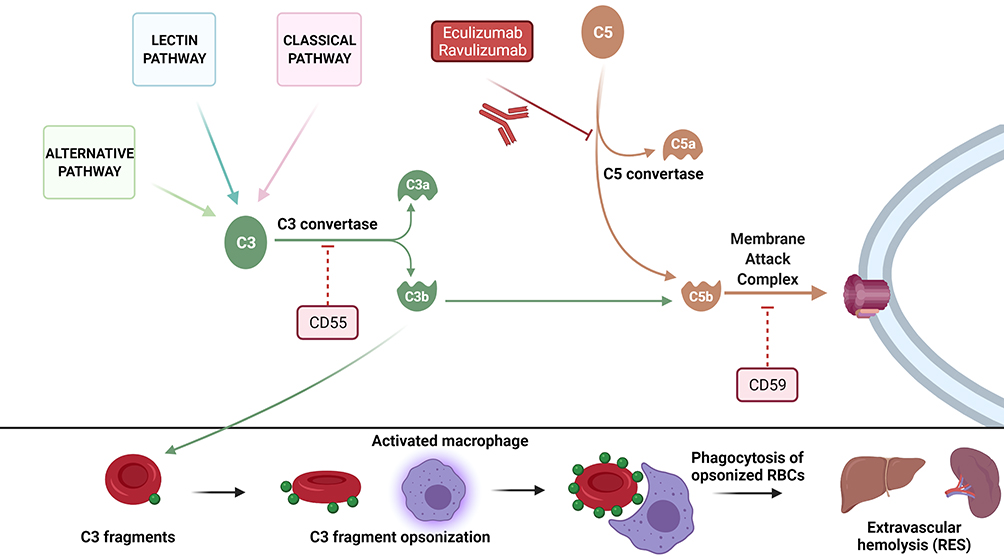 |
Figure 1 Pathogenesis of hemolysis in paroxysmal nocturnal hemoglobinuria. In the upper panel, the three complement pathways (classic, alternative, and lectin binding) converge to C3 whose activation following formation of C3 convertase and formation of C3b is regulated by CD55. C3b can then join C5b once cleaved from C5 via C5 convertase and form the membrane attack complex (MAC) along with C6, C7, C8 and C9 (a process inhibited by CD59). In PNH, the lack of CD55 and CD59 makes PNH cells (in particular erythrocytes) vulnerable to complement-mediated lysis because of the absence of inhibitory signals. Eculizumab and ravulizumab block C5 cleavage preventing the formation of the MAC. In the lower panel, the excess of C3 fragment in PNH patients treated with C5 inhibitors leads to opsonization of circulating erythrocytes and extravascular hemolysis via macrophage-mediated phagocytosis in the reticuloendothelial system (RES). |
Despite the aforementioned shortcomings, inhibitors targeting the terminal complement pathway at C5 (including the long-lasting ravulizumab, FDA-approved in 2019) are nowadays the most commonly used medication for patients with PNH.16 However, while effectively controlling the intravascular hemolytic process and reducing the thrombotic events inherent to the disease, the use of these inhibitors unmasked another mechanism of hemolysis mediated by the immune clearance of C3-opsonized erythrocytes operated by the reticuloendothelial system, contributing to the persistence of anemia due to extravascular hemolysis in some cases (easily detectable with the demonstration of C3d positivity/IgG negativity at a direct antiglobulin test)17 (Figure 1). In addition to the extravascular hemolysis, C5 rare germline polymorphisms at Arg885 identified in patients of Asian ancestry (i.e., Japanese, Han Chinese) are responsible for a genetic resistance to terminal complement inhibition.18
Ravulizumab: A Refined C5 Inhibitor
Ravulizumab (ALXN1210) is a humanized antibody targeting C5 specifically engineered from its precursor eculizumab to retain safety and efficacy but have better pharmacokinetic and pharmacodynamic profiles and, thereby, a longer half-life. To obtain these results, ravulizumab differs from eculizumab because of 4 amino acidic substitutions. In particular, two amino acid residues were replaced by histidine in order to enable the dissociation of the complex drug-C5 at the pH level of the endosomes and allowing drug recirculation instead of its lysosomal degradation after endothelial cell pinocytosis. In addition, other two amino acid substitutions to the neonatal Fc-binding portion enhance the recycling of the drug from the endosome to the intravascular space (Figure 2). These modifications generated a new agent, ravulizumab, with a half-life up to four times longer than that of the first-in-class eculizumab.19
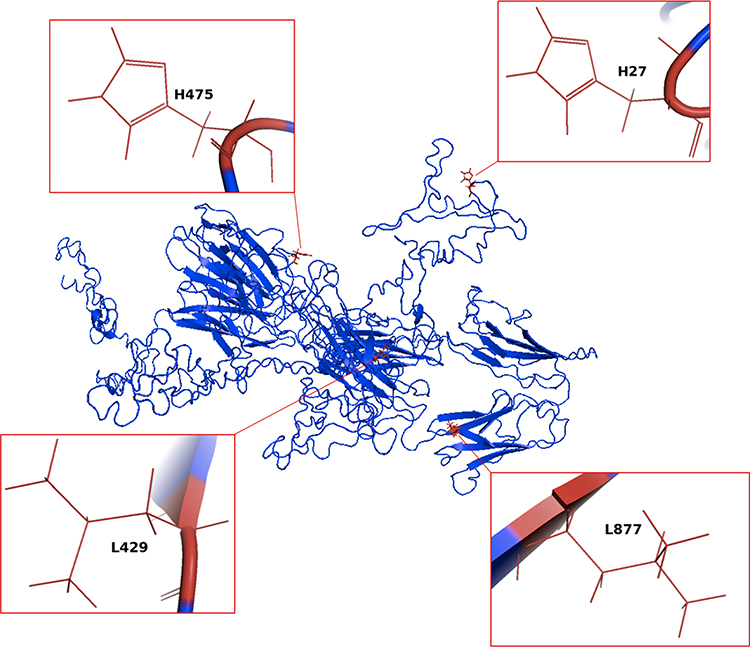 |
Figure 2 Generation of ravulizumab. The figure displays the amino acid sequence of eculizumab and highlights the amino acids substitutions responsible for the increased half-life of the second-generation C5 inhibitor ravulizumab. Figures were generated by means of I-Tasser47–49 and pyMOL Software (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC). |
After being tested in animal models, the safety and the pharmacokinetic and pharmacodynamic properties of ravulizumab were studied in a first-in-human single-ascending dose trial.20 Fourteen healthy subjects were enrolled in the study and randomized to receive 200 mg, 400 mg or placebo. Data from sera concentrations of ravulizumab showed a half-life of 32 days (compared with the previously reported 11 days of eculizumab) with a nearly complete (>99%) elimination of free C5. All subjects received vaccination against Neisseria meningitidis because of the known risk of infection in patients undergoing complement inhibitor treatment.10 Indeed, while being effective in controlling the disease manifestation, terminal complement inhibition has been associated with a risk of Neisseria infection >1000 fold that of healthy individuals with an overall rate of 0.25 meningococcal infections per 100 treated patients-years, regardless of vaccination status.21 Therefore, all PNH patients undergoing C5 inhibitors are required to receive appropriate immunization and in some cases (e.g., those younger than 45 years of age), or according to local epidemiology and health authority recommendations, additional penicillin prophylaxis (500 mg twice daily) may be considered.2
The main side effect reported after ALXN1210 administration was headache, a symptom related to vasodilation of cerebral blood circulation because of the transient surge in NO plasma levels subsequent to the drug administration, already known from the previous eculizumab experience.21
The following Phase 1b 103 study enrolled 13 PNH patients naïve to complement therapy to receive either 900 mg (lower trough exposure) or 1800 mg (higher trough exposure) of ravulizumab every 4 weeks.22 In parallel, study 201 was a Phase 2 trial testing longer dosing intervals compared with the Phase 1b 103 study (Table 1). In particular, the 26 PNH patients enrolled were divided into 4 cohorts receiving ravulizumab every 2, 3, or 4 weeks during induction and subsequently every 4 (1000 mg dosing), 6 (1600 mg), 8 (2400 mg) or 12 (5400 mg) weeks. The drug provided a significant, rapid and sustained reduction of LDH across all cohorts (primary endpoint) with a greater proportion of patients achieving normal levels when receiving higher doses. Episodes of breakthrough hemolysis were registered only in patients given lower trough exposure, possibly underlying a suboptimal drug dosing. Conversely, no episode was registered in those receiving higher trough exposures who also showed a better control of hemolytic parameters (haptoglobin, reticulocytes) and abatement of disease-related symptoms. Indeed, ravulizumab administration at dosing intervals up to 12 weeks was associated with improvement of FACIT-Fatigue Score (from 6.3 to 13.9) and EORTC QLQ-C30 for global health/QoL scale (by 8 to 28 points). In addition, no thrombotic events were registered during the study period.
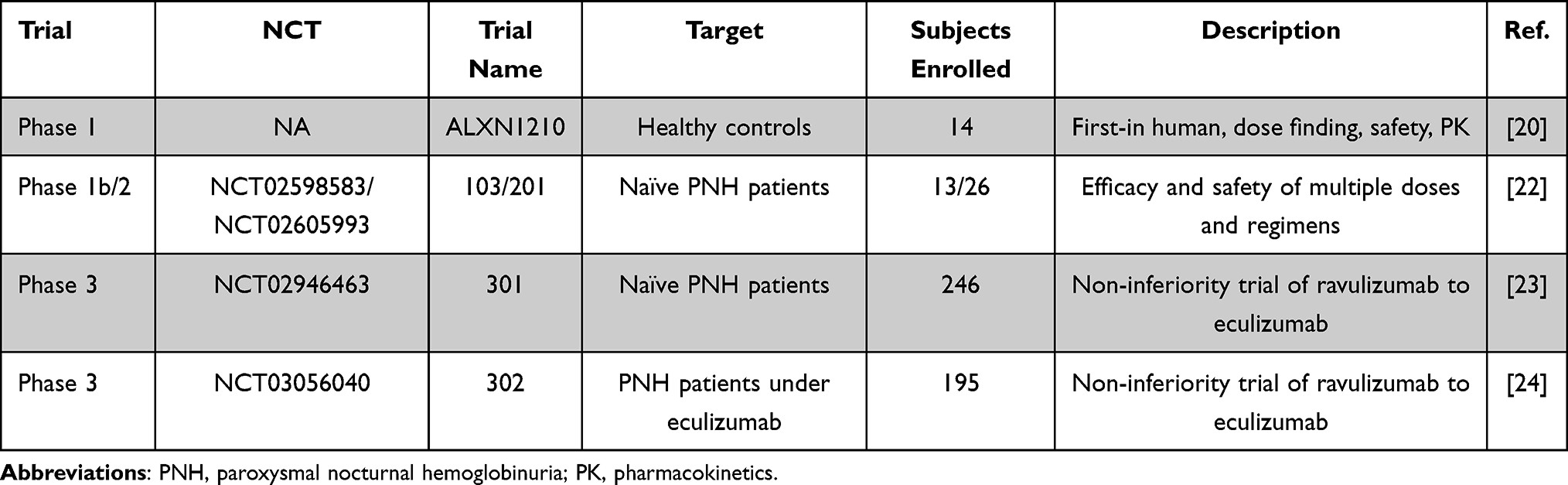 |
Table 1 Clinical Trials of Ravulizumab in PNH |
The subsequent non-inferiority Phase 3 trials compared head-to-head eculizumab and ravulizumab in PNH patients naïve to (study 301) and under eculizumab treatment (study 302) (Table 1).23,24 In both studies, ravulizumab was given intravenously after a loading dose according to body weight (2400 mg for patients weighing ≥40 to <60 kg, 2700 mg ≥60 to <100 and 3000 mg ≥100 kg) followed after 2 weeks and thereafter every 8 weeks by a maintenance dose (3000 mg, 3300 mg and 3600 mg, for the same weight subgroups, respectively). The 301 study enrolled 246 patients naïve to complement inhibitors in a randomized fashion 1:1 to receive either ravulizumab or eculizumab for a total of 183 days. Ravulizumab was shown to be non-inferior to eculizumab as to LDH normalization and transfusion-independency (primary endpoints, pinf <0.001). Moreover, the non-inferiority was demonstrated also for reduction in LDH levels, hemoglobin stabilization, number of breakthrough hemolytic episodes, C5 levels reduction and changes in disease-specific burden of symptoms.23 Importantly, the trial demonstrated the efficacy of a four-fold longer dosing interval of the new experimental drug as opposed to a 2 weeks dosing of eculizumab with a substantial improvement in patients reported QoL.
The 302 study enrolled instead eculizumab-experienced (> 6 months) PNH patients (n = 195) randomized to switch to ravulizumab or continue with eculizumab at a labeled-dose in a 1:1 fashion.24 In this study, the primary endpoint was the change in decrease of LDH from baseline to the end of study period (183 days) while secondary endpoints were similar to the previous 301 twin study. Similarly to the results in the complement inhibitor naïve setting, ravulizumab was non-inferior to eculizumab for all the primary and secondary endpoints (pinf <0.0006). Furthermore, the trial demonstrated the safety and efficacy of switching from the classical eculizumab schedule to the new ravulizumab dosing administered every 8 weeks.
As previously mentioned, the most common adverse event (expected for this class of drug) was headache, which occurred roughly in 30% of cases in both studies with no significant difference between the groups and tended to decrease over time.25 Of note, no episode of meningococcal infections was registered even after an updated analysis of both studies (one year follow-up results) but these data need to be interpreted cautiously and confirmed by more mature long-term results.26 Most importantly, no episode of breakthrough hemolysis in the ravulizumab arm was registered because of suboptimal C5 inhibition (free C5 levels ≥0.5 μg/mL), as demonstrated by a recent analysis of pharmacokinetics data of both trials showing that steady-state serum concentration and neutralization of free C5 levels were achieved immediately after the first dose of ravulizumab and were stably maintained over time as opposed to eculizumab.13 Indeed, in the majority of cases (80%) the observed breakthrough hemolytic episodes were found to be temporally associated with CAC such as infections and occurred less frequently in the ravulizumab vs the eculizumab arm (4% vs 10.7% in the 301 study; 0% vs 5.1% in the 302).27
It is noteworthy that the 301 and 302 trials excluded patients on non-standard treatment dosing of eculizumab, thus there is the potential that some patients will require reduced intervals between dosing also on ravulizumab, an aspect which is now evaluated in a Phase 4 clinical trial (NCT04320602) enrolling patients requiring eculizumab doses of more than 900 mg every 2 weeks.
Considerations on the Use of Ravulizumab in PNH
Ravulizumab has dramatically revolutionized PNH treatment. As discussed above, the better control of free C5 levels not only resulted in improvement of the hemolytic and thrombotic manifestations of the disease but also of the patient-reported symptoms, as demonstrated by the data derived from FACIT-Fatigue score and EORTC QLQ-C30 questionnaires of the one-year follow-up analysis.25,26 In addition to a better control of the disease manifestations and symptoms, ravulizumab substantially decreased the number of hospitalizations with a total of six infusions per year vs the 26 of the previous administration schedule of eculizumab. This dosing has been favorably welcomed by PNH patients who now can more easily plan vacations, work and family-related duties without the risk of incurring episodes of breakthrough hemolysis, as was happening previously when requesting a wider time frame of eculizumab administration. In a study enrolling patients participating in the phase 3 302 trial, the specific preferences for eculizumab or ravulizumab were evaluated using an 11-item PNH-specific Patient Preference Questionnaire.28 The analysis of the results obtained from the 95 subjects who completed the assessment clearly demonstrated a predilection (>90%) towards ravulizumab (p<0.001). In comparison to eculizumab, ravulizumab was preferred because of the lower infusion frequency, ability to plan activities and overall QoL, even considering its longer time for administration (3 hours vs 35 minutes for eculizumab).28
The lower number of hospital admissions has obvious repercussions also on costs for health care systems.29 In a cost-utility analysis from a US payer perspective with the application of a semi-Markov model, ravulizumab was associated with cost savings and health benefit (in terms of QALY, quality-adjusted life-year).30 Savings included a 10% lower labeled maintenance costs per year, lower hospital admissions and lower number of breakthrough hemolytic episodes.
It has to be noted that the current ravulizumab schedule provides mean serum concentrations 4 times higher than those ensuring a complete C5 inhibition (i.e., >100 μg/mL). Therefore, by applying a clinical trial simulation of 3 different strategies of ravulizumab dosing with the use of therapeutic drug monitoring (TDM) over a 1 year period, a recent study showed that the interval between doses may be potentially extended up to almost 13 weeks with a reduction of 37% in drug costs.31 Besides savings, which are of utmost importance especially for lower-income countries, the consequences of even more rarefied drug doses are obvious also in terms of patient-friendliness. Indeed, complement inhibition is still not available in a non-negligible fraction of countries around the world and the high costs of such treatments further contribute in creating inequality of patient care.32
Finally, the possible fear of hypogammaglobulinemia due to the long serum half-life of ravulizumab and its modified high affinity neonatal Fc receptor (possibly interfering with IgG homeostasis) has been removed by a recent study on 12 chronically treated PNH patients. Indeed, the results showed no changes in IgG concentrations throughout the observation time (median of 21.6 months).33 Once the efficacy of ravulizumab is established, the evaluation of its possible off-target effects are important as to the implications that they have in special PNH populations such as children, pregnant women and elderly patients.
A Glimpse into the Next Future: From Terminal to Proximal Complement Inhibition
Even though ravulizumab eliminated the breakthrough hemolytic events related to suboptimal free C5 inhibition, a non-negligible fraction of patients still experience various degrees of extravascular hemolysis because of C3 fragment opsonization (Figure 1).9 To overcome this problem, pegcetacoplan, a new drug inhibiting the complement pathway at the level of C3 (proximal inhibition) and thereby able to control both intravascular and extravascular hemolysis, has been recently FDA approved for the treatment of PNH.34 In a phase 3 open-label clinical trial, the new drug has been evaluated head-to-head with eculizumab in PNH patients registering suboptimal eculizumab responses (hemoglobin levels < 10.5 g/dL).35 Pegcetacoplan administered subcutaneously twice weekly reached the primary endpoint of increase in hemoglobin levels from baseline to week 16 (adjusted mean difference of 3.84 g/dL, p<0.001).34
Novel agents targeting other components of the complement system are currently under investigation in clinical trials with encouraging results.9 Danicopan is an orally available first-in-class factor D inhibitor blocking the alternative pathway and now evaluated in phase 3 trials (NCT04469465). Two open-label, single arm phase 2 studies of Danicopan alone in treatment-naïve PNH patients or as an add-on in eculizumab poor-responders showed its efficacy in reducing LDH and preventing both intravascular and extravascular hemolysis with improvement of hemoglobin levels.36,37 Furthermore, other factor D inhibitors (ALXN2050, BCX9930) are being currently tested in ongoing clinical trials (NCT04623710, NCT04702568).
Another oral inhibitor of the early phase of complement activation is Iptacopan, a first-in-class factor B inhibitor, which has been evaluated in a phase 2 trial as an add-on to eculizumab in treated patients presenting signs of uncontrolled hemolysis with promising results. Iptacopan is now under investigation in a phase 3 trial (NCT04558918).38
Although optimal results have been achieved with terminal complement inhibitors after the introduction of ravulizumab and a subcutaneous formulation of this drug is currently under investigation (EudraCT number: 2017-002370-39), a new long-acting (administered subcutaneously once a month) anti-C5 inhibitor, crovalimab, has been recently developed and is being tested in two phase 3 trials in treatment naïve (NCT04432584) and eculizumab-treated (NCT04434092) PNH patients after promising data derived from phase 1 and 2 studies.39,40 Of note, this drug is also effective in patients harboring C5 germline polymorphisms at Arg885. Finally, another subcutaneous C5 inhibitor (REGN3918, a human IgG4P antibody) also active in carriers of the Arg885 variant, is currently evaluated in a phase 3 clinical trial (NCT04162470) after showing encouraging results in previous studies (NCT03946748).41
Conclusions
PNH is a peculiar disorder whose natural history was revolutionized after the introduction of complement inhibitors. From a median survival of 15 years after diagnosis in the pre-eculizumab era, nowadays PNH patients have survival corresponding that of age-matched healthy individuals.12,42,43 The appropriate control of the underlying hemolytic process has indeed reduced the rate of thrombotic events, the main cause of PNH mortality, responsible for up to 50% of fatal events in the pre-eculizumab era.43
As of today, no curative treatment for PNH has been identified other than hematopoietic stem cell transplantation (HSCT), which is considered typically in young patients with concomitant severe aplastic anemia (overlap syndrome).2,44 Indeed, given the impressive long-term results of terminal complement inhibitors and a retrospective analysis, HSCT has been challenged as a suitable option for the treatment of classical hemolytic PNH in a study of the Severe Aplastic Anemia Working Party (SAAWP) of the European Group for Blood and Marrow Transplantation (EBMT).44
Currently, ravulizumab represents a very effective terminal complement inhibitor and the encouraging data deriving from its use as well as the possible cost-saving dosing individualization via TDM approaches make this drug an attractive option for the treatment of PNH.31,45
In the near future, PNH patients will also benefit from an expansion of the disease therapeutic armamentarium thanks to the availability of multiple options (oral and subcutaneous besides the classical intravenous formulations) and dosing schedules, possibly warranting a complete abatement of symptoms and a better control of the disease-specific manifestations along with optimization of patient treatment and convenience.46 Moreover, the increased number of therapeutic options will also help in reducing drugs costs, enabling more and more PNH patients worldwide to access the same care and treatment.
Acknowledgments
We thank the American-Italian Cancer Foundation Post-Doctoral Research Fellowship (to C.G.), the Italian Society of Hematology and Association HPN France – Aplasie medullaire and FFRD, Foundation For Rare Diseases (to S.P.).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no competing interests.
References
1. Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73(4):703–711. doi:10.1016/0092-8674(93)90250-T
2. Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2021;137(10):1304–1309. doi:10.1182/blood.2019003812
3. Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement-mediated hematological disorders. Immunobiology. 2012;217(11):1080–1087. doi:10.1016/j.imbio.2012.07.014
4. Gurnari C, Pagliuca S, Patel BJ, et al. Implication of PIGA genotype on erythrocytes phenotype in Paroxysmal Nocturnal Hemoglobinuria. Leukemia. 2021;35:2431–2434. doi:10.1038/s41375-020-01113-0
5. Sutherland DR, Keeney M, Illingworth A. Practical guidelines for the high-sensitivity detection and monitoring of paroxysmal nocturnal hemoglobinuria clones by flow cytometry. Cytometry B Clin Cytom. 2012;82(4):195–208. doi:10.1002/cyto.b.21023
6. Gurnari C, Colak C, Zawit M, Maciejewski JP. Chronic Budd-Chiari syndrome in paroxysmal nocturnal haemoglobinuria. Lancet. 2021;398(10304):e14. doi:10.1016/S0140-6736(21)01542-7
7. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin a novel mechanism of human disease. JAMA. 2005;293(13):1653–1662. doi:10.1001/jama.293.13.1653
8. Risitano AM, Peffault de Latour R. How we (‘ll) treat paroxysmal nocturnal haemoglobinuria: diving into the future. Br J Haematol. 2021. doi:10.1111/bjh.17753
9. Gavriilaki E, Peffault de Latour R, Risitano AM. Advancing therapeutic complement inhibition in hematologic diseases: PNH and beyond. Blood. 2021. doi:10.1182/blood.2021012860
10. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–1243. doi:10.1056/NEJMoa061648
11. Gurnari C, Graham AC, Efanov A, et al. Frequency and perturbations of various peripheral blood cell populations before and after eculizumab treatment in paroxysmal nocturnal hemoglobinuria. Blood Cells Mol Dis. 2020;87:102528. doi:10.1016/j.bcmd.2020.102528
12. Hillmen P, Muus P, Röth A, et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162(1):62–73. doi:10.1111/bjh.12347
13. Peffault de Latour R, Brodsky RA, Ortiz S, et al. Pharmacokinetic and pharmacodynamic effects of ravulizumab and eculizumab on complement component 5 in adults with paroxysmal nocturnal haemoglobinuria: results of two phase 3 randomised, multicentre studies. Br J Haematol. 2020;191(3):476–485. doi:10.1111/bjh.16711
14. Harder MJ, Kuhn N, Schrezenmeier H, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129(8):970–980. doi:10.1182/blood-2016-08-732800
15. Harder MJ, Höchsmann B, Dopler A, et al. Different levels of incomplete terminal pathway inhibition by eculizumab and the clinical response of PNH patients. Front Immunol. 2019;10:1639. doi:10.3389/fimmu.2019.01639
16. McKeage K. Ravulizumab: first global approval. Drugs. 2019;79(3):347–352. doi:10.1007/s40265-019-01068-2
17. Risitano AM, Notaro R, Marando L, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–4100. doi:10.1182/blood-2008-11-189944
18. Nishimura JI, Yamamoto M, Hayashi S, et al. Genetic variants in C5 and poor response to eculizumab. New Engl J Med. 2014;370(7):632–639. doi:10.1056/NEJMoa1311084
19. Sheridan D, Yu ZX, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti-C5 antibody with extended duration of action. PLoS One. 2018;13(4):e0195909. doi:10.1371/journal.pone.0195909
20. Sahelijo L, Mujeebuddin A, Mitchell D, et al. First in human single-ascending dose study: safety, biomarker, pharmacokinetics and exposure-response relationships of ALXN1210, a humanized monoclonal antibody to C5, with marked half-life extension and potential for significantly longer dosing intervals. Blood. 2015;126(23):4777. doi:10.1182/blood.V126.23.4777.4777
21. Socié G, Caby-Tosi M-P, Marantz JL, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol. 2019;185(2):297–310. doi:10.1111/bjh.15790
22. Röth A, Rottinghaus ST, Hill A, et al. Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of 2 phase 1b/2 studies. Blood Adv. 2018;2(17):2176–2185. doi:10.1182/bloodadvances.2018020644
23. Lee JW, Sicre de Fontbrune F, Wong lee lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539. doi:10.1182/blood-2018-09-876136
24. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549. doi:10.1182/blood-2018-09-876805
25. Schrezenmeier H, Kulasekararaj A, Mitchell L, et al. One-year efficacy and safety of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria naïve to complement inhibitor therapy: open-label extension of a randomized study. Ther Adv Hematol. 2020;11:2040620720966137. doi:10.1177/2040620720966137
26. Kulasekararaj AG, Hill A, Langemeijer S, et al. One-year outcomes from a phase 3 randomized trial of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria who received prior eculizumab. Eur J Haematol. 2021;106(3):389–397. doi:10.1111/ejh.13564
27. Brodsky RA, Peffault de Latour R, Rottinghaus ST, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(1):230–237. doi:10.3324/haematol.2019.236877
28. Peipert JD, Kulasekararaj AG, Gaya A, et al. Patient preferences and quality of life implications of ravulizumab (every 8 weeks) and eculizumab (every 2 weeks) for the treatment of paroxysmal nocturnal hemoglobinuria. PLoS One. 2020;15(9):e0237497. doi:10.1371/journal.pone.0237497
29. Connell NT. Ravulizumab: a complementary option for PNH. Blood. 2019;133(6):503–504. doi:10.1182/blood-2018-12-891499
30. O’Connell T, Buessing M, Johnson S, Tu L, Thomas SK, Tomazos I. Cost-utility analysis of ravulizumab compared with eculizumab in adult patients with Paroxysmal Nocturnal Hemoglobinuria. PharmacoEconomics. 2020;38(9):981–994. doi:10.1007/s40273-020-00929-z
31. Ter Avest M, Langemeijer SMC, Schols SEM, et al. The potential of individualized dosing of ravulizumab to improve patient-friendliness of paroxysmal nocturnal haemoglobinuria treatment at reduced costs. Br J Clin Pharmacol. 2021;87(8):3359–3363. doi:10.1111/bcp.14748
32. Griffin M, Kelly R, Pike A. A review of the treatment landscape in paroxysmal nocturnal haemoglobinuria: where are we now and where are we going? Therap Adv Rare Dis. 2020;1:2633004020959349.
33. Alashkar F, Rottinghaus S, Vance C, et al. No evidence for hypogammaglobulinemia in patients with paroxysmal nocturnal hemoglobinuria (PNH) chronically treated with ravulizumab. PLoS One. 2020;15(3):e0230869. doi:10.1371/journal.pone.0230869
34. Hillmen P, Szer J, Weitz I, et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2021;384(11):1028–1037. doi:10.1056/NEJMoa2029073
35. Gurnari C, Pagliuca S, Maciejewski JP. Pegcetacoplan versus Eculizumab in PNH. N Engl J Med. 2021;385(18):1725. doi:10.1056/NEJMc2106424
36. Risitano AM, Kulasekararaj AG, Lee JW, et al. Danicopan: an oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica;2020. doi:10.3324/haematol.2020.261826
37. Kulesekararaj A, Risitano AM, Maciejewski JP, et al. Phase 2 study of danicopan in paroxysmal nocturnal hemoglobinuria patients with an inadequate response to Eculizumab. Blood. 2021;138:1928–1938. doi:10.1182/blood.2021011388
38. Risitano AM, Röth A, Soret J, et al. Addition of iptacopan, an oral factor B inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: an open-label, single-arm, phase 2, proof-of-concept trial. Lancet Haematol. 2021;8(5):e344–e354. doi:10.1016/S2352-3026(21)00028-4
39. Röth A, Nishimura JI, Nagy Z, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood. 2020;135(12):912–920. doi:10.1182/blood.2019003399
40. Fukuzawa T, Sampei Z, Haraya K, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci Rep. 2017;7(1):1080. doi:10.1038/s41598-017-01087-7
41. Weyne J, Ni Y, DelGizzi R, et al. A randomized, double-blind, placebo-controlled phase 1 study of the pharmacokinetics and pharmacodynamics of REGN3918, a human antibody against complement factor C5, in healthy volunteers. Blood. 2018;132(Supplement 1):1039. doi:10.1182/blood-2018-99-112262
42. Socié G, Mary JY, de Gramont A, et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet. 1996;348(9027):573–577. doi:10.1016/S0140-6736(95)12360-1
43. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. New Engl J Med. 1995;333(19):1253–1258. doi:10.1056/NEJM199511093331904
44. Régis Peffault de L, Hubert S, Andrea B, et al. Allogeneic stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Haematologica. 2012;97(11):1666–1673.
45. Risitano AM, Marotta S, Ricci P, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. 2019;10:1157. doi:10.3389/fimmu.2019.01157
46. Stern RM, Connell NT. Ravulizumab: a novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Ther Adv Hematol. 2019;10(2040620719874728). doi:10.1177/2040620719874728
47. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. doi:10.1038/nprot.2010.5
48. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi:10.1038/nmeth.3213
49. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9(1):40. doi:10.1186/1471-2105-9-40
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.