")
Back to Archived Journals » Metalloproteinases In Medicine » Volume 5
Current insights into matrix metalloproteinases and glioma progression: transcending the degradation boundary
Authors Pullen NA , Pickford AR , Perry MM , Jaworski DM, Loveson KF, Arthur DJ , Holliday JR , Van Meter TE , Peckham R , Younas W, Briggs SEJ, MacDonald S, Butterfield T, Constantinou M , Fillmore HL
Received 9 April 2018
Accepted for publication 11 June 2018
Published 17 September 2018 Volume 2018:5 Pages 13—30
DOI https://doi.org/10.2147/MNM.S105123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Nicholas A Pullen,1 Andrew R Pickford,2 Mark M Perry,3 Diane M Jaworski,4 Katie F Loveson,3 Daniel J Arthur,3 Jonathan R Holliday,3 Timothy E Van Meter,5 Ritchie Peckham,3 Waqar Younas,3 Sophie EJ Briggs,6 Sophie MacDonald,3 Thomas Butterfield,3 Myrianni Constantinou,3 Helen L Fillmore3
1School of Biological Sciences, University of Northern Colorado, Greeley, CO, USA; 2Biophysics Laboratories, School of Biological Sciences, Institute of Biological and Biomedical Sciences, University of Portsmouth, 3School of Pharmacy and Biomedical Sciences, University of Portsmouth, Portsmouth, UK; 4Department of Neurological Sciences, Robert Larner College of Medicine, University of Vermont, Burlington, VT, 5Department of Pediatrics, Pediatric Hematology–Oncology, VCU School of Medicine, Richmond, VA, USA; 6Arnie Charbonneau Cancer Institute, University of Calgary, Calgary, AB, Canada
Abstract: Glioblastoma multiforme (GBM) remains one of the most deadly cancers, with modest advances in overall survival despite significant improvements in imaging, surgery, and molecular genomic understanding. The highest-grade glioma, GBM is a primary brain cancer that is molecularly heterogeneous among patients and even within the same patient. Key hallmarks include glioma-cell invasion, angiogenesis, and therapeutic resistance. While once considered a major player in glioma invasion, members of the MMP family are also associated with other key pathological hallmarks of glioma. Investigations into understanding MMP function in GBM were slowed due to the failed MMP-inhibitor trials for GBM in the 2000s. In contrast, the field of MMPs in other brain pathologies has flourished in such areas as traumatic brain injury, multiple sclerosis, and stroke. In the past decade, the increase in publicly available data sets documenting patient-biopsy molecular information has empowered laboratory investigations into the spectrum of genomic, transcriptomic, and proteomic changes associated with glioma, including MMPs. In this review, we selected one of these data sets to illustrate a small sample of information that can be obtained from such analyses. Combined with recent reports on the use of MMP-cleavable peptides for imaging and the multifunctionality of MMPs, including intracellular nonproteolytic actions in various cell types, this paves the way for new avenues of MMP research. Understanding the function of MMPs in host–tumor interactions both spatially and temporally during tumor progression and in response to treatment will be crucial for the advancement of targeting specific MMPs in GBM. The opportunities to explore MMP regulation, expression, and function further in GBM have never been so great with progress in modern bioinformatics and molecular techniques, and it is hoped that advancements will translate in some way to patients diagnosed with GBM.
Keywords: glioblastoma, matrix metalloproteinase, glioma, brain
Introduction
Although there are over 120 types of primary brain cancer, the most deadly and among the most common is glioblastoma multiforme (GBM).1 In 2016, the World Health Organization (WHO) classification of central nervous system (CNS) tumors was updated to incorporate recognized molecular indicators with the classical histopathological categories.1 By integrating molecular genetics with classical pathology, it is believed that this will help guide treatment. GBM (WHO grade IV astrocytoma) can emerge de novo or develop from a malignant progression of a lower-grade diffuse astrocytoma (WHO grade II) or anaplastic astrocytoma (WHO grade III). The progression of a lower grade to a high grade is associated with mutations in IDH1 and IDH2.2
GBM produces heterogeneous tumors even within the same patient, with intratumoral heterogeneity presenting regional differences in critical drives of tumor growth and treatment response.3 Prevalent vascular disorganization, angiogenesis, and invasion are key features. Despite advances in conventional therapies over the past few decades, there is no cure.4 Initial treatment usually includes surgical resection of the tumor mass, followed by radiation and chemotherapy (temozolomide [Tmz]).5,6 In virtually all cases, the tumor recurs, usually near the margins of the previous resection, frequently in a form that is more resistant to subsequent therapies. Less than 4% of patients with GBM survive 5 years following diagnosis.7
While considered a major contributor to glioma invasion, members of the MMP family are also associated with other key pathological hallmarks of glioma. Recent reports on the multifunctionality of MMPs, including intracellular nonproteolytic actions in various cell types, are intriguing8,9 and pave the way for new avenues of MMP research built upon an already solid foundation.10 This review provides an overview on the structure/function and regulation of MMPs, but will primarily focus on relating data on MMP gene expression obtained from publicly available data sets and our reverse-transcription quantitative polymerase chain reaction (RT-qPCR) data demonstrating unique glioma-expression patterns of MMPs to proteolytic and recently identified novel, nonproteolytic MMP functions. Although previous MMP-inhibitory trials were largely unsuccessful,11 these recently identified functions not only open up new research avenues but may also lead to the identification of potentially targetable MMPs for therapeutics.
Overview
The 23 MMPs (also known as matrixins) are part of a larger protease family, the metzincins, and include the astacins, adamlysins, pappalysins and bacterial serralysins,12 some which have been implicated in glioma.13–15 MMPs are zinc-dependent, calcium-containing endopeptidases responsible for a wide range of biological processes, including angiogenesis, migration, growth, metabolism, and cell survival. Therefore, MMP activity must be tightly regulated at multiple levels:16 transcriptionally by multiple factors and posttranscriptionally by microRNAs, compartmentalization of the inactive proenzyme (zymogen), zymogen activation, and subsequent inhibition of the activated enzymes by endogenous inhibitors.17–20
MMPs are multidomain endopeptidases translated as latent proenzyme or zymogens (proMMPs) with an archetypal domain arrangement, including a secretion signal (removed upon entry into the endoplasmic reticulum) followed by an inhibitory propeptide (Pro) domain, a catalytic (Cat) domain, a proline-rich linker (or “hinge” region), and a hemopexin-like (Hpx) domain (Figure 1A). This organization accurately describes approximately half the family members; the others either lack the Hpx domain or contain additional ancillary domains important for cellular localization, substrate recognition, and/or inhibitor binding. An extensive account of the structure–function relationship in MMPs is beyond the scope of this review; therefore, only a brief synopsis is provided in the following section. Interested readers are directed to excellent accounts of MMP structure elsewhere.21,22
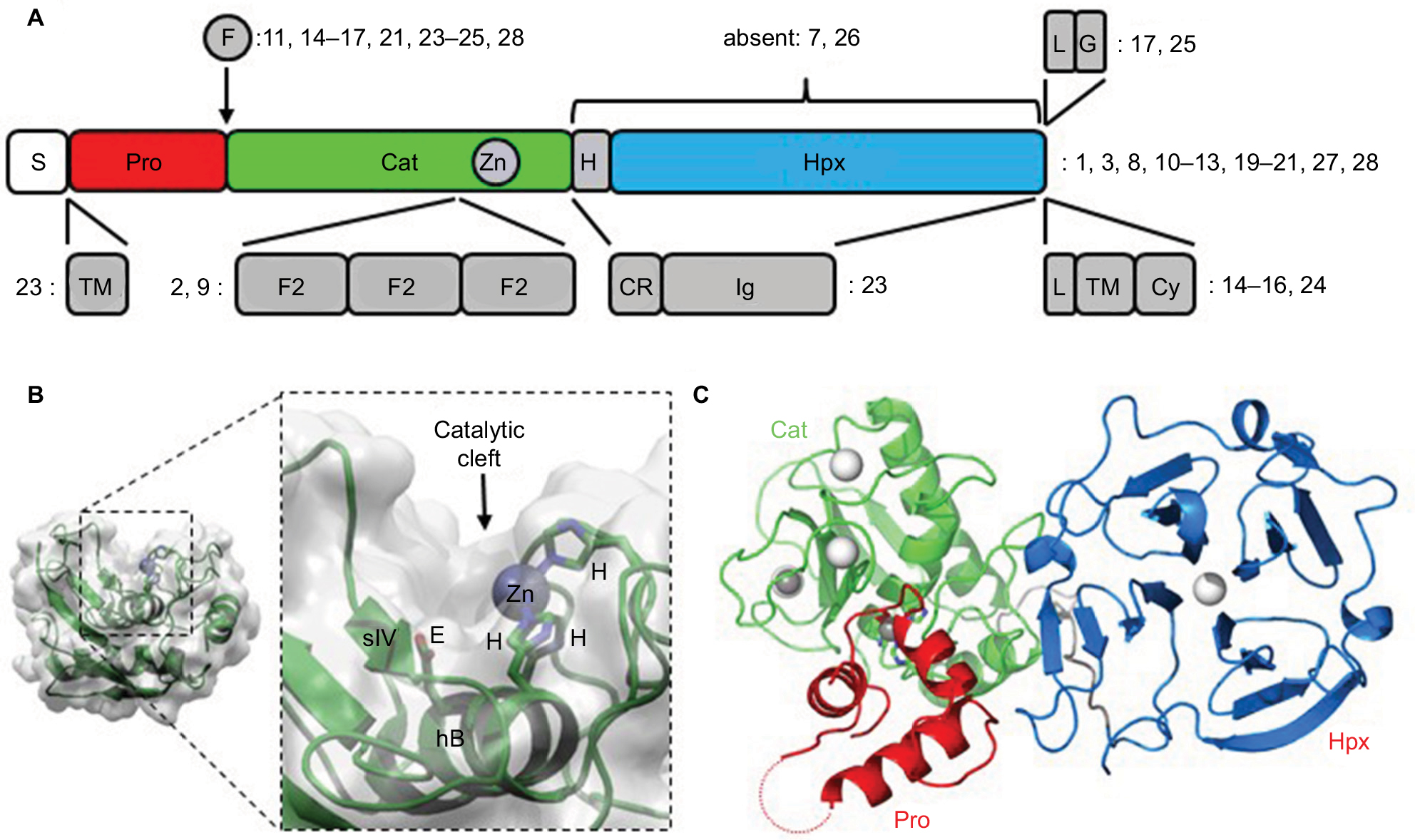 | Figure 1 Domain organization in MMPs. Notes: (A) Overall schematic. Numbers indicate in which MMP family members the various features occur. (B) Representation of the Cat domain from MMP1 depicting the secondary-structure elements within the molecular envelope, and (magnified) details of the catalytic cleft depicting the roles of the His and Glu residues in the characteristic HEXGHXXGXXH motif. (C) Crystal structure of human proMMP1211 showing the relative arrangement of the Pro, Cat, and Hpx domains. Bound calcium and zinc ions are shown as white and gray spheres, respectively. The dotted red line indicates the approximate location of the unstructured “bait region” within the Pro domain. Abbreviations: S, secretion signal; Pro, propeptide; Cat, catalytic; H, hinge region; Hpx, hemopexin; F, furin-recognition sequence; L, linker; G, glycosylphosphatidylinositol anchor; TM, transmembrane; F2, fibronectin type II; CR, cysteine-rich; Cy, cytoplasmic; Zn, zinc. |
Importance of structure–function relationship
The hydrolytic apparatus of the ~170-residue Cat domain resides within a shallow crevice of what is otherwise a roughly spherical tertiary structure combining three α-helices (hA–hC) and a five-stranded (sI–sV) mixed β-sheet (Figure 1B). The second of these helices (ie, hB) provides the base of the active-site cleft, and together with the subsequent loop region (hB–hC) contains the characteristic Cat-domain motif HEXGHXXGXXH, which harbors the residues involved in substrate hydrolysis: the three histidine side chains chelate the active-site Zn2+ ion, which together with the invariant glutamate supercharges a bound water molecule to perform a nucleophilic attack on the carbonyl carbon of the substrate’s scissile peptide bond.
Although originally named for their breakdown of the extracellular matrix (ECM), MMPs are now known to hydrolyze multiple substrate families, including growth factors, cytokines, cell-surface receptors, and even intracellular components.23–25 The precise shape and character of the active-site cleft (determined by the amino-acid residues that line it) largely dictate each MMP’s substrate preference; notable regions include the so-called wall-forming segment (strand sIV; Figure 1B) and subsequent S1’ specificity loop (hB–hC).22 However, additional regions, more distant from the active site and often in ancillary (noncatalytic) domains, provide additional determinants of substrate specificity.26,27 These regions are termed “exosites”.28 For example, in MMP2 and -9, three tandem fibronectin type 2 (F2) domains, each ~60 residues, are inserted into the sV–hB loop within the Cat domain (Figure 1A). These F2 domains provide supplementary binding sites for their substrates.
With the exception of MMPs 7, 23, and 26, the Cat domain is linked to an Hpx domain via a proline-rich linker 10–70 amino acids in length (Figure 1A). The Hpx domain is a ~190 residue, four-bladed β-propeller with a bound Ca2+ ion along its axis. For many MMPs and their substrates, the precise role of the Hpx domain in hydrolysis has not been investigated comprehensively. However, for others, it is an exosite location, eg, the domain is required for the collagenolytic activity of MMP1, -2, -8, -9, -13, and -14.29 Although type I collagen may not be a relevant MMP substrate in glioma, this dependence illustrates the potential for the Hpx domain to direct proteolysis by the Cat domain. The Hpx domain can also influence nonproteolytic interactions of MMPs within the ECM. For example, the Hpx domain is known to mediate the interactions of MMP1, -2, and -9 with heparin,30 the binding of MMP1 to the I-domain of integrin α2β1,31 and in MMP9 and -14 it is the site of enzyme dimerization.32 In place of an Hpx domain, MMP23 possesses cysteine-rich and immunoglobulin-like domains.33
The inactivating Pro domain, a cluster of three α-helices with connecting loops, docks into the Cat domain’s active-site cleft (Figure 1C), maintaining latency by chemically neutralizing the reactive Zn2+ ion with an invariant Cys residue and sterically restricting substrate access.21 Activation from this hydrolytically-inert, proMMP state to the mature, active enzyme requires disruption of this Cys–Zn interaction, a mechanism known as the “cysteine switch”.34 In those MMPs possessing a furin-recognition sequence in the Pro–Cat linker (Figure 1A), the zymogen is activated by this proprotein convertase during transit through the Golgi.35 In the other proMMPs, activation typically occurs after secretion and involves removal of the entire Pro domain through serial proteolytic truncations. The initial trigger for this may itself be a proteolytic event, such as cleavage of the flexible, protease-sensitive “bait region” within the Pro domain of many proMMPs. This may be elicited by an activated molecule of the same MMP (a process termed “autolysis”) by other MMPs or certain serine proteases, such as plasmin.36 Alternatively, the initial triggering event may be an allosteric structural change (eg, membrane-association-triggered activation of proMMP7)37 or chemical oxidation of the Zn2+-chelating Cys thiol by reactive oxygen species.36
Six MMPs are localized to the cell surface: four are integral membrane-type enzymes (MMPs 14, 15, 16, and 24) with a type I transmembrane helix C-terminal to the Hpx domain and two (MMPs 17 and 25) have a glycosylphosphatidylinositol (GPI) anchor toward the C-terminus.38 Each membrane-type MMP has a furin recognition sequence (RX[R/K]R) in the Pro–Cat linker (Figure 1A), and is thus activated by proprotein convertase during passage to the cell surface. Similarly, the secreted MMPs 11, 21, 23, and 28 can also be activated by furin.21,38 In the case of MMP23, the zymogen is anchored to the membrane by a type II transmembrane helix N-terminal to the Pro domain. It is simultaneously activated and shed from the membrane upon furin cleavage.33
Once activated, MMPs are susceptible to strong inhibition by α2-macroglobulin and TIMPs, a family of four endogenous proteins, each of which is capable of forming a binary complex with an MMP Cat domain.39,40 In general, TIMPs show broad cross-reactivity with MMPs, but sensitivity varies for different MMP–TIMP pairings (eg, TIMP1 is only a weak inhibitor of MMPs 14, 16, 19, and 24). TIMPs are extensively disulfide-bonded and contain two domains with the N-terminal domain providing the inhibitory activity, the wedge-shaped structure of which docks into the active-site cleft, with the N-terminal Cys residue chelating the catalytic zinc.40 In addition to blocking MMP hydrolytic activity, TIMPs display other, noninhibitory roles.21,39 For example, TIMP2 recruits proMMP2 for activation at the cell surface by an MMP14 dimer and a ternary complex forms in which a TIMP2 molecule, already inhibiting one monomer of an MMP14 dimer via its inhibitory N-terminal domain, binds through its (noninhibitory) C-terminal domain to the proMMP2 Hpx domain, orienting the zymogen for proteolysis of its Pro domain by the second (uninhibited) MMP14 monomer.32 Intriguingly, chlorotoxin (a scorpion toxin that has anti-invasive effects on glioma cells) interacts specifically with MMP2, both reducing its cell-surface expression and inhibiting its hydrolytic activity.41
MMPs in gliomas: past and present
There is ample evidence suggesting a correlation between specific MMPs and glioma progression.42–47 Data from both in vitro and in vivo studies using glioma models point to a role of MMPs in glioma pathobiology. In addition, this field has gained insight from other neuropathology, including traumatic brain injury, stroke, neurodegenerative diseases, and immunorelated pathologies of the CNS, such as multiple sclerosis.48–52 While most of the published data focus on glioma-cell invasion, there is growing evidence of MMP involvement in other hallmarks of cancer, eg, angiogenesis.53–55 As in many fields of medical research, lessons were learned from failed clinical trials. Excitement over the ability to inhibit glioma invasion by targeting MMPs in patients with GBM was welcomed. Marimastat, a broad-spectrum MMP inhibitor developed in the late 1990s/early 2000s was the first orally bioavailable MMP inhibitor to reach clinical trials following in vitro and in vivo research showing high levels of efficacy in multiple cancer types.56,57 However, once clinical trials began, it became clear that one of the major adverse effects in humans was moderate–severe joint and muscle pain, which was present in up to 60% of patients. Unfortunately, the impact on progression-free and overall survival was limited, and in some cases treatment with marimastat led to negative patient outcomes. Marimastat was tested alone and in conjunction with other chemotherapeutic agents and radiation, demonstrating benefit in only a few cases, but more often leading to severe musculoskeletal toxicity. It became apparent quite quickly from these trials that many MMPs have functions essential for normal physiological functions, and developing selective MMP inhibitors would be essential for effective therapeutic targeting of MMPs. In hindsight, the absence of selectivity and the importance of appropriately timing therapy in relation to tumor growth are only two hurdles of many that must be overcome. The heterogeneous cellular composition within GBM is a major barrier to successful intervention, as is distinct host–tumor interactions that change and develop over time and in response to treatment. The relatively recent revelation that certain MMPs have intracellular nonproteolytic functions contributes to this complexity.8,9,58 Until we learn more about the temporal and spatial expression and activity of MMPs in GBM and develop MMP-specific inhibitors, it will be difficult to foresee successful GBM treatment utilizing MMP-based therapies.
MMP gene expression in GBM
To provide some context for this review, we performed bioinformatic analyses using a publicly available data set – R2: Genomics Analysis and Visualization (http://r2.amc.nl) – to assess MMP-mRNA expression. This is a valuable resource, and contains over 50,000 human samples from a wide range of tumors, tissues, and diseases, including GBM, many containing treatment and survival information. The French – 284 – MAS5.0 – u133p2 data set was interrogated to obtain relative MMP gene expression in GBM and determine the relationship of expression with Kaplan–Meier survival-probability estimates. This data set contains 276 histologically confirmed samples: GBM; 159 astrocytomas, eight grade I, 13 grade II, and 16 grade III; eight normal brain; 28 mixed oligoastrocytomas; and 52 oligodendrogliomas.59 For our analyses, we first surveyed the relationship of MMP-mRNA expression with the probability of overall survival across all the glioma types represented in this patient data set (Figure 2). Nine MMPs had low mRNA-expression levels in glioma that correlated with poor survival (MMPs 3, 12, 13, 16, 20, 21, 24, 25, and 28; P<0.02), while eleven MMPs had high mRNA-expression levels that correlated with poor survival (MMPs 1, 2, 7, 8, 9, 10, 11, 14, 17, and 23; P<0.02). When data were partitioned as GBM vs grade II and III and control tissue, MMP1, -2, -7, -9, -14, and -19 mRNA levels were significantly higher in GBM. MMP1 was significantly higher in GBM compared to control and anaplastic astrocytoma (P=<0.0001). MMP2 was significantly higher in both anaplastic astrocytoma and GBM compared to control (P=0.0006 and 0.0001, respectively; Figure 3A). However in this data set, MMP2 levels did not associate with increasing grade and were not consistent with a recent report by Ramachandran et al in which more detailed analysis using a different data set was conducted, demonstrating MMP2 expression increases with increasing grade.60 Based on previous reports, it was anticipated that MMP1, MMP2, and MMP9 mRNA levels would be high in GBM.53,61–65 Surprisingly, MMP19 mRNA was significantly higher in GBM, not only compared to control (P<0.0001) but also relative to lower-grade gliomas (P<0.0006) (Figure 3A). Interestingly, only one MMP, MMP24, was significantly lower in GBM compared to controls and other gliomas (P<0.0001). When survival probability was assessed in just the GBM samples, high mRNA-expression levels of MMPs 1, 2, 7, 9, 12, and 19 were associated with poor survival (Figure 3B), whereas low levels of MMP24 were associated with poor survival in this data set (Figure 3C). MMP24, identified by and characterized by Pei et al in 2000, is temporally and spatially regulated during neurodevelopment,66 associated with neurons,67 and reported to be associated with synaptic reorganization following traumatic brain injury.51 This current analysis of MMP24 expression in GBM appears to be in conflict with a previous study, where MMP24 was reported to be overexpressed in brain tumors.68 However, when GBM samples were examined, only two of seven demonstrated MMP24 transcripts. It is also not known if the MMP24 transcript variant described by Ross and Fillmore is associated with GBM.69 In this study, the authors report the upregulation of MMP24 in differentiated multipotent NT2 cells (a neurogenic in vitro model for differentiation). Based on our improved knowledge of MMPs, it may not be surprising that not all MMPs will follow the same pattern in gliomas. In the context of the tumor microenvironment, where there is multidirectional and dynamic communication between host cells (including neurons) and glioma cells, we would predict that neurally associated MMPs may have differential expression and functional patterns.70
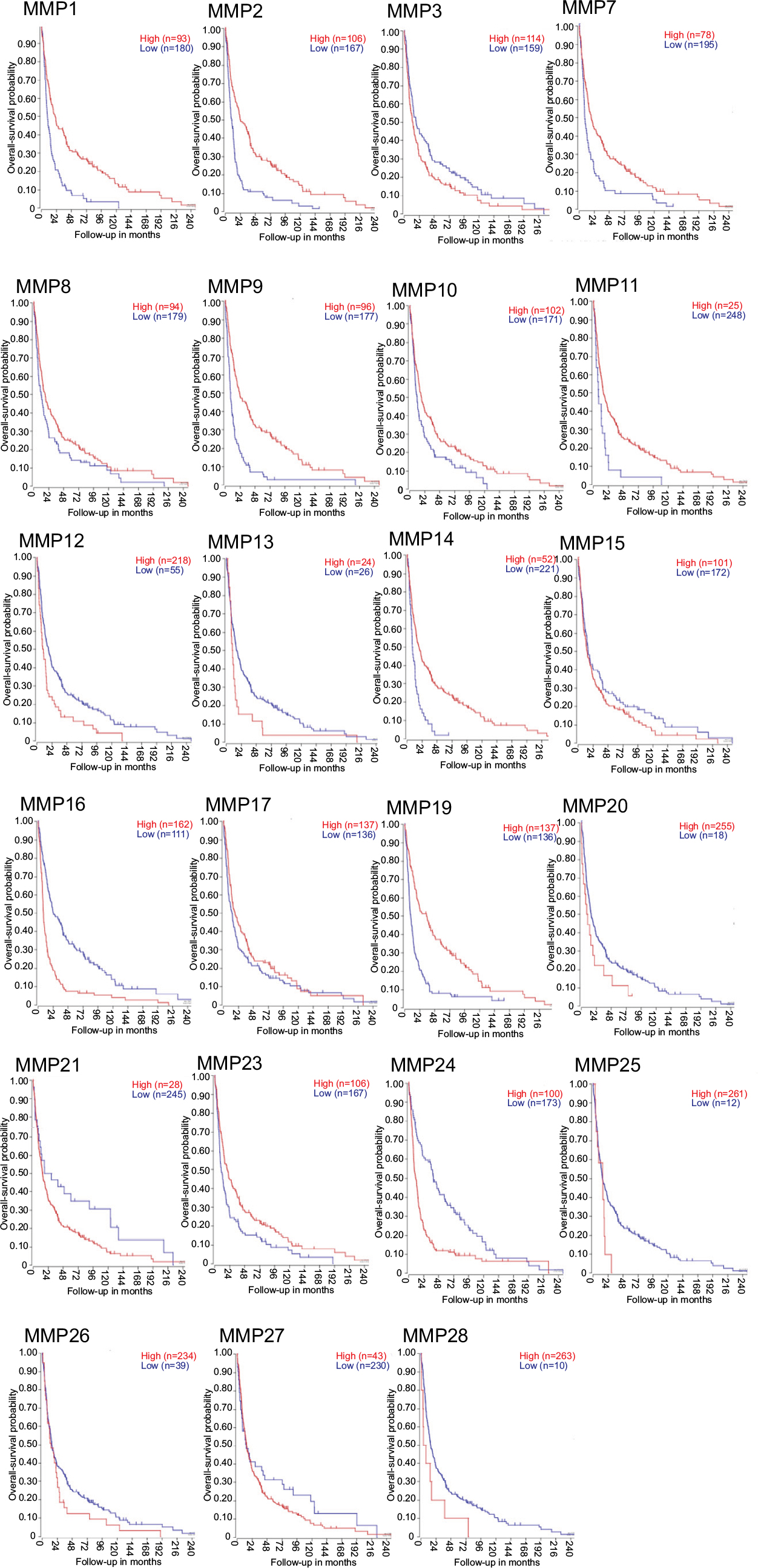 | Figure 2 Kaplan–Meier curves of overall-survival probability across all glioma-tumor types in French data set separated based on MMP gene expression. Notes: The Kaplan Scan function of R2 was used to display Kaplan–Meier information with the cutoff modus set at “scan”. This function finds the best-possible curve based on log-rank test to find the most significant expression cutoff for survival. |
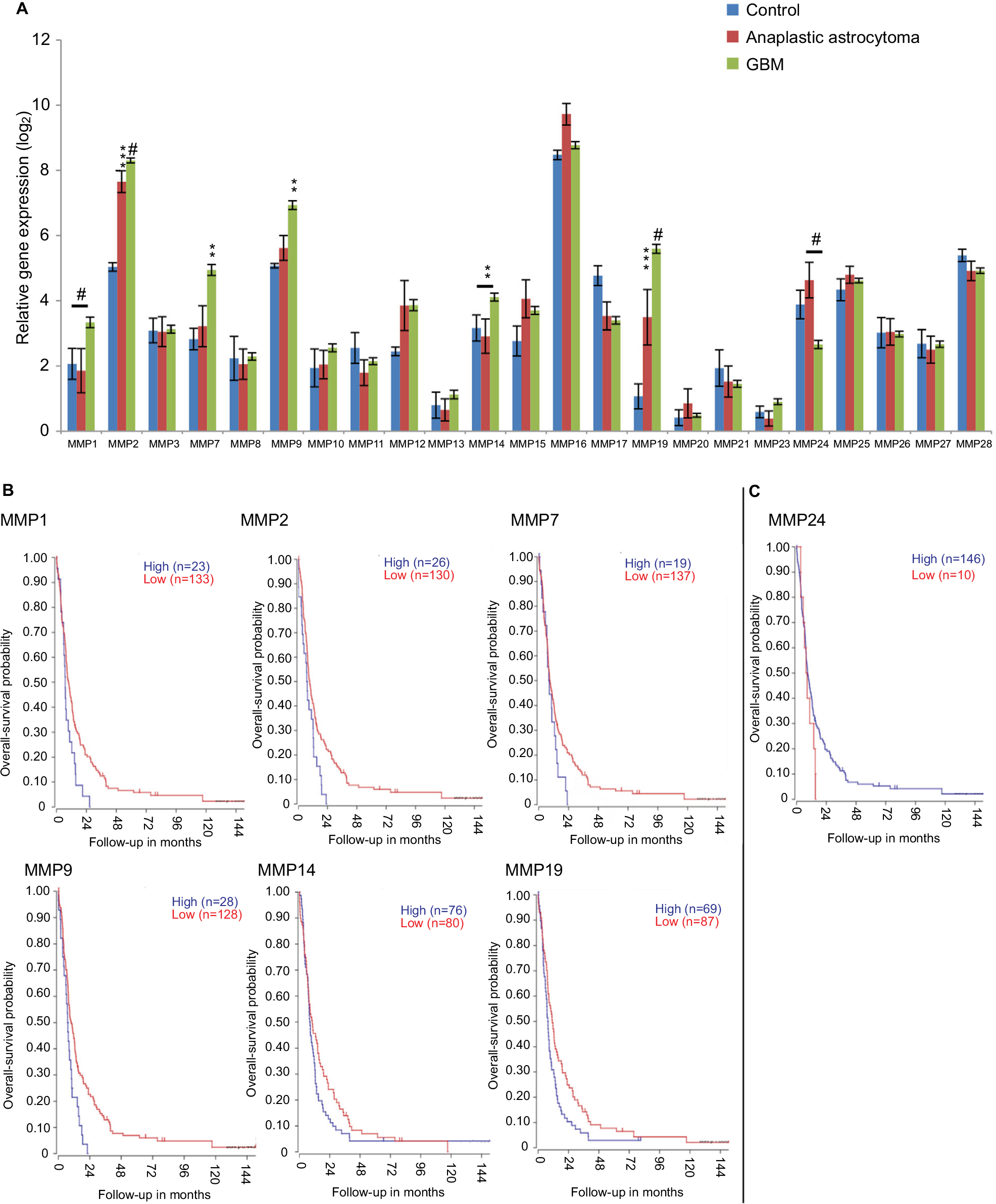 | Figure 3 Gene-expression analysis of MMPs of 276 glioma samples of all histologies, with eight control samples. Notes: Data set (tumor glioma – French – 284 – MAS5.0 – u133p2) retrieved from R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl). (A). Data reported as mean ± SEM. P values: #<0.0001; ***0.0006; **0.0039. Two-way ANOVA was performed using GraphPad Prism. (B) Kaplan–Meier curves of overall-survival probability of 156 GBM patients in French data set. MMPs significantly overexpressed in GBM patients compared to control, and patients had worse overall-survival probability. (C) MMP24 was significantly underexpressed in GBM patients, and patients with lower expression had worse overall-survival probability. Abbreviations: SEM, standard error of the mean; ANOVA, analysis of variance; GBM, glioblastoma multiforme. |
The bioinformatic data obtained for MMPs 1, 2, 7, 9, 19, and 24 are consistent with data obtained from RT-qPCR (Figure 4). mRNA-expression levels in control tissue (ie, non-pathological tissue obtained during surgical resection of epilepsy patients), grade III anaplastic astrocytoma, and GBM were assessed using custom-designed TaqMan low-density arrays containing primer and probe sets for 384 proteases. Transcript levels of MMPs 1, 2, 7, 9, 10, 11, 12, 14, 19, and 21 were significantly increased in GBM relative to control. Notably, MMP19 was also increased in anaplastic astrocytoma relative to control, but decreased in grade IV GBM relative to grade III anaplastic gliomas.
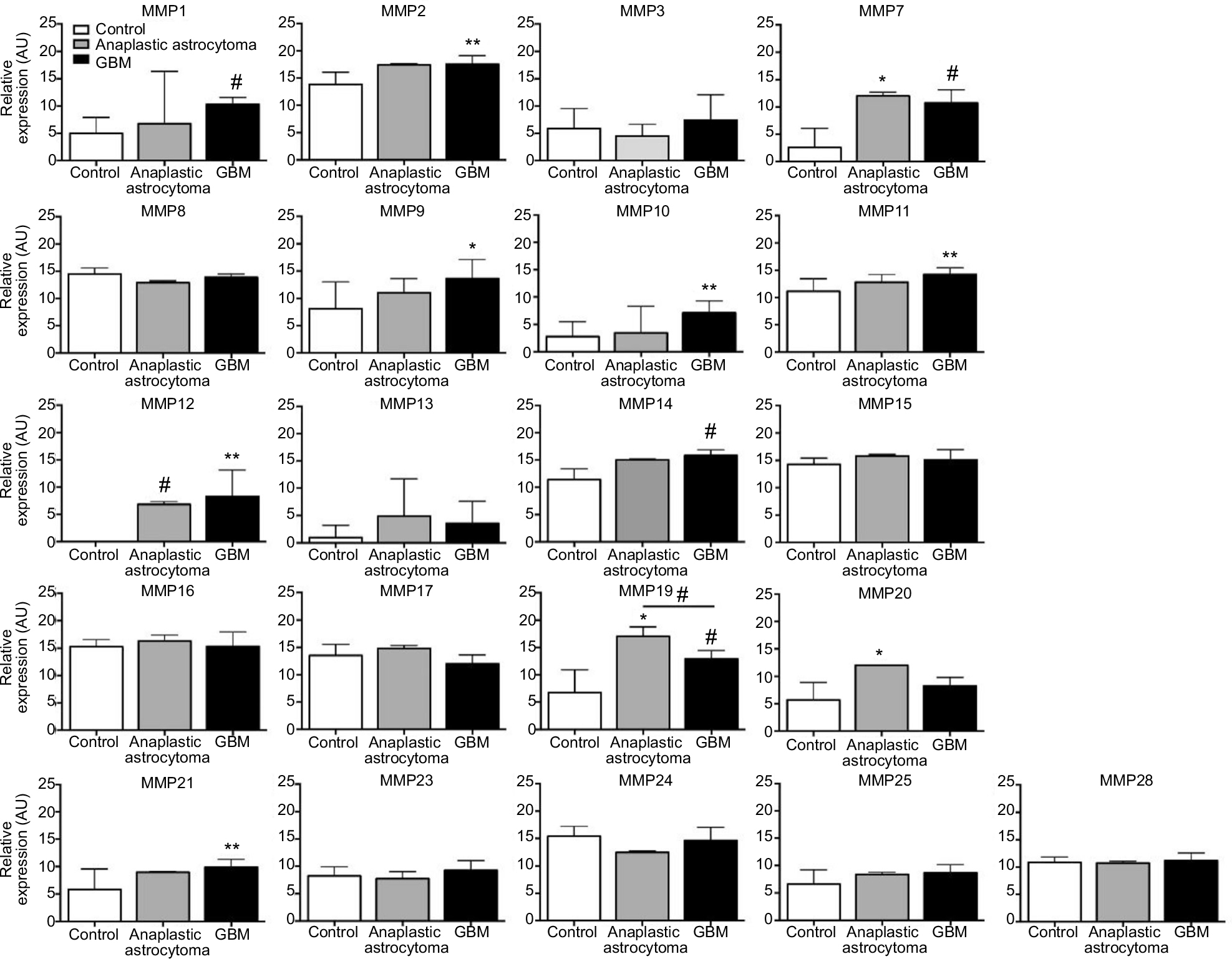 | Figure 4 Quantitative reverse-transcription PCR. Notes: PCR revealed significantly increased mRNA expression of MMPs 1, 2, 7, 9, 10, 11, 12, 14, 19, and 21 in GBM tumors relative to control tissue (ie, nonpathological tissue obtained during surgical resection in epilepsy patients). *P<0.05; **P<0.01; #P<0.001. mRNA-expression levels in control tissue (ie, nonpathological tissue obtained during surgical resection in epilepsy patients), grade III anaplastic astrocytoma, and GBM were assessed using custom-designed TaqMan low-density arrays containing primer and probe sets for 384 proteases, as previously described.195,196 Abbreviations: PCR, polymerase chain reaction; GBM, glioblastoma multiforme. |
Insofar as RNA levels are dependent on transcript stability and degradation by miRNAs, we surveyed in silico which miRNAs may be likely MMP-mRNA regulators. miRNA-expression signatures characterize and contribute to the phenotypic diversity of GBM subclasses through their ability to regulate developmental growth and differentiation. Furthermore, they have been identified as diagnostic and prognostic biomarkers for patient stratification, and may also serve as therapeutic targets and agents.71 We utilized the miRNA-target-prediction program TargetScan (http://www.targetscan.org) to review the miRNA-target sites of these MMPs (Table 1). Not surprisingly, all but two MMPs (MMP12 and MMP23A/B) contained binding sites for at least one miRNA, but many of these sites were poorly conserved. More importantly, nine MMPs (MMPs 2, 3, 11, 13, 14, 15, 16, 19, and 24) also demonstrated conserved miRNA-binding sites. Several of these sites have been previously described in GBM (miR587, -223, -377, -29c, -544, -136, -339, and -140).71–75 miR27b and miR29c have been experimentally validated to target their respective MMPs (MMP13 and MMP2) in other pathologies.76–86
 | Table 1 Conserved and poorly conserved sites at the 3′-end of matrix metalloproteinases, for miRNA binding predicted by TargetScan |
Breaking out of the invasion box
While the majority of reports on MMP and GBM have focused on their role in glioma invasion, over the past few years it has become abundantly clear that MMPs are more than just “machetes” clearing ECM molecules out of the way for glioma cells to move.53 Their influences on cell growth, apoptosis, and inflammation are just a few of their biological roles that impact tumorigenicity. Elucidation of these mechanisms has not only generated intriguing results in this area but also opened new potential therapeutic avenues for GBM treatment.
Angiogenesis
In addition to being a highly invasive tumor, GBM is known to exhibit a high degree of vascularity. Indeed, one of the determining pathological hallmarks is extensive vascular hyperplasia, in addition to areas of necrosis and pseudopalisading zones demarcating areas of low oxygen. As oxygen is essential for the survival and proliferation of tumor cells, molecular cross talk between the tumor and the host microenvironment plays a crucial role in the activation of molecular pathways that affect endothelial cells and promote abnormal vessel formation.87,88 One of the key molecules involved in angiogenesis is VEGF, released by tumor cells and overexpressed in most malignancies, including brain tumors.89 Clinical trials using a humanized monoclonal antibody to VEGFA (Avastin [bevacizumab]), while revealing improvement in progression-free survival, often fail to demonstrate improvement in overall survival.90 Reports from both human and animal studies support the mechanism of bevacizumab GBM vascular normalization).90,91 Even though bevacizumab shows improved progression-free survival, the tumor adapts and becomes more aggressive (invasive) and more resistant to treatment.92,93
Historically, MMPs have been linked to angiogenesis mostly by their ability to degrade the basal lamina (BL) components that support the blood–brain barrier and by their regulation via cytokines and growth factors that play major roles in angiogenesis. The blood–brain barrier dynamically protects the brain, and is composed of what is called the “neurovascular unit”. For this review, we want to highlight the major cell types and unique BL ECM molecules. Brain endothelial cells are specialized and characterized by a complex network of adhesion molecules, tight-junction proteins, and associated cytoplasmic proteins. Brain endothelial cells compose the capillary lumen and are surrounded by the BL and pericytes. Pericytes, critical in vascular homeostasis and stabilization, are typically embedded within the BL, which is composed of collagens I and IV, fibronectin, laminin, thrombospondin, and proteoglycans (agrin and perlecan).94–96
As with GBM being heterogeneous in nature, so are the vasculature structures generated to supply the tumor. There are at least five documented mechanisms of neovascularization theorized in GBM pathobiology: vessel co-opting,97,98 angiogenesis,99,100 vasculogenesis,101 vascular mimicry,102 and transdifferentiation of glioma cells to endothelial cells.103,104 In an elegant (and highly recommended) review, Hardee and Zagzag105 described each of these processes and provided a review of published reports that support each mechanism in GBM. They also noted that these putative mechanisms to provide oxygen to the tumor are not always exclusive, but are molecularly linked to one another and overlap under certain situations. Rather than correlating specific MMPs with angiogenesis, it would be beneficial to interrogate the specific mechanistic roles of MMPs in each of these angiogenic processes.
MMPs have been shown to have both proangiogenic and antiangiogenic roles in tumor angiogenesis106–108 and specifically in GBM.105 MMP2 and MMP9 play a critical role in the angiogenic switch when a tumor is initially vascularized.109–111 Numerous in vitro and in vivo studies have shown the angiogenic implications of MMP2 and MMP9 in retinoblastoma, lung cancer, breast cancer, ovarian cancer, gastric cancer, and gliomas.112–121 Interestingly, studies have shown correlated expression of MMP2, MMP9, and MMP14 with VEGF in GBM.122 Pullen et al showed the induction of angiogenesis in MMP1-overexpressing GBM cells in an in vivo model.53 MMP1 has also been shown to have a potential role in tumor vascularization through PAR1.13,53,123 MMP7 induces endothelial cell proliferation by triggering MMP1 and MMP2 endothelial cell expression.124,125
Certain MMPs can promote angiogenesis in different ways: pericyte detachment from sprouting vessels, while MMP9 plays an important role in recruiting pericytes;126 induction of ECM-bound angiogenic growth factors, such as FGF, PDGF, VEGF, and TNFα;127,128 proteolytic cleavage of type IV collagen reveals increased cryptic αvβ3 proangiogenic integrin-binding sites and correlates with increased expression and activation of MMP2;129,130 and cleavage of the ectodomain of vascular–endothelial–cadherin cell–cell adhesions.108,131 MMPs also have antiangiogenic properties by cleaving plasma proteins and ECM components. MMP2, -7, -9, and -12 can inhibit angiogenesis by inducing plasminogen proteolytic cleavage and produce angiostatin.132–135 MMP3, -9, -12, -13, and -20 can release endostatin, resulting in endothelial cell migration and apoptosis.135–137
In light of recent reports of “moonlighting” jobs, additional functions may exist for MMPs within mechanisms tumors use to obtain oxygen, as well as recruitment of other cell types, such as pericytes,138 neutrophils, bone-marrow derived endothelial cells, or macrophages and other myeloid cells. Historically, the investigation of MMPs in the CNS has been limited, due to the focus on documented ECM substrates and the lack of “appropriate” substrates for specific MMPs. As discussed by Jobin et al,8 experimental rigor and criteria are needed for studies designed to investigate these potentially “novel” roles for MMPs.
Immune-system functions
It has long been understood that a variety of immune-system cells, beyond microglia, are effectors of neuroinflammation by trafficking into and out of brain parenchyma in response to neuroinflammatory insults, such as viral or bacterial infections, and this has been recently supported by the confirmation of brain lymphatics.139 For gliomas, involvement of extra-CNS cells is likely more acute, because of a combination of signals produced by tumor cells and disruption of localized blood–brain barrier function.140 Here, we focus on the involvement of MMPs in glioma-mediated immune-system responses. We begin with a discussion of specific cytokines, followed by a discussion of myeloid-lineage cells, specifically microglia/macrophages, mast cells, and neutrophils.
Cytokines
It is well known that GBM cells create an immunosuppressive microenvironment and utilize multiple methods to escape immunosurveillance. However, there is also evidence that GBM cells express antigens that are recognized by a patient’s immune system. Given the current clinical emphasis on immunotherapy for GBM and that GBM stem-like cells are prone to evading the immunoresponse, understanding the roles MMPs play in immunomodulation is essential. A great deal of the putative neuroimmune impacts of MMPs have been studied in the context of autoimmune disorders, such as multiple sclerosis (MS). MMP2 and -9 have been suggested as differential biomarkers for MS.141,142 Notably, MMP9 expression is suppressed moderately by IFNβ143,144 and IgG, two commonly employed therapeutic approaches for MS.145 Additionally, T-helper 1 (TH1) cells, which produce IFNγ, IL2, and TNFβ, which activate macrophages and are responsible for cell-mediated immunity, are the highest expressers of MMP2 and -9 among their CD4 cohorts. This could contribute to greater motility and homing in an autoimmune setting.146 With the emergent interest in checkpoint-inhibitor therapeutics and more targeted therapies inherent to human leukocyte-antigen typing, it is worth examining the roles of MMPs in glioma-associated T cells, specifically CD8+, as well as other innate lymphoid-cell varieties.
With guidance from the MS field, IFNβ as a combination therapy for GBM has also been explored. The antitumor effect of Tmz on malignant glioma cells in vitro is improved with IFNβ.147 MMP2 induction by IFNβ treatment has also been reported in an in vivo mouse glioma model.148 However, unlike work done with respect to MS, there appears to be no effect of IFNβ treatment on MMP9 or TIMP1 expression, at least in cell lines.149 The latter study reported IL10 upregulation as a potential mechanism for immunomodulation of the tumor microenvironment. This is especially interesting in light of recent studies from hypersensitivity research demonstrating that IL10 has pro-TH2 inflammatory effects,150,151 which are in contrast to IL10 being thought of as an immunosuppressive cytokine. Could IL10 application be utilized to enhance antitumor immune-system responses, specifically T-cell and innate lymphoid-cell programming? In vitro, IL10 has no significant effect on expression levels of MMP2, -3, -7, -9, or -12,152 yet paradoxically in the same study, it was noted that IL10 enhanced glioma cell-line invasion through mock basement membrane, an effect mitigated by marimastat. This begs the question as to the activation state of these MMPs and/or the expression of others. However, in the presence of IL10, activation of MMP2 or MMP9 was unaffected.152 Recent evidence from clinical studies of IFNβ–Tmz combination therapy have demonstrated that this therapeutic approach has limited benefit.153 Furthermore, a small retrospective analysis demonstrated median survival of 19.9 months for IFNβ–Tmz combination therapy vs 12.7 months for Tmz alone.154 However, more recent molecular research suggests that IFNβ therapy enhances innate immunoeffectors in glioma stem-like cells,155 and thus additional research into IL10 and IFNβ therapy and MMP expression is warranted.
The effects of other cytokines on MMP expression in glioma cells have been investigated, including combinatorial responses to TNFα and IFNβ. Cheng et al reported that TNFα induced glioma-cell migration in vitro, and ascribed this to increased MMP3 expression.156 Transient MMP3 knockdown reduced invasion across mock basement membrane in the presence of TNFα. In addition, they showed that IFNβ suppressed MMP3 transcription in a dose-dependent manner. The proposed mechanism involves inhibition of Ets1 and NFκB binding to DNA.156 However, it is unknown what NFκB subunits are affected, the role of TIMP3 (whose transcription was also suppressed), or whether IFNβ directly or indirectly inhibits glioma cell invasion. Conversely, TNFα and IFNβ both individually inhibit MMP2 expression and together impart an additive suppression of MMP2.157 Therefore, the effects of cytokines on MMP expression is MMP-specific.
IL6 and its major intracellular effector, STAT3, have been linked to enhanced MMP2 expression from glioma cell lines.158 A deeper study into the relationships among MMP2, IL6, and STAT3 revealed a novel role for MMP2 as a coeffector of the fibronectin receptor, α5β1 integrin, whereby MMP2 interacts directly with the receptor.159 Removal of MMP2 inhibited glioma-cell and xenograft proliferation, inhibited production of multiple cytokines, including IL6, and inhibited STAT3 phosphorylation.159 IL6 promotes MMP14 expression in glioma cell lines, and expressions of both IL6 and MMP14 are increased in patient tumor samples.160 However, the localization of MMP14 is confounding, given other research161 showing that MMP14 imparts negative effects on glioma cells, yet it is a clear marker and effector of associated protumor microglia. Therefore, observations of MMP14 on glioma cell lines in vitro likely do not replicate actual glioma cells in vivo. Nevertheless, the presence of MMP14 in patient samples (likely in an immune-cell compartment) alongside IL6 is an impactive observation. Tocilizumab, an anti-IL6-receptor biologic, was US Food and Drug Administration-approved for moderate–severe rheumatoid arthritis this decade. The potential for off-label anti-IL6 therapies is becoming more attractive, given this cytokine’s role in cancer pathophysiology, which we submit might include MMP expression. Observations of IL6-receptor localization on glioma-associated endothelia from patient samples, along with inhibition of glioma cell line proliferation by tocilizumab, have been reported.162 More mechanistic details utilizing xenograft models, which demonstrated hypoxia as a major inducer of IL6, showed that treatment with tocilizumab induced glioma cells to shift from an autophagic to apoptotic phenotype, thus increasing TMZ efficacy.163 Taken together, IL6 and MMP expressions in gliomas require further detailed study. While recent approaches to targeting IL6 itself have been scuttled (eg, sirukumab) due to adverse side effects, targeting the IL6 receptor has proven effective in other pathology, and other work using small-molecule inhibitors of the IL6–STAT3 axis show promise, such as work using stattic and cucurbitacin I.164
Glioma-associated microglia/macrophages
Most high-grade tumors, regardless of tissue origin, accumulate mutations related to TGFβ overexpression, and gliomas are no exception to this. In turn TGFβ, along with other factors, such as GM-CSF, is a major recruitment and differentiation factor for immunosuppressive myeloid cells. Among those recently studied are glioma-associated microglia/macrophages (GAMs), a niche-specific nomenclature for tumor-associated macrophages (TAMs). Recent extensive reviews, including therapeutic approaches targeting microglia, are explained by Roesch et al165 and Hambardzumyan et al.166 Recent work has sought to dissect further the pathological molecular mechanisms behind VEGF receptor–TGFβ receptor complex activities,167 and indeed it has been shown that neuropilin 1 is a critical coreceptor in the induction of GAMs in a mouse model of glioma.168 Furthermore, GAMs are thought to contribute to the poor efficacy of some oncolytic viruses by sequestering and inhibiting the replication of the therapeutic virus.169 Specific to MMPs, the hypoxic environment characteristic of high-grade gliomas is known to induce MMP expression, and macrophages can independently induce MMP9 expression from glioma cells, with a slight additive effect in a hypoxic setting.170 Seminal work by Markovic et al demonstrated the key role of MMP14 on GAMs, which in turn activate glioma-derived MMP2, thus enhancing the invasive capability of the tumor cells.161 Using primary microglia from rats, Ellert-Miklaszewska et al171 broke ground in defining the transcriptomic network programmed into GAMs, which notably includes cementing MMP14 as a definitive marker of GAM activation. Further studies clearly defined the following: TLR2 (and to some extent TLRs 1 and 6) as mediators of the GAM MMP14 conversion/phenotype,172 activation of TLR2 inducing these cells to express MMP9,173 and that the likely major stimulus for this TLR2-dependent activation was versican produced by glioma cells.174 Off-label use of atorvastatin has attracted attention in a variety of pathologies, including glioma, where it has been reported that it specifically suppresses MMP14 on in vitro cultured GAM-like cells,175 and thus potentially inhibits glioma invasion by reducing MMP2 activation. Atorvastatin enhances Tmz-induced glioma-cell death in vitro and in xenograft models.176 Additionally, atorvastatin inhibits IL17R expression on glioma cell lines in vitro,177 which is thought to be a keystone proglioma stem-cell effector.178 TGFβ (specifically TGFβ1) is also produced by GAMs/TAMs, and this source of cytokines has been shown to induce MMP9 expression and invasiveness of glioma stem cells in their local niche.179 Finally, investigators are cautioned to interpret semantics carefully when reading such reports as Bayat et al’s description of IL17RA inhibition as “anti-inflammatory”:177 proinflammatory is likely the desired effect in this specific setting, as it is immunosuppression and escape from immunosurveillance effected by GAMs/TAMs that supports progression of malignant cancers, including high-grade gliomas.
Mast cells
Packed with proteases including MMPs, mast cells are known to accumulate around tumor margins and within some tumor tissue, including high-grade gliomas. Surprisingly, the study of the roles of mast cells in CNS neoplasia is a neglected area. Typically, mast cells are considered only in the context of IgE-mediated hypersensitivity. However, in recent years these myeloid granulocytes have gained attention in other areas, especially due to proven alternative, IgE-independent activation schemes, notably through IL33/ST2, and for their potent nociceptive effects on the somatosensory system (though mechanistic details are scant). A timely review of mast cells in neuroinflammation is explained by Skaper et al.180 Astrocytes are known to activate microglia through IL33,181 and furthermore there are reports linking IL33 induction with decreased survival time182 and IL33 stimulating expression of MMP2 and MMP9 from immortal cell cultures.183 Of note is that IL33 positively regulates mast-cell survival and inflammatory functions,184–186 and thus it will be important to investigate a potential pathological link between gliomas and mast cells via IL33 signaling. Mast cells decrease the efficacy of antiangiogenic therapies,187 which have historically been a focus of glioma-therapy research. Glioma cells also secrete factors that serve as signals for mast-cell homing in vivo, which include PAI1188 and CXCL12/SDF1.189 One in vitro study has reported detailed observations regarding mast cell–glioma cell coculture.190 Interestingly, glioma cell line-conditioned media alone were enough to stimulate IL6 secretion from mast cells, though the specific stimulus remains unknown. Paradoxically, the authors reported inhibition of glioma-cell STAT3 signaling despite enhanced IL6 production, and termed these mast cells “tumor-educated”. Protease involvement was not assessed, and it will be important to replicate this work with in vivo modeling.
Neutrophils
Neutrophils are the most abundant leukocytes in circulation and the namesake of MMP8 (neutrophil [polymorphonuclear] collagenase) – release of which can drive matrix-degradative pathology – such as seen in tuberculosis.191 Neutrophils invade gliomas, eg, a study of 105 patient samples reported that neutrophil infiltration was observed across all grades, and suggested that the degree of infiltration increased with grade, though without correlative statistical modeling.192 Systemic neutrophilia with malignant disease is well known, and a recent meta-analysis reported that higher-than-normal circulating neutrophil:lymphocyte ratios were an indicator of poor prognosis for glioma patients.193 Treatment with bis-chloroethylnitrosourea or Tmz might induce prolonged posttreatment neutropenia in some glioma patients, with one case report of persistently dysfunctional neutrophils (specifically, deficient reactive oxygen species production) from patients treated with Tmz.194 An underlying mechanism linking Tmz to chronic neutropenia (and/or long-term neutrophil dysfunction) in a select patient subset has not been clearly defined. In a rat-flank glioma model, Graf et al195 showed that IL6 could be important in the chemotaxis of antitumor neutrophils. Specifically, tumor-cell-derived IL6 elicited neutrophil infiltration into the tumors and aspecific depletion of neutrophils in vivo was tumorigenic. Later work on mouse models of lung cancer showed distinct phenotypes of tumor-associated neutrophils, termed N1 and N2, that are tumoricidal and tumor-promoting, respectively, with the latter being TGFβ-dependent.196 A detailed review is provided by Uribe-Querol and Rosales.197 Therefore, neutrophilia associated with high-grade neoplasia might be skewing toward an N2 phenotype. N2 neutrophils overlap functionally with but are likely a distinct population from polymorphonuclear myeloid-derived suppressor cells (MDSCs), which are also found in abundance in high-grade cancer cases.198 Harnessing the fact that regardless of phenotype, neutrophils potently invade brain tumors, Xue et al recently demonstrated proof of principle for using these cells to transport paclitaxel to brain tumors in an orthotopic mouse model noninvasively.199 Phagocytic and degranulatory responses of neutrophils are long established, with the latter being a likely mechanism for neutrophil-mediated drug delivery. Furthermore, significant interest is garnered from the unique ability of neutrophils to eject sticky chromatin complexes as what are termed neutrophil extracellular traps (NETs). These were originally described as an aspecific antibacterial immunoresponse.200 However, there is accumulating evidence that NETs may fulfill a protumor role and be activated more specifically through Fc receptors.197 In systemic lupus erythematosus, MMP9 released with NETs activates MMP2.201 This report raises interest because of the predominance of gelatinases in past glioma studies. Additionally, it is notable that another recent report detailed a mechanism whereby MMP9-cleaved osteopontin induced MDSC proliferation in a mouse lung cancer model. Altogether, there is need for much more research on the interplay of neutrophils with gliomas. Neutrophil-effector functions in the glioma setting hypothetically might include NET-associated MMP9 delivery, downstream activation of other pathways because of MMP9, induced release of MMP8, and expansion and recruitment of MDSCs, thus compounding issues of immunosuppression and tumor immunoevasion, and in turn MDSCs might support angiogenesis into tumors.202
Recent advances harnessing the immune system
Other recent emphases on the neuroimmunology of high-grade glioma have obviously included development and trials with dendritic-cell and CAR T-cell therapies, but the research here has not yet focused on potential roles of MMPs or much on proteases more generally (a review of the dendritic-cell field is provided by Reardon et al,203 and a timely perspective from leaders in the CAR T-cell field is provided by Migliorini et al204). As mentioned already, GAMs might cordon off certain oncolytic therapies and MMPs are deeply involved in GAM activation and function. Safety trials of oncolytic therapies are just now being released, eg, with DNX2401,205 and the PVSRIPO Phase I trial has recently concluded.206 A recent major collaboration among several US labs also concluded that Zika virus has oncolytic potential, specifically targeting glioma stem cells.207 Clearly, the importance of diverse glioma-targeting immunotherapies has been demonstrated, and our knowledge of the roles of metalloproteinases in these areas is constantly evolving.
Conclusion
While this review was not meant to cover all aspects of MMP function in glioma, our goal was to provide examples that would help free MMPs from the limitations they have been ascribed and to share our excitement for the future in terms of learning more about MMPs and better understanding the underlying pathobiology of GBM. The opportunities to explore MMP regulation, expression and function further in the CNS have never been so great. The public availability and size of patient-data platforms will only grow and become better. In addition to future exploration in terms of abnormal vessel formation, immunoresponse, and other host–tumor interactions, the use of MMPs for targeting and imaging of brain cancers is very encouraging.208,209 Other areas that lack information are MMP epigenetic and metabolic effector functions (including the epigenetic induction of MMPs). An almost uncharted territory in the brain for MMP enthusiasts are the unique mechanical properties of the brain parenchyma and the complicated changes that take place with the ECM during brain diseases.210 Much of what is known about MMP structure/function comes from studies in arthritis,10 and while Young’s modulus of brain tissue is quite different from bone, we believe there is a vast amount of information concerning mechanotransduction regulation of MMPs in gliomas. Little is known about the influence of fluid dynamics and sheer stress on temporal and spatial MMP functions in gliomas.
Acknowledgments
This article is dedicated to all MMP scientists past and present, for providing an incredible foundation for us and others to build upon. Special thanks to Brain Tumour Research UK and Professor Geoffrey Pilkington. The authors thank Barbara Zvar Baskovic for her editorial assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. | ||
Mansouri A, Karamchandani J, Das S. Molecular genetics of secondary glioblastoma. 2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK469981. Accessed July 18, 2018. | ||
van Meter T, Dumur C, Hafez N, Garrett C, Fillmore H, Broaddus WC. Microarray analysis of MRI-defined tissue samples in glioblastoma reveals differences in regional expression of therapeutic targets. Diagn Mol Pathol. 2006;15(4):195–205. | ||
Shergalis A, Bankhead A, Luesakul U, Muangsin N, Neamati N. Current challenges and opportunities in treating glioblastoma. Pharmacol Rev. 2018;70(3):412–445. | ||
Pellerino A, Franchino F, Soffietti R, Rudà R. Overview on current treatment standards in high-grade gliomas. Q J Nucl Med Mol Imaging. Epub 2018 Apr 26. | ||
Fernandes C, Costa A, Osório L. Current Standards of Care in Glioblastoma Therapy. 2017. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29251860. Accessed May 25, 2018. | ||
Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017;19 Suppl 5:v1–v88. | ||
Jobin PG, Butler GS, Overall CM. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim Biophys Acta. 2017;1864(11 Pt A):2043–2055. | ||
Xie Y, Mustafa A, Yerzhan A, et al. Nuclear matrix metalloproteinases: functions resemble the evolution from the intracellular to the extracellular compartment. Cell Death Discov. 2017;3:17036. | ||
Murphy G. Riding the metalloproteinase roller coaster. J Biol Chem. 2017;292(19):7708–7718. | ||
Groves MD, Puduvalli VK, Conrad CA, et al. Phase II trial of temozolomide plus marimastat for recurrent anaplastic gliomas: a relationship among efficacy, joint toxicity and anticonvulsant status. J Neurooncol. 2006;80(1):83–90. | ||
Brzdak P, Nowak D, Wiera G, Mozrzymas JW. Multifaceted roles of metzincins in CNS physiology and pathology: from synaptic plasticity and cognition to neurodegenerative disorders. Front Cell Neurosci. 2017;11:178. | ||
Hagemann C, Anacker J, Ernestus RI, Vince GH. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J Clin Oncol. 2012;3(5):67–79. | ||
Vanmeter TE, Rooprai HK, Kibble MM, Fillmore HL, Broaddus WC, Pilkington GJ. The role of matrix metalloproteinase genes in glioma invasion: co-dependent and interactive proteolysis. J Neurooncol. 2001;53(2):213–235. | ||
Fillmore HL, Vanmeter TE, Broaddus WC. Membrane-type matrix metalloproteinases (MT-MMPs): expression and function during glioma invasion. J Neurooncol. 2001;53(2):187–202. | ||
Fanjul-Fernández M, Folgueras AR, Cabrera S, López-Otín C. Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta. 2010;1803(1):3–19. | ||
Overall CM, López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2(9):657–672. | ||
Page-Mccaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–233. | ||
Loffek S, Schilling O, Franzke CW. Biological role of matrix metalloproteinases: a critical balance. Eur Respir J. 2011;38(1):191–208. | ||
Gaffney J, Solomonov I, Zehorai E, Sagi I. Multilevel regulation of matrix metalloproteinases in tissue homeostasis indicates their molecular specificity in vivo. Matrix Biol. 2015;44–46:191–199. | ||
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. | ||
Bode W, Maskos K. Structural studies on MMPs and TIMPs. Methods Mol Biol. 2001;151:45–77. | ||
Wells EM, Packer RJ. Pediatric brain tumors. Continuum (Minneap Minn). 2015;21(2):373–396. | ||
Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16(5):558–564. | ||
Mccawley LJ, Matrisian LM. Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol. 2001;13(5):534–540. | ||
Chaudhary A, Singh M, Bharti AC, Asotra K, Sundaram S, Mehrotra R. Genetic polymorphisms of matrix metalloproteinases and their inhibitors in potentially malignant and malignant lesions of the head and neck. J Biomed Sci. 2010;17:10. | ||
Kadoglou NP, Liapis CD. Matrix metalloproteinases: contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms. Curr Med Res Opin. 2004;20(4):419–432. | ||
Overall CM. Molecular determinants of metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol Biotechnol. 2002;22(1):51–86. | ||
Fields GB. Interstitial collagen catabolism. J Biol Chem. 2013;288(13):8785–8793. | ||
Crabbe T, Ioannou C, Docherty AJP. Human progelatinase A can be activated by autolysis at a rate that is concentration-dependent and enhanced by heparin bound to the C-terminal domain. Eur J Biochem. 1993;218(2):431–438. | ||
Dumin JA, Dickeson SK, Stricker TP, et al. Pro-collagenase-1 (matrix metalloproteinase-1) binds the α2β1 integrin upon release from keratinocytes migrating on type I collagen. J Biol Chem. 2001;276(31):29368–29374. | ||
Itoh Y, Takamura A, Ito N, et al. Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J. 2001;20(17):4782–4793. | ||
Pei D, Kang T, Qi H. Cysteine array matrix metalloproteinase (CA-MMP)/MMP-23 is a type II transmembrane matrix metalloproteinase regulated by a single cleavage for both secretion and activation. J Biol Chem. 2000;275(43):33988–33997. | ||
van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci U S A. 1990;87(14):5578–5582. | ||
Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, Winberg JO. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 2011;278(1):28–45. | ||
Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26(8):587–596. | ||
Prior SH, Fulcher YG, Koppisetti RK, Jurkevich A, van Doren SR. Charge-triggered membrane insertion of matrix metalloproteinase-7, supporter of innate immunity and tumors. Structure. 2015;23(11):2099–2110. | ||
Murphy G, Nagase H. Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. 2011;278(1):2–15. | ||
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 1803;2010(1):55–71. | ||
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–839. | ||
Deshane J, Garner CC, Sontheimer H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J Biol Chem. 2003;278(6):4135–4144. | ||
Nakano A, Tani E, Miyazaki K, Yamamoto Y, Furuyama J. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in human gliomas. J Neurosurg. 1995;83(2):298–307. | ||
Lampert K, Machein U, Machein MR, Conca W, Peter HH, Volk B. Expression of matrix metalloproteinases and their tissue inhibitors in human brain tumors. Am J Pathol. 1998;153(2):429–437. | ||
Kachra Z, Beaulieu E, Delbecchi L, et al. Expression of matrix metalloproteinases and their inhibitors in human brain tumors. Clin Exp Metastasis. 1999;17(7):555–566. | ||
Pagenstecher A, Wussler EM, Opdenakker G, Volk B, Campbell IL. Distinct expression patterns and levels of enzymatic activity of matrix metalloproteinases and their inhibitors in primary brain tumors. J Neuropathol Exp Neurol. 2001;60(6):598–612. | ||
Gabelloni P, da Pozzo E, Bendinelli S, et al. Inhibition of metalloproteinases derived from tumours: new insights in the treatment of human glioblastoma. Neuroscience. 2010;168(2):514–522. | ||
Hagemann C, Anacker J, Haas S, et al. Comparative expression pattern of matrix-metalloproteinases in human glioblastoma cell-lines and primary cultures. BMC Res Notes. 2010;3:293. | ||
Hannocks MJ, Zhang X, Gerwien H, et al. The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes. Matrix Biol. Epub 2017 Nov 17. | ||
Rempe RG, Hartz AM, Bauer B. Matrix metalloproteinases in the brain and blood–brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36(9):1481–1507. | ||
Brkic M, Balusu S, Libert C, Vandenbroucke RE. Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators Inflamm. 2015;2015:620581. | ||
Warren KM, Reeves TM, Phillips LL. MT5-MMP, ADAM-10, and N-cadherin act in concert to facilitate synapse reorganization after traumatic brain injury. J Neurotrauma. 2012;29(10):1922–1940. | ||
Yong VW, Agrawal SM, Stirling DP. Targeting MMPs in acute and chronic neurological conditions. Neurotherapeutics. 2007;4(4):580–589. | ||
Pullen NA, Anand M, Cooper PS, Fillmore HL. Matrix metalloproteinase-1 expression enhances tumorigenicity as well as tumor-related angiogenesis and is inversely associated with TIMP-4 expression in a model of glioblastoma. J Neurooncol. 2012;106(3):461–471. | ||
Ezhilarasan R, Jadhav U, Mohanam I, Rao JS, Gujrati M, Mohanam S. The hemopexin domain of MMP-9 inhibits angiogenesis and retards the growth of intracranial glioblastoma xenograft in nude mice. Int J Cancer. 2009;124(2):306–315. | ||
Lakka SS, Gondi CS, Dinh DH, et al. Specific Interference of urokinase-type plasminogen activator receptor and matrix metalloproteinase-9 gene expression induced by double-stranded RNA results in decreased invasion, tumor growth, and angiogenesis in gliomas. J Biol Chem. 2005;280(23):21882–21892. | ||
Ramnath N, Creaven PJ. Matrix metalloproteinase inhibitors. Curr Oncol Rep. 2004;6(2):96–102. | ||
Steward WP, Thomas AL. Marimastat: the clinical development of a matrix metalloproteinase inhibitor. Expert Opin Investig Drugs. 2000;9(12):2913–2922. | ||
Butler GS, Overall CM. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry. 2009;48(46):10830–10845. | ||
Gravendeel LA, Kouwenhoven MC, Gevaert O, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69(23):9065–9072. | ||
Ramachandran RK, Sørensen MD, Aaberg-Jessen C, Hermansen SK, Kristensen BW. Expression and prognostic impact of matrix metalloproteinase-2 (MMP-2) in astrocytomas. PLoS One. 2017;12(2):e0172234. | ||
Anand M, van Meter TE, Fillmore HL. Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J Neurooncol. 2011;104(3):679–687. | ||
Thorns V, Walter GF, Thorns C. Expression of MMP-2, MMP-7, MMP-9, MMP-10 and MMP-11 in human astrocytic and oligodendroglial gliomas. Anticancer Res. 2003;23(5A):3937–3944. | ||
Deryugina EI, Bourdon MA, Luo GX, Reisfeld RA, Strongin A. Matrix metalloproteinase-2 activation modulates glioma cell migration. J Cell Sci. 1997;110(Pt 1):2473–2482. | ||
Musumeci G, Magro G, Cardile V, et al. Characterization of matrix metalloproteinase-2 and -9, ADAM-10 and N-cadherin expression in human glioblastoma multiforme. Cell Tissue Res. 2015;362(1):45–60. | ||
Komatsu K, Nakanishi Y, Nemoto N, Hori T, Sawada T, Kobayashi M. Expression and quantitative analysis of matrix metalloproteinase-2 and -9 in human gliomas. Brain Tumor Pathol. 2004;21(3):105–112. | ||
Jaworski DM. Developmental regulation of membrane type-5 matrix metalloproteinase (MT5-MMP) expression in the rat nervous system. Brain Res. 2000;860(1–2):174–177. | ||
Hayashita-Kinoh H, Kinoh H, Okada A, et al. Membrane-type 5 matrix metalloproteinase is expressed in differentiated neurons and regulates axonal growth. Cell Growth Differ. 2001;12(11):573–580. | ||
Llano E, Pendás AM, Freije JP, et al. Identification and characterization of human MT5-MMP, a new membrane-bound activator of progelatinase A overexpressed in brain tumors. Cancer Res. 1999;59(11):2570–2576. | ||
Ross HH, Fillmore HL. Identification of a novel human MT5-MMP transcript variant in multipotent NT2 cells. FEBS Lett. 2007;581(30):5923–5928. | ||
Venkatesh HS, Tam LT, Woo PJ, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature. 2017;549(7673):533–537. | ||
Ahir BK, Ozer H, Engelhard HH, Lakka SS. MicroRNAs in glioblastoma pathogenesis and therapy: a comprehensive review. Crit Rev Oncol Hematol. 2017;120:22–33. | ||
Huang SW, Ali ND, Zhong L, Shi J. MicroRNAs as biomarkers for human glioblastoma: progress and potential. Acta Pharmacol Sin. Epub 2018 Feb 8. | ||
Qu K, Lin T, Pang Q, et al. Extracellular miRNA-21 as a novel biomarker in glioma: evidence from meta-analysis, clinical validation and experimental investigations. Oncotarget. 2016;7(23):33994–34010. | ||
Wang H, Xu T, Jiang Y, Yan Y, Qin R, Chen J. MicroRNAs in human glioblastoma: from bench to beside. Front Biosci (Landmark Ed). 2015;20:105–118. | ||
Brower JV, Clark PA, Lyon W, Kuo JS. MicroRNAs in cancer: glioblastoma and glioblastoma cancer stem cells. Neurochem Int. 2014;77:68–77. | ||
Akhtar N, Rasheed Z, Ramamurthy S, Anbazhagan AN, Voss FR, Haqqi TM. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010;62(5):1361–1371. | ||
Wang J, Song Y, Zhang Y, et al. Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell Res. 2012;22(3):516–527. | ||
Li HR, Cui Q, Dong ZY, Zhang JH, Li HQ, Zhao L. Downregulation of MIR-27b is involved in loss of type II collagen by directly targeting matrix metalloproteinase 13 (MMP13) in human intervertebral disc degeneration. Spine. 2016;41(3):E116–E123. | ||
Li YF, Li SH, Liu Y, Luo YT. Long noncoding RNA CIR promotes chondrocyte extracellular matrix degradation in osteoarthritis by acting as a sponge for miR-27b. Cell Physiol Biochem. 2017;43(2):602–610. | ||
Pastuszak-Lewandoska D, Kordiak J, Czarnecka KH, et al. Expression analysis of three miRNAs, miR-26a, miR-29b and miR-519d, in relation to MMP-2 expression level in non-small cell lung cancer patients: a pilot study. Med Oncol. 2016;33(8):96. | ||
Wang H, Zhu Y, Zhao M, et al. miRNA-29c suppresses lung cancer cell adhesion to extracellular matrix and metastasis by targeting integrin β1 and matrix metalloproteinase 2 (MMP2). PLoS One. 2013;8(8):e70192. | ||
Wang H, Guan X, Tu Y, et al. MicroRNA-29b attenuates non-small cell lung cancer metastasis by targeting matrix metalloproteinase 2 and PTEN. J Exp Clin Cancer Res. 2015;34:59. | ||
Tang W, Zhu Y, Gao J, et al. MicroRNA-29a promotes colorectal cancer metastasis by regulating matrix metalloproteinase 2 and E-cadherin via KLF4. Br J Cancer. 2014;110(2):450–458. | ||
Lu L, Xue X, Lan J, et al. MicroRNA-29a upregulates MMP2 in oral squamous cell carcinoma to promote cancer invasion and anti-apoptosis. Biomed Pharmacother. 2014;68(1):13–19. | ||
Jones JA, Stroud RE, O’Quinn EC, et al. Selective microRNA suppression in human thoracic aneurysms: relationship of miR-29a to aortic size and proteolytic induction. Circulation. 2011;4(6):605–613. | ||
Kim JH, Jeon S, Shin BA. MicroRNA-29 family suppresses the invasion of HT1080 human fibrosarcoma cells by regulating matrix metalloproteinase 2 expression. Chonnam Med J. 2017;53(2):161–167. | ||
Bougnaud S, Golebiewska A, Oudin A, et al. Molecular crosstalk between tumour and brain parenchyma instructs histopathological features in glioblastoma. Oncotarget. 2016;7(22):31955–31971. | ||
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8(8):610–622. | ||
Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273(2):114–127. | ||
Mahase S, Rattenni RN, Wesseling P, et al. Hypoxia-mediated mechanisms associated with antiangiogenic treatment resistance in glioblastomas. Am J Pathol. 2017;187(5):940–953. | ||
Thompson EM, Frenkel EP, Neuwelt EA. The paradoxical effect of bevacizumab in the therapy of malignant gliomas. Neurology. 2011;76(1):87–93. | ||
Chen C, Huang R, Maclean A, et al. Recurrent high-grade glioma treated with bevacizumab: prognostic value of MGMT methylation, EGFR status and pretreatment MRI in determining response and survival. J Neurooncol. 2013;115(2):267–276. | ||
Flanigan PM, Aghi MK. Adaptation to antiangiogenic therapy in neurological tumors. Cell Mol Life Sci. 2015;72(16):3069–3082. | ||
Maherally Z, Fillmore HL, Tan SL, et al. Real-time acquisition of transendothelial electrical resistance in an all-human, in vitro, 3-dimensional, blood–brain barrier model exemplifies tight-junction integrity. FASEB J. 2018;32(1):168–182. | ||
Miner JH, Li C, Mudd JL, Go G, Sutherland AE. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development. 2004;131(10):2247–2256. | ||
Tilling T, Korte D, Hoheisel D, Galla HJ. Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J Neurochem. 1998;71(3):1151–1157. | ||
Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL. Expression of hypoxia-inducible factor 1α in brain tumors: association with angiogenesis, invasion, and progression. Cancer. 2000;88(11):2606–2618. | ||
Caspani EM, Crossley PH, Redondo-Garcia C, Martinez S. Glioblastoma: a pathogenic crosstalk between tumor cells and pericytes. PLoS One. 2014;9(7):e101402. | ||
von Baumgarten L, Brucker D, Tirniceru A, et al. Bevacizumab has differential and dose-dependent effects on glioma blood vessels and tumor cells. Clin Cancer Res. 2011;17(19):6192–6205. | ||
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. | ||
Machein MR, Renninger S, Lima-Hahn E, Plate KH. Minor contribution of bone marrow-derived endothelial progenitors to the vascularization of murine gliomas. Brain Pathol. 2003;13(4):582–597. | ||
Yue WY, Chen ZP. Does vasculogenic mimicry exist in astrocytoma? J Histochem Cytochem. 2005;53(8):997–1002. | ||
Soda Y, Marumoto T, Friedmann-Morvinski D, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A. 2011;108(11):4274–4280. | ||
Ricci-Vitiani L, Pallini R, Biffoni M, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468(7325):824–828. | ||
Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012;181(4):1126–1141. | ||
Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015;44–46:94–112. | ||
Handsley MM, Edwards DR. Metalloproteinases and their inhibitors in tumor angiogenesis. Int J Cancer. 2005;115(6):849–860. | ||
Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9(2):267–285. | ||
Fang J, Shing Y, Wiederschain D, et al. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc Natl Acad Sci U S A. 2000;97(8):3884–3889. | ||
Bergers G, Brekken R, Mcmahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–744. | ||
Jadhav U, Chigurupati S, Lakka SS, Mohanam S. Inhibition of matrix metalloproteinase-9 reduces in vitro invasion and angiogenesis in human microvascular endothelial cells. Int J Oncol. 2004;25(5):1407–1414. | ||
Webb AH, Gao BT, Goldsmith ZK, et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer. 2017;17:434. | ||
Chetty C, Lakka SS, Bhoopathi P, Kunigal S, Geiss R, Rao JS. Tissue inhibitor of metalloproteinase 3 suppresses tumor angiogenesis in matrix metalloproteinase 2-down-regulated lung cancer. Cancer Res. 2008;68(12):4736–4745. | ||
Rojiani MV, Alidina J, Esposito N, Rojiani AM. Expression of MMP-2 correlates with increased angiogenesis in CNS metastasis of lung carcinoma. Int J Clin Exp Pathol. 2010;3(8):775–781. | ||
Weng Y, Cai M, Zhu J, et al. Matrix metalloproteinase activity in early-stage lung cancer. Onkologie. 2013;36(5):256–259. | ||
Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5(9):2736–2749. | ||
Belotti D, Calcagno C, Garofalo A, et al. Vascular endothelial growth factor stimulates organ-specific host matrix metalloproteinase-9 expression and ovarian cancer invasion. Mol Cancer Res. 2008;6(4):525–534. | ||
Belotti D, Paganoni P, Manenti L, et al. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res. 2003;63(17):5224–5229. | ||
Zheng H, Takahashi H, Murai Y, et al. Expressions of MMP-2, MMP-9 and VEGF are closely linked to growth, invasion, metastasis and angiogenesis of gastric carcinoma. Anticancer Res. 2006;26(5 A):3579–3583. | ||
Du R, Petritsch C, Lu K, et al. Matrix metalloproteinase-2 regulates vascular patterning and growth affecting tumor cell survival and invasion in GBM. Neuro Oncol. 2008;10(3):254–264. | ||
Choe G, Park JK, Jouben-Steele L, et al. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin Cancer Res. 2002;8(9):2894–2901. | ||
Munaut C, Noël A, Hougrand O, Foidart JM, Boniver J, Deprez M. Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1-MMP, MMP-2 and MMP-9 in human glioblastomas. Int J Cancer. 2003;106(6):848–855. | ||
Kim SJ, Shin JY, Lee KD, et al. Galectin-3 Facilitates cell motility in gastric cancer by up-regulating protease-activated receptor-1 (PAR-1) and matrix metalloproteinase-1 (MMP-1). PLoS One. 2011;6(9):e25103. | ||
Huo N, Ichikawa Y, Kamiyama M, et al. MMP-7 (matrilysin) accelerated growth of human umbilical vein endothelial cells. Cancer Lett. 2002;177(1):95–100. | ||
Nishizuka I, Ichikawa Y, Ishikawa T, et al. Matrilysin stimulates DNA synthesis of cultured vascular endothelial cells and induces angiogenesis in vivo. Cancer Lett. 2001;173(2):175–182. | ||
Chantrain CF, Shimada H, Jodele S, et al. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res. 2004;64(5):1675–1686. | ||
Li J, Zhang YP, Kirsner RS. Angiogenesis in wound repair: angiogenic growth factors and the extracellular matrix. Microsc Res Tech. 2003;60(1):107–114. | ||
Beck L, d’Amore PA. Vascular development: cellular and molecular regulation. FASEB J. 1997;11(5):365–373. | ||
Xu J, Rodriguez D, Petitclerc E, et al. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol. 2001;154(5):1069–1080. | ||
Silletti S, Kessler T, Goldberg J, Boger DL, Cheresh DA. Disruption of matrix metalloproteinase 2 binding to integrin αvβ3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc Natl Acad Sci U S A. 2001;98(1):119–124. | ||
Herren B, Levkau B, Raines EW, Ross R. Cleavage of β-catenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: evidence for a role for caspases and metalloproteinases. Mol Biol Cell. 1998;9(6):1589–1601. | ||
Patterson BC, Sang QA. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase B/type IV collagenase (MMP-9). J Biol Chem. 1997;272(46):28823–28825. | ||
O’Reilly MS, Wiederschain D, Stetler-Stevenson WG, Folkman J, Moses MA. Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem. 1999;274(41):29568–29571. | ||
Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin α1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000;97(5):2202–2207. | ||
Pozzi A, Levine WF, Gardner HA. Low plasma levels of matrix metalloproteinase 9 permit increased tumor angiogenesis. Oncogene. 2002;21(2):272–281. | ||
Ferreras M, Felbor U, Lenhard T, Olsen BR, Delaissé J-M. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett. 2000;486(3):247–251. | ||
Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by αvβ3 and α5β1 integrins. Proc Natl Acad Sci U S A. 2003;100(8):4766–4771. | ||
Chantrain CF, Henriet P, Jodele S, et al. Mechanisms of pericyte recruitment in tumour angiogenesis: a new role for metalloproteinases. Eur J Cancer. 2006;42(3):310–318. | ||
Absinta M, Ha SK, Nair G, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. 2017;6:e29738. | ||
Sowers JL, Johnson KM, Conrad C, Patterson JT, Sowers LC. The role of inflammation in brain cancer. Adv Exp Med Biol. 2014;816:75–105. | ||
Benešová Y, Vašků A, Novotná H, et al. Matrix metalloproteinase-9 and matrix metalloproteinase-2 as biomarkers of various courses in multiple sclerosis. Mult Scler. 2009;15(3):316–322. | ||
Avolio C, Ruggieri M, Giuliani F, et al. Serum MMP-2 and MMP-9 are elevated in different multiple sclerosis subtypes. J Neuroimmunol. 2003;136(1–2):46–53. | ||
Waubant E, Goodkin D, Bostrom A, et al. IFN lowers MMP-9/TIMP-1 ratio, which predicts new enhancing lesions in patients with SPMS. Neurology. 2003;60(1):52–57. | ||
Boz C, Ozmenoglu M, Velioglu S, et al. Matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteinase (TIMP-1) in patients with relapsing–remitting multiple sclerosis treated with interferon beta. Clin Neurol Neurosurg. 2006;108(2):124–128. | ||
Nakada M, Okada Y, Yamashita J. The role of matrix metalloproteinases in glioma invasion. Front Biosci. 2003;8(5):e261–e269. | ||
Abraham M, Shapiro S, Karni A, Weiner HL, Miller A. Gelatinases (MMP-2 and MMP-9) are preferentially expressed by Th1 vs. Th2 cells. J Neuroimmunol. 2005;163(1–2):157–164. | ||
Yoshino et al. Effect of IFN-β on human glioma cell lines with temozolomide resistance. Int J Oncol. 2009;35(01):139–148. | ||
Wiranowska M, Rojiani AM, Gottschall PE, Moscinski LC, Johnson J, Saporta S. CD44 expression and MMP-2 secretion by mouse glioma cells: effect of interferon and anti-CD44 antibody. Anticancer Res. 2000;20(6B):4301–4306. | ||
Mohsenzadegan M, Fayazi MR, Abdolmaleki M, Bakhshayesh M, Seif F, Mousavizadeh K. Direct immunomodulatory influence of IFN-β on human astrocytoma cells. Immunopharmacol Immunotoxicol. 2015;37(2):214–219. | ||
Polukort SH, Rovatti J, Carlson L, et al. IL-10 enhances IgE-mediated mast cell responses and is essential for the development of experimental food allergy in IL-10-deficient mice. J Immunol. 2016;196(12):4865–4876. | ||
Qayum AA, Paranjape A, Abebayehu D, et al. IL-10-induced miR-155 targets SOCS1 to enhance IgE-mediated mast cell function. J Immunol. 2016;196(11):4457–4467. | ||
Wagner S, Stegen C, Bouterfa H, et al. Expression of matrix metalloproteinases in human glioma cell lines in the presence of IL-10. J Neurooncol. 1998;40(2):113–122. | ||
Kawaji H, Tokuyama T, Yamasaki T, Amano S, Sakai N, Namba H. Interferon-β and temozolomide combination therapy for temozolomide monotherapy-refractory malignant gliomas. Mol Clin Oncol. 2015;3(4):909–913. | ||
Motomura K, Natsume A, Kishida Y, et al. Benefits of interferon-β and temozolomide combination therapy for newly diagnosed primary glioblastoma with the unmethylated MGMT promoter. Cancer. 2011;117(8):1721–1730. | ||
Wolpert F, Happold C, Reifenberger G, et al. Interferon-β Modulates the Innate immune response against glioblastoma initiating cells. PLoS One. 2015;10(10):e0139603. | ||
Cheng SM, Xing B, Li JC, Cheung BK, Lau AS. Interferon-γ regulation of TNFα-induced matrix metalloproteinase 3 expression and migration of human glioma T98G cells. Int J Cancer. 2007;121(6):1190–1196. | ||
Qin H, Moellinger JD, Wells A, Windsor LJ, Sun Y, Benveniste EN. Transcriptional suppression of matrix metalloproteinase-2 gene expression in human astroglioma cells by TNF-α and IFN-γ. J Immunol. 1998;161(12):6664–6673. | ||
Li R, Li G, Deng L, et al. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol Rep. 2010;23(6):1553–1559. | ||
Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with α5β1 integrin in glioma. Oncogene. 2013;32(3):327–340. | ||
Chen W, Xia T, Wang D, et al. Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget. 2016;7(38):62425–62438. | ||
Markovic DS, Vinnakota K, Chirasani S, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. 2009;106(30):12530–12535. | ||
Kudo M, Jono H, Shinriki S, et al. Antitumor effect of humanized anti-interleukin-6 receptor antibody (tocilizumab) on glioma cell proliferation. J Neurosurg. 2009;111(2):219–225. | ||
Xue H, Yuan G, Guo X, et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy. 2016;12(7):1129–1152. | ||
Wang H, Lathia JD, Wu Q, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27(10):2393–2404. | ||
Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad: tumor-associated microglia/macrophages in glioma. Int J Mol Sci. 2018;19(2):E436. | ||
Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–27. | ||
Mangani D, Weller M, Sadr ES, et al. Limited role for transforming growth factor-β pathway activation-mediated escape from VEGF inhibition in murine glioma models. Neuro Oncol. 2016;18(12):1610–1621. | ||
Miyauchi JT, Caponegro MD, Chen D, Choi MK, Li M, Tsirka SE. Deletion of neuropilin 1 from microglia or bone marrow-derived macrophages slows glioma progression. Cancer Res. 2018;78(3):685–694. | ||
Delwar ZM, Kuo Y, Wen YH, Rennie PS, Jia W. Oncolytic virotherapy blockade by microglia and macrophages requires STAT1/3. Cancer Res. 2018;78(3):718–730. | ||
Wang Y, Liu T, Yang N, Xu S, Li X, Wang D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol Rep. 2016;36(6):3522–3528. | ||
Ellert-Miklaszewska A, Dabrowski M, Lipko M, Sliwa M, Maleszewska M, Kaminska B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia. 2013;61(7):1178–1190. | ||
Vinnakota K, Hu F, Ku MC, et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol. 2013;15(11):1457–1468. | ||
Hu F, Ku MC, Markovic D, et al. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int J Cancer. 2014;135(11):2569–2578. | ||
Hu F, a Dzaye OD, Hahn A, et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol. 2015;17(2):200–210. | ||
Yi YJ, Huang SY, Chen L, Chen XR, Yang ZL, Ke YQ. Atorvastatin suppresses glioma invasion and migration by reducing microglial MT1-MMP expression. J Neuroimmunol. 2013;260(1–2):1–8. | ||
Peng P, Wei W, Long C, Li J. Atorvastatin augments temozolomide’s efficacy in glioblastoma via prenylation-dependent inhibition of Ras signaling. Biochem Biophys Res Commun. 2017;489(3):293–298. | ||
Bayat N, Ebrahimi-Barough S, Norouzi-Javidan A, et al. Anti-inflammatory effects of atorvastatin by suppressing TRAF3IP2 and IL-17RA in human glioblastoma spheroids cultured in a three-dimensional model: possible relevance to glioblastoma treatment. Mol Neurobiol. 2018;55(3):2102–2110. | ||
Parajuli P, Anand R, Mandalaparty C, et al. Preferential expression of functional IL-17R in glioma stem cells: potential role in self-renewal. Oncotarget. 2016;7(5):6121–6135. | ||
Ye XZ, Xu SL, Xin YH, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-1 signaling pathway. J Immunol. 2012;189(1):444–453. | ||
Skaper SD, Facci L, Zusso M, Giusti P. Neuroinflammation, mast cells, and glia: dangerous liaisons. Neuroscientist. 2017;23(5):478–498. | ||
Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359(6381):1269–1273. | ||
Zhang J, Wang P, Ji W, Ding Y, Lu X. Overexpression of interleukin-33 is associated with poor prognosis of patients with glioma. Int J Neurosci. 2017;127(3):210–217. | ||
Zhang JF, Wang P, Yan YJ, et al. IL-33 enhances glioma cell migration and invasion by upregulation of MMP2 and MMP9 via the ST2-NF-κB pathway. Oncol Rep. 2017;38(4):2033–2042. | ||
Hsu CL, Neilsen CV, Bryce PJ. IL-33 Is produced by mast cells and regulates IgE-dependent inflammation. PLoS One. 2010;5(8):e11944. | ||
Wang JX, Kaieda S, Ameri S, et al. IL-33/ST2 axis promotes mast cell survival via BCLXL. Proc Natl Acad Sci U S A. 2014;111(28):10281–10286. | ||
Joulia R, L’Faqihi FE, Valitutti S, Espinosa E. IL-33 fine tunes mast cell degranulation and chemokine production at the single-cell level. J Allergy Clin Immunol. 2017;140(2):497–509. | ||
Wroblewski M, Bauer R, Córdova MC, et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat Commun. 2017;8(1):269. | ||
Roy A, Coum A, Marinescu VD, et al. Glioma-derived plasminogen activator inhibitor-1 (PAI-1) regulates the recruitment of LRP1 positive mast cells. Oncotarget. 2015;6(27):23647–23661. | ||
Põlajeva J, Sjösten AM, Lager N, et al. Mast cell accumulation in glioblastoma with a potential role for stem cell factor and chemokine CXCL12. PLoS One. 2011;6(9):e25222. | ||
Attarha S, Roy A, Westermark B, Tchougounova E. Mast cells modulate proliferation, migration and stemness of glioma cells through downregulation of GSK3β expression and inhibition of STAT3 activation. Cell Signal. 2017;37:81–92. | ||
Cwm O, Elkington PT, Brilha S, et al. Neutrophil-derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog. 2015;11(5):e1004917. | ||
Fossati G, Ricevuti G, Edwards SW, Walker C, Dalton A, Rossi ML. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999;98(4):349–354. | ||
Zhang J, Zhang S, Song Y, et al. Prognostic role of neutrophil lymphocyte ratio in patients with glioma. Oncotarget. 2017;8(35):59217–59224. | ||
Hartmann P, Herholz K, Salzberger B, Petereit HF. Unusual and severe symptomatic impairment of neutrophil function after one cycle of temozolomide in patients with malignant glioma. Ann Hematol. 2004;83(4):212–217. | ||
Graf MR, Prins RM, Merchant RE. IL-6 secretion by a rat T9 glioma clone induces a neutrophil-dependent antitumor response with resultant cellular, antiglioma immunity. J Immunol. 2001;166(1):121–129. | ||
Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–194. | ||
Uribe-Querol E, Rosales C. Neutrophils in cancer: two sides of the same coin. J Immunol Res. 2015;2015:983698. | ||
Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200(2):422–431. | ||
Xue J, Zhao Z, Zhang L, et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat Nanotechnol. 2017;12(7):692–700. | ||
Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. | ||
Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. 2015;74(7):1417–1424. | ||
Huang Y, Rajappa P, Hu W, et al. A proangiogenic signaling axis in myeloid cells promotes malignant progression of glioma. J Clin Invest. 2017;127(5):1826–1838. | ||
Reardon DA, Mitchell DA. The development of dendritic cell vaccine-based immunotherapies for glioblastoma. Semin Immunopathol. 2017;39(2):225–239. | ||
Migliorini D, Dietrich PY, Stupp R, Linette GP, Posey AD, June CH. CAR T-cell therapies in glioblastoma: A first look. Clin Cancer Res. 2018;24(3):535–540. | ||
Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419–1427. | ||
Gromeier M, Nair SK. Recombinant poliovirus for cancer immunotherapy. Annu Rev Med. 2018;69:289–299. | ||
Zhu Z, Gorman MJ, Mckenzie LD, et al. Zika virus has oncolytic activity against glioblastoma stem cells. J Exp Med. 2017;214(10):2843–2857. | ||
Mohanty S, Chen Z, Li K, et al. A novel theranostic strategy for MMP-14-expressing glioblastomas impacts survival. Mol Cancer Ther. 2017;16(9):1909–1921. | ||
Daldrup-Link HE. Rethinking brain cancer therapy: tumor enzyme activatable theranostic nanoparticles. Mol Imaging. 2017;16:1536012117730950. | ||
Barnes JM, Przybyla L, Weaver VM. Tissue mechanics regulate brain development, homeostasis and disease. J Cell Sci. 2017;130(1):71–82. | ||
Jozic D, Bourenkov G, Lim NH, et al. X-ray structure of human proMMP-1: new insights into procollagenase activation and collagen binding. J Biol Chem. 2005;280(10):9578–9585. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.