")
Back to Journals » Research and Reports in Urology » Volume 11
Current evidence for the involvement of sex steroid receptors and sex hormones in benign prostatic hyperplasia
Authors Da Silva MHA , De Souza DB
Received 11 October 2018
Accepted for publication 7 December 2018
Published 7 January 2019 Volume 2019:11 Pages 1—8
DOI https://doi.org/10.2147/RRU.S155609
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jan Colli
Marcello Henrique Araujo Da Silva, Diogo Benchimol De Souza
Urogenital Research Unit, Rio de Janeiro State University, Rio de Janeiro, RJ, Brazil
Abstract: Benign prostatic hyperplasia (BPH) is a pathology that affects 50% of men over 50 years of age and 90% of men develop BPH in their eighth decade of life. In 2018, more than 1 billion men will be affected by this disease worldwide. However, the progression of BPH is highly complex and has been debated and studied for approximately four decades. Recent studies indicate that BPH can originate from the alteration of different hormone synthesis pathways, and that it is also linked to the function of hormone receptors. There is a close relationship between the progression of BPH and sexual hormones, such as progesterone, testosterone, dihydrotestosterone, and estrogen. The focus of this study was to characterize the interactions of these hormones and investigate the direct or indirect role of each sex hormone receptor in the progression of BPH. Although several studies have described the effects of these hormones on BPH, no conclusions have been drawn regarding their role in disease progression. Here, we present a literature review on the sexual receptors possibly involved in the progression of BPH.
Keywords: benign prostatic hyperplasia, testosterone, progesterone, estrogen, dihydrotestosterone
Introduction
Benign prostatic hyperplasia (BPH) is a non-malignant disease characterized by the enlargement of prostatic epithelial and stromal tissues and reduced urinary flow, causing disturbances commonly known as lower urinary tract syndrome.1
Recent evidences have provided new insights into the mechanisms that govern or generate BPH. In addition, we know that aging is correlated with BPH. BPH currently affects 50% of men over 50 years of age and 90% of men in their eighth decade of life.2–4 It is estimated that, in 2018, 1 billion new BPH cases will be diagnosed worldwide.5
The prostate is a typical androgen-dependent gland and is highly reactive to sex hormones.1,6–8 In the 1970s, sex hormones were recognized as potential promoting factors of BPH. Among them, the most studied are testosterone (TT), dihydrotestosterone (DHT), progesterone (PGR), and estrogen.9–11 However, the influence of each sex hormone on the progression and development of BPH and the pathogenetic role of complex hormone/receptor interactions have not yet been fully clarified.
Androgenic receptors (ARs)
AR is present at high concentrations in the prostate gland10 in two isoforms, namely AR-A and AR-B.12 These receptors are encoded by the AR gene, located on the X chromosome at the locus Xq11-Xq12, and comprising eight exons, for a total length of about 90 kb.13–15
The production of AR is particularly important because this protein activates the expression of other genes. The transactivation ability of AR lies in its N-terminal domain, which is encoded by exon 1. In this region, the mutation of two genes CAG and GGC also occurs.16
An excessive of polymorphisms is present in the CAG repeat, while few repetition polymorphisms are present in GGC.17–20 Giovannucci et al21 showed that the CAG polymorphism in exon 1 is 25-fold higher than GGC, and that CAG is the main cause of BPH progression. However, Biolchi et al18,19 reported that the CAG polymorphism does not directly influence the serum levels of TT or the development of BPH, whereas the GGC polymorphism increases the risk of BPH and prostate cancer.
Thus, AR gene polymorphism may be one of the causes of BPH development and progression. In addition, the binding of sex hormones to AR increases the likelihood of developing BPH. AR not only regulates the expression of hundreds of genes, but it is also involved in androgen binding and mediates the effects of TT and its most active metabolite, DHT.22
Prostate growth is stimulated by DHT, the active metabolite generated from TT by the enzyme 5-alpha-reductase.6 This enzyme is present in three different isoforms: the type I isoform is ubiquitously expressed but present at low concentrations in the organs of the urogenital system; the type II isoform is enriched in organs of the urogenital system and the hair follicles; the type III isoform is especially abundant in the epidermis and in some organs such as the pancreas.23–26
The inhibition of the TT-to-DHT conversion reduces the levels of intraprostatic DHT6,24,25 and prevents the enlargement of the prostatic epithelium, resulting in a 20%–30% decrease in prostate volume after 12 months of treatment.27
This dramatic reduction reflects the high proportion of AR, accounting for ~90% of all prostate sex receptors. The reduction of the intraprostatic TT levels decreases the conversion into DHT, and, thus, the hormone-receptor binding.28–33 The impaired binding of androgens to their receptors prevents the translocation of AR from the cytoplasm to the nucleus of the prostate cells, making activation of specific pathways theoretically impossible.34
In addition, patients with BPH present an extremely high DHT plasma concentration, compared to age-matched healthy subjects.35 Also of note is the reported decline in the level of TT in BPH patients,30 reinforcing the theory that DHT is largely responsible for the growth and proliferation of prostatic components. However, the reason for the DHT increase is not clear, as both serum and intraprostatic levels of TT are reduced with age.
A possible explanation for the increased serum and intraprostatic DHT levels observed in patients with BPH may reside in the conversion of 5-α-androstenedione to DHT by 17β-hydroxysteroid dehydrogenases (17β-HSDs). 17β-HSDs belong to a family of enzymes that catalyze the reduction of stereospecific oxides of alcohols or carbonyls using NAD(P)H or NAD(P+) as cofactors. HSDs include five primordial subtypes: 3α-, 3β-, 11β-, 17β-, and 20α-HSDs.36
Fundamentally, 17β-HSDs catalyze the synthesis of estrone (E1), estradiol (E2), 5-androstene-3β, 17β-diol, TT, and DHT. They also convert inactive or low-activity steroid hormones into more potent hormones, and vice versa. 17β-HSDs are key regulators in humans. 17β-HSDs comprise eight isoforms, each responsible for catalyzing the synthesis of a steroid or androgen (Figure 1).12,36–38
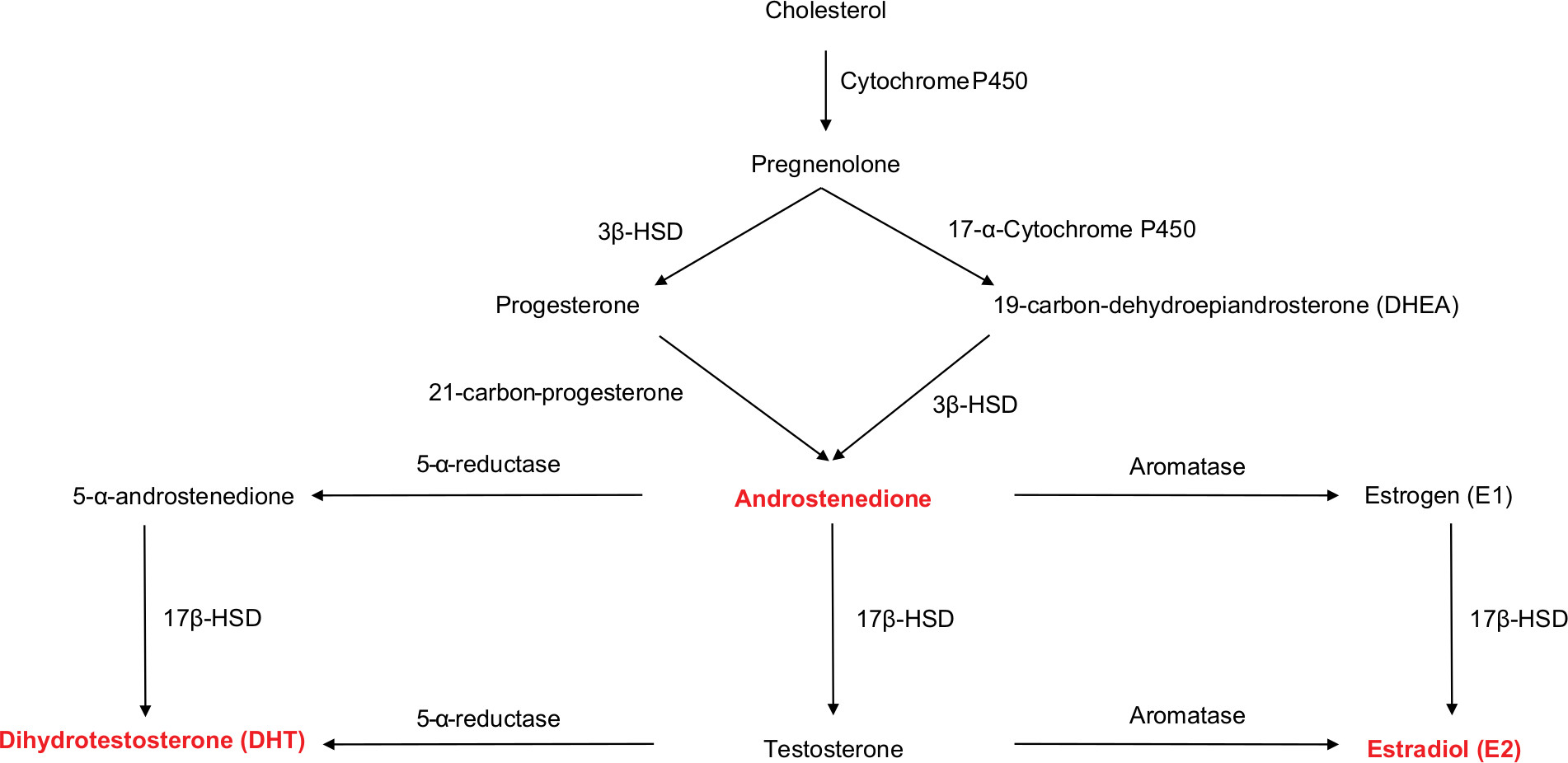 | Figure 1 Schematic drawing of the reaction cascade leading to the synthesis of testosterone, dihydrotestosterone, and estradiol from cholesterol. Abbreviations: 3β-HSD, 3β-hydroxysteroid dehydrogenases; 5-α-reductase, 5-alpha-reductase; 17β-HSD, 17β-hydroxysteroid dehydrogenases. |
Estrogen receptors (ERs)
In the 1970s, a direct link between prostate enlargement and the increase in intraprostatic estrogen was hypothesized.39 Moreover, a crucial role of estrogen in the development of BPH has been described. However, the importance of AR and in the progression of BPH is indisputable.40
We currently know that, in men, 75%–90% of the circulating estrogen is produced in adipose tissue, brain, bone, and other tissues. Estrogen synthesis is mainly controlled by the enzyme aromatase, which converts androstenedione to E1 and TT to E2. Aromatase is a member of the CYP/CYP450 superfamily.7,41
Aromatase is expressed in the gonad gland of both sexes. Approximately 10%–25% of TT is converted in the testicles, generating E2. This maintains the levels of E2 required for normal spermatogenesis, sperm maturation, and sperm motility. In addition, 17β-HSD converts the excess of E2 into E1 (Figure 2).12,42
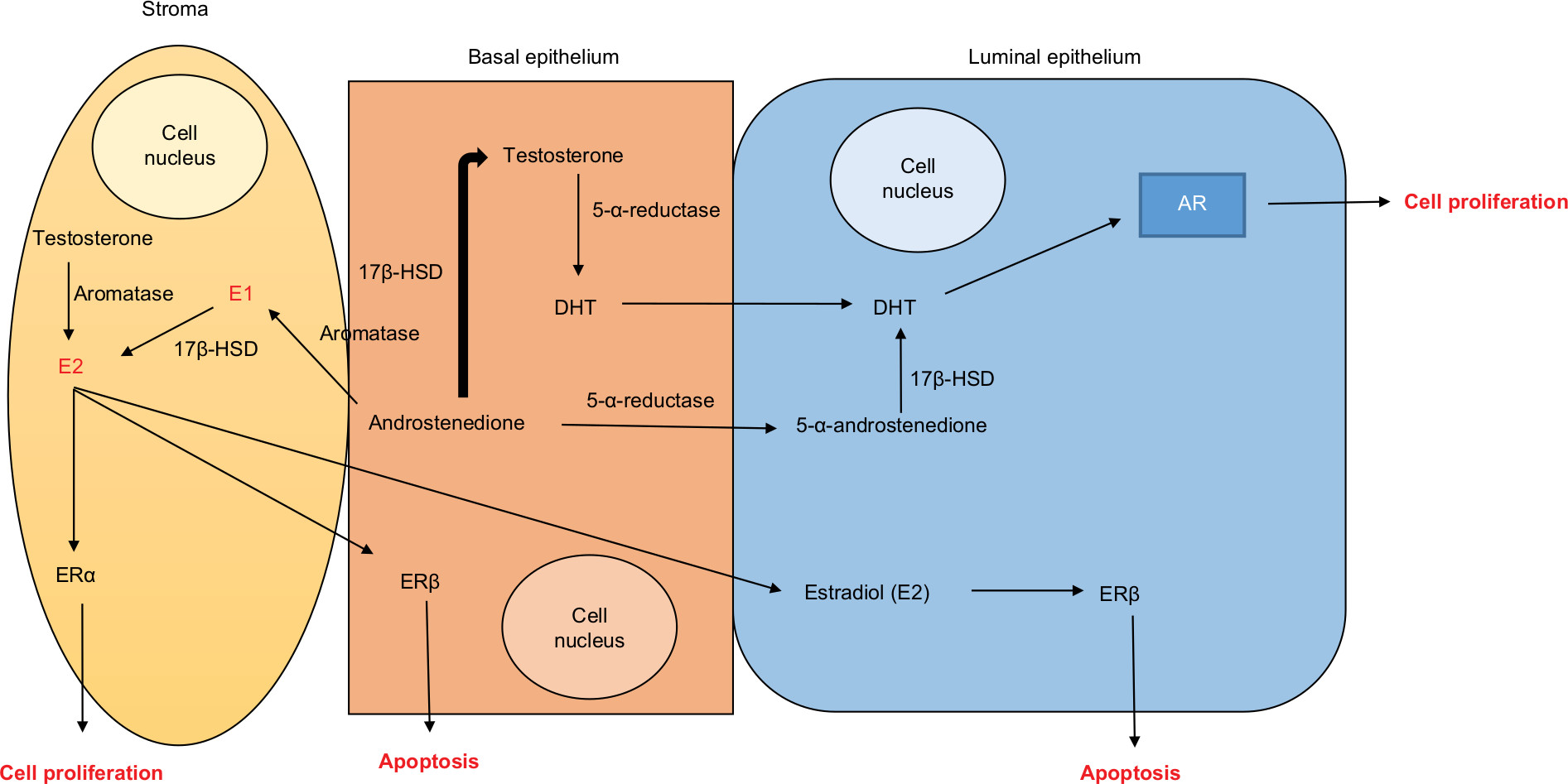 | Figure 2 Schematic drawing of the synthesis of the active hormones dihydrotestosterone and estradiol from testosterone and androstenedione and binding of sex hormones with their receptors in the prostatic tissue. Abbreviations: AR, androgen receptors; DHT, dihydrotestosterone; E1, estrogen; E2, estradiol; ERα, estrogen receptor alpha; ERβ, estrogen receptor beta; 5-α-androstenedione, 5-alpha-androstenedione; 5-α-reductase, 5-alpha-reductase; 17β-HSD, 17β-hydroxysteroid dehydrogenases. |
The hormones E1 and E2 exhibit high affinity and specificity for an intranuclear protein in target cells, termed ER. After binding of the hormone to ER, the latter undergoes a conformational change, allowing it to bind to chromatin with high affinity and, consequently, modulate the transcription of target genes. ER consists of an N-terminal hypervariable domain that contributes to the transactivation of a central domain responsible for a specific binding to DNA. In addition, ER activates the dimerization and nuclear localization of the C-terminal domain involved in ligand binding and ligand-dependent transactivation, ultimately inducing cell mitosis.41,43,44
There are two types of ER, ERα and ERβ, encoded by ESR1 and ESR2, respectively.7,41,44 In the prostate, ERα is expressed in a few stromal cells, whereas ERβ is highly expressed in the epithelium and some stromal cells.45
Upon activation, ERα and ERβ translocate from the cytoplasm to the nucleus, where they activate DNA transcription by recruiting cofactors such as specific protein 1 (SP-1), activator protein 1 (AP-1), and nuclear factor κB.41,46 In addition, such binding may stimulate further production of E1 and E2, inducing cell proliferation or apoptosis.
During this new transcription tape, SP-1 becomes the predominant mediator of the ER–DNA interaction and stimulates the expression of ESR2.41 In addition, E2 can activate the production of endothelial nitric oxide,47 which leads to relaxation of the arterial tunica media via increased blood perfusion of the prostatic tissues.
A number of studies have shown that AP-1 increases the binding of ERα and ERβ to DNA.48–50 Moreover, the association of ERα and AP-1 promotes cell proliferation and can promote neoplasias by E2F1 and cyclin D1 co-regulation. Notably, ERβ activation by AP-1 only results in cyclin D1 transition and activation.41
A study conducted in rats reported an alternative ERα-dependent activation mechanism, consisting in direct E2 binding to ERα, at the plasma membrane or in the cytoplasm, forming a complex that functions as a transcriptional activator. This pathway generates a faster DNA transcription response and plays a crucial physiological role.51
In addition, unlike Erβ, which can induce apoptosis, ERα activates the proliferation of prostatic cells.44,52 There is a clear difference in the actions of ERα and ERβ, as well as in the adverse events possibly caused by estrogen binding to the two receptors. Estrogen binding to ERα is associated with cell proliferation and inflammation of the prostate, whereas estrogen interaction with ERβ appears to mediate antiproliferative, anti-inflammatory, and potentially anticarcinogenic effects.8 Zhang et al53 reported that patients with BPH exhibit a decreased ERα expression, whereas ERβ expression is not affected.
PGR receptors
PGR is a typical female hormone, although it is also synthesized in the adrenal glands and the Leydig cells through the action of 3β-HSD. Its production occurs through the classical pathway of cholesterol synthesis and can generate TT and estrogen.54–56
PGR binds PR, which belongs to the superfamily of steroid receptors.57 Three different human PR isoforms exist, ie, PRa, PRb, and PRc. However, only PRa and PRb are functional.58–61
Compared to PRb, PRa contains 164 extra amino acids at its amino terminus.62 PRa regulates the transcription of the gene encoding PRb. The two isoforms have been associated with distinct physiological functions and are present in different intracellular concentrations. Moreover, studies have reported that PRa can inhibit the production of PRb.63,64
In 1970, E2 was proposed to bind to PR by virtue of its higher specificity for this receptor, compared to PGR.65 Furthermore, Kastner et al66 described that PRa expression is controlled by ERα and PRa controls the intracellular concentrations of PRb of both receptors.
Cell culture studies reported that ERα interacts with SP-1 and AP-1, thereby controlling the expression of PR.67,68 However, other studies using the same cell line suggested that ERα binds to regulatory regions of the PR gene rather than acting directly on the PR synthesis and did not affect the production of the PR isoforms.69,70
Therefore, ERα is associated with the transcription of the PR gene only as a recruiter of transitional co-regulators that are dependent on the PR gene.70 These co-regulators alter the chromatin and activate the DNA transition. However, the transcriptional activation of the PR gene isoforms depends primarily on the status of DNA methylation and chromatin as their promoters.71
Thus, it demonstrates that the synthesis of PR isoforms is much more complex than the ER synthesis, as it encompasses several mechanisms of PR gene regulation.62 Some studies have indicated that, in humans and rats with BPH, a higher expression of PGR is associated with an increased proliferation of prostatic stromal cells.1,57
Clinical correlation and the role of AR, ER, and PR in BPH
The role of sex receptors and hormones in the progression and development of BPH has been discussed for decades and pharmacological treatments targeting the hormone-receptor interaction have been developed and described. The most effective and utilized treatment worldwide is based on 5-alpha-reductase enzyme inhibitors.3,25,28 However, various studies have reported that this treatment generates changes in the morphology of the corpus cavernosum that are linked to erectile dysfunction, after or during therapy.3,72,73
Despite the availability of effective pharmacological treatments, little is known about the progression of BPH. One hypothesis is that the abnormal expression of AR receptors in the prostate stroma leads to enhanced DHT binding to AR, thereby resulting in increased cell proliferation.12 However, in patients with BPH, the levels of TT are low, contradicting the theory that TT is responsible for the progression of BPH.
An abnormal expression of AR in the prostate could derive from CAG and GGC repeat polymorphism in the AR gene.18,19,21 However, Weng et al74 showed that CAG is linked to prostate cancer but not to BPH in Caucasian men.
The observation that serum levels of TT in patients with BPH are reduced generates uncertainty about the biogenesis of intraprostatic DHT. We believe that this phenomenon could be related to the reaction converting androstenedione to α-androstenedione, catalyzed by 5-alpha-reductase, followed by 17β-HSD-mediated α-androstenedione conversion to DHT. This may explain the presence of intraprostatic DHT in the serum of patients with BPH, even in the absence of high levels of TT.
17β-HSD is extremely important as it converts androstenedione into TT, α-androstenedione in DHT, and E1 into E2. Thus, this enzyme is considered a potential therapeutic target for BPH. However, the efficacy of 17β-HSD inhibitors, as well as their possible adverse effects, are still a matter of intense debate, and additional studies are needed to definitively clarify this issue.12,20
Another question that should be addressed is the role of aromatase, which is expressed in the prostate stroma and can convert TT to E2 and androstenedione to E1. Increased aromatase activity in the stroma can affect the local production of precursor estrogens, leading to increased estrogen and disproportionate levels of androgens.12
The inhibition of aromatase activity reduces intracellular E2 concentrations, and may allow control of the synthesis ER receptor, representing another potential therapeutic strategy for BPH.12 However, further studies are needed to determine whether the inhibition of aromatase can reduce prostate cell proliferation in patients with BPH.
Another “hot topic” is the abnormal overexpression of ER in the prostate stroma and the epithelium of BPH patients. Several studies indicate that ERα and ERβ control proliferation and apoptosis in the prostatic tissue.12,41,75
ERα may associate with excessive proliferation, inflammation, and, possibly, carcinogenic effects. In contrast, ERβ appears to be responsible for antiproliferative, anti-inflammatory, and anticancer effects. At puberty, the serum levels of testosterone are elevated over time but decreasing levels may correlate with the action of aromatases. By raising the E2-to-TT ratio, aromatase causes an increase in serum estradiol levels.76 Men with BPH have higher levels of E2 than patients without BPH77,78 and, surprisingly, ERβ was found to be linked to prostate cancer.76 This reinforces the theory that ERα receptors are responsible for the development of BPH.45,76
Circulating and intraprostatic E2 levels increase with age, eventually activating extracellular signal-regulated kinases 1/2 (ERK1/2), responsible for cell proliferation.77 Increased differentiation and proliferation of smooth muscle cells.78,79 Cell replication triggers a series of changes in stromal cells, including loss of expression of G protein-coupled receptor 30 (GPR30), a proliferation factor downstream of E2, ERα, and ERβ. It is believed that, as a consequence of ERK1/2 activation, an epigenetic inhibitory change occurs in the promoter of the GPR30 gene.80,81 With the activation of ERK1/2 and cell differentiation, there is an increase in the expression of TGFβ1, which is responsible for stromal cell growth and differentiation.82
Increased production of ERα is related to high concentrations of E2 and ERα controls the synthesis of PR.62,67,68 A study of patients with BPH showed an increase of PR in the prostate stroma, demonstrating that the expression of ERα and PR receptors were tightly linked and were linked to complex mechanisms of receptor synthesis,83 and no PR was found in the prostate stroma of patients with BPH.84 Rather than reducing its expression with the ERα demonstrating that PGR may be a promoter of BPH.85–87
A new pharmacological strategy against BPH may be based on the modulation of specific ERs. For instance, the use of ERα antagonists and ERβ agonists has been proposed.8 Moreover, the inhibition of intraprostatic E2 synthesis can be achieved through modulation of 17β-HSD, resulting in a decreased rate of E1-to-E2 conversion and reduced prostatic cell proliferation.88 In accordance with these findings, ERβ agonists have been reported to decrease prostate cancer and BPH in humans.52
However, other studies found higher expression of AR and PR, compared to ERα, in the prostate stroma of animals with BPH. This may indicate that AR and PR play a role in the pathogenesis of BPH, whereas ERα only regulates PR production.1,44,53,76,89,90
In normal prostate and BPH samples, ARs are located in the nuclei of stromal, as well as luminal and basal epithelial cells. TT and DHT bind to ARs, influencing the BPH growth factors. The increased expression of prostatic epithelial AR might contribute to enhanced cell proliferation and is one of the possible causes of BPH.91–93
Androgens play several roles in prostate maintenance. Notably, TT supplementation to castrated animals restored the prostate architecture.94
Patients with deficiency of the enzyme 5-alpha-reductase exhibit reduced serum and intraprostatic levels of DHT and normal growth and development of the prostate gland.20 DHT deficiency can be associated with a decline in serum androgen levels and is characterized by numerous clinical symptoms.95
Conclusion
E2 has a major influence on the expression of the ERα-encoding gene, ESR1. We conclude that ERα controls the synthesis of ERβ and has an impact on cell proliferation and apoptosis, thereby affecting the risk of BPH and prostate cancer. In addition, ERα can stimulate the synthesis of PR, exerting proliferative effects on the prostate gland and possibly causing BPH. Thus, alterations in E2 levels may represent a major cause of BPH. However, robust evidence indicates that a major pathogenetic factor for BPH is represented by ARs. Notably, the binding of DHT to AR can stimulate cellular proliferation factors. The origin of DHT in men with BPH is unclear, as these patients have low serum and intraprostatic levels of TT. We believe that a possible explanation may reside in the 17β-HSD-mediated conversion of 5-α-androstenedione to DHT. In line with this hypothesis, 17β-HSD inhibitors interfere with BPH progression. However, additional studies on the role of 17β-HSD and E2 in the prostate of patients with BPH are necessary to definitively solve this issue.
Disclosure
The authors report no conflicts of interest in this work.
References
Song L, Shen W, Zhang H, Wang Q, Wang Y, Zhou Z. Differential expression of androgen, estrogen, and progesterone receptors in benign prostatic hyperplasia. Bosn J Basic Med Sci. 2016;16(3):201–208. | ||
Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol. 1984;132(3):474–479. | ||
Erdemir F, Harbin A, Hellstrom WJ. 5-Alpha reductase inhibitors and erectile dysfunction: the connection. J Sex Med. 2008;5(12):2917–2924. | ||
Lepor H. Management of clinically localized prostate cancer. Rev Urol. 2004;6(Suppl 2):S3–S12. | ||
Casabé A, Roehrborn CG, da Pozzo LF, et al. Efficacy and safety of the coadministration of tadalafil once daily with finasteride for 6 months in men with lower urinary tract symptoms and prostatic enlargement secondary to benign prostatic hyperplasia. J Urol. 2014;191(3):727–733. | ||
Wilson JD. The critical role of androgens in prostate development. Endocrinol Metab Clin North Am. 2011;40(3):577–590. | ||
McPherson SJ, Ellem SJ, Risbridger GP. Estrogen-regulated development and differentiation of the prostate. Differentiation. 2008;76(6):660–670. | ||
Ellem SJ, Risbridger GP. The dual, opposing roles of estrogen in the prostate. Ann N Y Acad Sci. 2009;1155(1):174–186. | ||
Robel P, Eychenne B, Blondeau JP, et al. Androgen receptors in rat and human prostate. Horm Res. 1983;18(1-3):28–36. | ||
Robel P, Eychenne B, Blondeau JP, Baulieu EE, Hechter O. Sex steroid receptors in normal and hyperplastic human prostate. Prostate. 1985;6(3):255–267. | ||
Balistreri CR, Caruso C, Carruba G, Miceli V, Candore G. Genotyping of sex hormone-related pathways in benign and malignant human prostate tissues: data of a preliminary study. OMICS. 2011;15(6):369–374. | ||
Ho CK, Habib FK. Estrogen and androgen signaling in the pathogenesis of BPH. Nat Rev Urol. 2011;8(1):29–41. | ||
Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. 1988;240(4850):327–330. | ||
Chamberlain NL, Driver ED, Miesfeld RL. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res. 1994;22(15):3181–3186. | ||
Loughlin KR. The testosterone conundrum: the putative relationship between testosterone levels and prostate cancer. Urol Oncol. 2016;34(11):482.e1–e482.e4. | ||
Sleddens HF, Oostra BA, Brinkmann AO, Trapman J. Trinucleotide (GGN) repeat polymorphism in the human androgen receptor (AR) gene. Hum Mol Genet. 1993;2(4):493. | ||
Edwards A, Hammond HA, Jin L, Caskey CT, Chakraborty R. Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics. 1992;12(2):241–253. | ||
Biolchi V, Silva Neto B, Koff W, Brum IS. Androgen receptor CAG polymorphism and the risk of benign prostatic hyperplasia in a Brazilian population. Int Braz J Urol. 2012;38(3):373–379. | ||
Biolchi V, Silva Neto B, Pianta DB, Koff WJ, Berger M, Brum IS. Androgen receptor GGC polymorphism and testosterone levels associated with high risk of prostate cancer and benign prostatic hyperplasia. Mol Biol Rep. 2013;40(3):2749–2756. | ||
Zhu YS, Katz MD, Imperato-McGinley J. Natural potent androgens: lessons from human genetic models. Baillières Clin Endocrinol Metab. 1998;12(1):83–113. | ||
Giovannucci E, Platz EA, Stampfer MJ, et al. The CAG repeat within the androgen receptor gene and benign prostatic hyperplasia. Urology. 1999;53(1):121–125. | ||
Ito Y, Sadar MD. Enzalutamide and blocking androgen receptor in advanced prostate cancer: lessons learnt from the history of drug development of antiandrogens. Res Rep Urol. 2018;10:23–32. | ||
Jarvis TR, Chughtai B, Kaplan SA. Testosterone and benign prostatic hyperplasia. Asian J Androl. 2015;17(2):212–216. | ||
Andriole G, Bruchovsky N, Chung L, et al. Dihydrotestosterone and the prostate: the scientific rationale for 5alpha-reductase inhibitors in the treatment of benign prostatic hyperplasia. J Urol. 2004;172(4):1399–1403. | ||
Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63(1):25–61. | ||
Wu XJ, Zhi Y, Zheng J, et al. Dutasteride on benign prostatic hyperplasia: a meta-analysis on randomized clinical trials in 6460 patients. Urology. 2014;83(3):539–543. | ||
Nickel JC, Gilling P, Tammela TL, Morrill B, Wilson TH, Rittmaster RS. Comparison of dutasteride and finasteride for treating benign prostatic hyperplasia: the Enlarged Prostate International Comparator Study (EPICS). BJU Int. 2011;108(3):388–394. | ||
Roehrborn CG. Pathology of benign prostatic hyperplasia. Int J Impot Res. 2008;20(S3):S11–S18. | ||
Yatkin E, Bernoulli J, Talvitie EM, Santti R. Inflammation and epithelial alterations in rat prostate: impact of the androgen to oestrogen ratio. Int J Androl. 2009;32(4):399–410. | ||
Ryl A, Rotter I, Slojewski M, et al. Hormone concentration, metabolic disorders and immunoexpression of androgen and estrogen-alpha receptors in men with benign prostatic hyperplasia and testosterone deficiency syndrome. Folia Histochem Cytobiol. 2015;53(3):227–235. | ||
Sayed RH, Saad MA, El-Sahar AE. Dapoxetine attenuates testosterone-induced prostatic hyperplasia in rats by the regulation of inflammatory and apoptotic proteins. Toxicol Appl Pharmacol. 2016;311:52–60. | ||
Yarrow JF, Conover CF, McCoy SC, et al. 17β-Hydroxyestra-4,9,11-trien-3-one (trenbolone) exhibits tissue selective anabolic activity: effects on muscle, bone, adiposity, hemoglobin, and prostate. Am J Physiol Endocrinol Metab. 2011;300(4):E650–E660. | ||
Windahl SH, Andersson N, Börjesson AE, et al. Reduced bone mass and muscle strength in male 5α-reductase type 1 inactivated mice. PLoS One. 2011;6(6):e21402. | ||
Roy AK, Lavrovsky Y, Song CS, et al. Regulation of androgen action. Vitam Horm. 1999;55:309–352. | ||
Hammond GL, Kontturi M, Vihko P, Vihko R. Serum steroids in normal males and patients with prostatic diseases. Clin Endocrinol (Oxf). 1978;9(2):113–121. | ||
Poirier D. Inhibitors of 17 beta-hydroxysteroid dehydrogenases. Curr Med Chem. 2003;10(6):453–477. | ||
Engel LL, Groman EV. Human placental 17beta-estradiol dehydrogenase: characterization and structural studies. Recent Prog Horm Res. 1974;30(0):139–169. | ||
Ryan KJ, Engel LL. The interconversion of estrone and estradiol by human tissue slices. Endocrinology. 1953;52(3):287–291. | ||
Hansson V, Tveter KJ, Attramadal A, Torgersen O. Androgenic receptors in human benign nodular prostatic hyperplasia. Eur J Endocrinol (Copenh). 1971;68(1):79–88. | ||
Snochowski M, Pousette A, Ekman P, et al. Characterization and measurement of the androgen receptor in human benign prostatic hyperplasia and prostatic hyperplasia and prostatic carcinoma. J Clin Endocrinol Metab. 1977;45(5):920–930. | ||
Cui J, Shen Y, Li R. Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends Mol Med. 2013;19(3):197–209. | ||
Carreau S, de Vienne C, Galeraud-Denis I. Aromatase and estrogens in man reproduction: a review and latest advances. Adv Med Sci. 2008;53(2):139–144. | ||
Kuiper GG, Carlsson B, Grandien K, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138(3):863–870. | ||
Morais-Santos M, Nunes AE, Oliveira AG, et al. Changes in estrogen receptor ERβ (ESR2) expression without changes in the estradiol levels in the prostate of aging rats. PLoS One. 2015;10(7):e0131901. | ||
Tsurusaki T, Aoki D, Kanetake H, et al. Zone-dependent expression of estrogen receptors alpha and beta in human benign prostatic hyperplasia. J Clin Endocrinol Metab. 2003;88(3):1333–1340. | ||
Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem. 2010;285(4):2676–2685. | ||
Chambliss KL, Shaul PW. Rapid activation of endothelial NO synthase by estrogen: evidence for a steroid receptor fast-action complex (SRFC) in caveolae. Steroids. 2002;67(6):413–419. | ||
Zhao C, Gao H, Liu Y, et al. Genome-wide mapping of estrogen receptor-beta-binding regions reveals extensive cross-talk with transcription factor activator protein-1. Cancer Res. 2010;70(12):5174–5183. | ||
Liu Y, Gao H, Marstrand TT, et al. The genome landscape of ERalpha- and ERbeta-binding DNA regions. Proc Natl Acad Sci U S A. 2008;105(7):2604–2609. | ||
Carroll JS, Meyer CA, Song J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38(11):1289–1297. | ||
Chimento A, Sirianni R, Delalande C, et al. 17 Beta-estradiol activates rapid signaling pathways involved in rat pachytene spermatocytes apoptosis through GPR30 and ER alpha. Mol Cell Endocrinol. 2010;320(1–2):136–144. | ||
McPherson SJ, Hussain S, Balanathan P, et al. Estrogen receptor- activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNF mediated. Proc Natl Acad Sci U S A. 2010;107(7):3123–3128. | ||
Zhang P, Hu WL, Cheng B, et al. Which play a more important role in the development of large-sized prostates (≥80 ml), androgen receptors or oestrogen receptors? A comparative study. Int Urol Nephrol. 2016;48(3):325–333. | ||
Fajer AB, Holzbauer M, Newport HM. The contribution of the adrenal gland to the total amount of progesterone produced in the female rat. J Physiol. 1971;214(1):115–126. | ||
Pelletier G, Li S, Luu-The V, Tremblay Y, Bélanger A, Labrie F. Immunoelectron microscopic localization of three key steroidogenic enzymes (cytochrome P450(scc), 3 beta-hydroxysteroid dehydrogenase and cytochrome P450(c17)) in rat adrenal cortex and gonads. J Endocrinol. 2001;171(2):373–383. | ||
Andersen ML, Tufik S. Does male sexual behavior require progesterone? Brain Res Rev. 2006;51(1):136–143. | ||
O’Malley BW, Sherman MR, Toft DO. Progesterone “receptors” in the cytoplasm and nucleus of chick oviduct target tissue. Proc Natl Acad Sci U S A. 1970;67(2):501–508. | ||
Chen R, Yu Y, Dong X. Progesterone receptor in the prostate: a potential suppressor for benign prostatic hyperplasia and prostate cancer. J Steroid Biochem Mol Biol. 2017;166:91–96. | ||
Luetjens CM, Didolkar A, Kliesch S, et al. Tissue expression of the nuclear progesterone receptor in male non-human primates and men. J Endocrinol. 2006;189(3):529–539. | ||
Wang DF, Minoura H, Sugiyama T, et al. Analysis on the promoter region of human decidual prolactin gene in the progesterone-induced decidualization and cAMP-induced decidualization of human endometrial stromal cells. Mol Cell Biochem. 2007;300(1–2):239–247. | ||
Kraus WL, Montano MM, Katzenellenbogen BS. Cloning of the rat progesterone receptor gene 5’-region and identification of two functionally distinct promoters. Mol Endocrinol. 1993;7(12):1603–1616. | ||
Vázquez-Martínez ER, Mendoza-Garcés L, Vergara-Castañeda E, Cerbón M. Epigenetic regulation of progesterone receptor isoforms: from classical models to the sexual brain. Mol Cell Endocrinol. 2014;392(1–2):115–124. | ||
Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277(7):5209–5218. | ||
Camacho-Arroyo I, González-Arenas A, González-Morán G. Ontogenic variations in the content and distribution of progesterone receptor isoforms in the reproductive tract and brain of chicks. Comp Biochem Physiol A Mol Integr Physiol. 2007;146(4):644–652. | ||
Asselin J, Labrie F, Gourdeau Y, Bonne C, Raynaud J-P. Binding of [3H] methyltrienolone (R 1881) in rat prostate and human benign prostatic hypertrophy (BPH). Steroids. 1976;28(4):449–459. | ||
Kastner P, Krust A, Turcotte B, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9(5):1603–1614. | ||
Petz LN, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002;143(12):4583–4591. | ||
Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004;88(2):113–122. | ||
Bonéy-Montoya J, Ziegler YS, Curtis CD, Montoya JA, Nardulli AM. Long-range transcriptional control of progesterone receptor gene expression. Mol Endocrinol. 2010;24(2):346–358. | ||
Won Jeong K, Chodankar R, Purcell DJ, Bittencourt D, Stallcup MR. Gene-specific patterns of coregulator requirements by estrogen receptor-α in breast cancer cells. Mol Endocrinol. 2012;26(6):955–966. | ||
Fleury L, Gerus M, Lavigne AC, Richard-Foy H, Bystricky K. Eliminating epigenetic barriers induces transient hormone-regulated gene expression in estrogen receptor negative breast cancer cells. Oncogene. 2008;27(29):4075–4085. | ||
Da Silva MHA, Costa WS, B Sampaio FJ, De Souza DB. The corpus cavernosum after treatment with dutasteride or finasteride: a histomorphometric study in a benign prostatic hyperplasia rodent model. Asian J Androl. 2018;20(5):505–510. | ||
Pinsky MR, Gur S, Tracey AJ, Harbin A, Hellstrom WJ. The effects of chronic 5-alpha-reductase inhibitor (dutasteride) treatment on rat erectile function. J Sex Med. 2011;8(11):3066–3074. | ||
Weng H, Li S, Huang JY, et al. Androgen receptor gene polymorphisms and risk of prostate cancer: a meta-analysis. Sci Rep. 2017;7(1):40554. | ||
Mäkelä S, Strauss L, Kuiper G, et al. Differential expression of estrogen receptors alpha and beta in adult rat accessory sex glands and lower urinary tract. Mol Cell Endocrinol. 2000;164(1–2):109–116. | ||
Pasquali D, Rossi V, Esposito D, et al. Loss of estrogen receptor beta expression in malignant human prostate cells in primary cultures and in prostate cancer tissues. J Clin Endocrinol Metab. 2001;86(5):2051–2055. | ||
Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. 2009;8(8):1168–1175. | ||
Pellegrino MJ, Stork PJ. Sustained activation of extracellular signal-regulated kinase by nerve growth factor regulates c-fos protein stabilization and transactivation in PC12 cells. J Neurochem. 2006;99(6):1480–1493. | ||
Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol. 2006;16(12):1171–1182. | ||
Pruitt K, Ulkü AS, Frantz K, et al. Ras-mediated loss of the pro-apoptotic response protein Par-4 is mediated by DNA hypermethylation through Raf-independent and Raf-dependent signaling cascades in epithelial cells. J Biol Chem. 2005;280(24):23363–23370. | ||
Lu R, Wang X, Chen ZF, Sun DF, Tian XQ, Fang JY. Inhibition of the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway decreases DNA methylation in colon cancer cells. J Biol Chem. 2007;282(16):12249–12259. | ||
Zhou W, Park I, Pins M, et al. Dual regulation of proliferation and growth arrest in prostatic stromal cells by transforming growth factor-beta1. Endocrinology. 2003;144(10):4280–4284. | ||
Mobbs BG, Johnson IE, Liu Y. Quantitation of cytosolic and nuclear estrogen and progesterone receptor in benign, untreated, and treated malignant human prostatic tissue by radioligand binding and enzyme-immunoassays. Prostate. 1990;16(3):235–244. | ||
Mobbs BG, Liu Y. Immunohistochemical localization of progesterone receptor in benign and malignant human prostate. Prostate. 1990;16(3):245–251. | ||
Bonkhoff H, Fixemer T, Hunsicker I, Remberger K. Progesterone receptor expression in human prostate cancer: correlation with tumor progression. Prostate. 2001;48(4):285–291. | ||
Yu Y, Liu L, Xie N, et al. Expression and function of the progesterone receptor in human prostate stroma provide novel insights to cell proliferation control. J Clin Endocrinol Metab. 2013;98(7):2887–2896. | ||
Yu Y, Lee JS, Xie N, et al. Prostate stromal cells express the progesterone receptor to control cancer cell mobility. PLoS One. 2014;9(3):e92714. | ||
Hill M, Bílek R, Safarík L, Stárka L. Analysis of relations between serum levels of epitestosterone, estradiol, testosterone, IGF-1 and prostatic specific antigen in men with benign prostatic hyperplasia and carcinoma of the prostate. Physiol Res. 2000;49(Suppl 1):S113–118. | ||
Pravettoni A, Mornati O, Martini PG, et al. Estrogen receptor beta (ERbeta) and inhibition of prostate cancer cell proliferation: studies on the possible mechanism of action in DU145 cells. Mol Cell Endocrinol. 2007;263(1–2):46–54. | ||
Yang GS, Wang Y, Wang P, Chen ZD. Expression of oestrogen receptor-alpha and oestrogen receptor-beta in prostate cancer. Chin Med J (Engl). 2007;120(18):1611–1615. | ||
Stanbrough M, Leav I, Kwan PWL, Bubley GJ, Balk SP. Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc Natl Acad Sci U S A. 2001;98(19):10823–10828. | ||
Pelletier G. Expression of steroidogenic enzymes and sex-steroid receptors in human prostate. Best Pract Res Clin Endocrinol Metab. 2008;22(2):223–228. | ||
Wen S, Chang HC, Tian J, Shang Z, Niu Y, Chang C. Stromal androgen receptor roles in the development of normal prostate, benign prostate hyperplasia, and prostate cancer. Am J Pathol. 2015;185(2):293–301. | ||
Felix-Patrício B, Miranda AF, Medeiros JL, et al. The prostate after castration and hormone replacement in a rat model: structural and ultrastructural analysis. Int Braz J Urol. 2017;43(5):957–965. | ||
Nieschlag E, Swerdloff R, Behre HM, et al. Investigation, treatment and monitoring of late-onset hypogonadism in males. ISA, ISSAM, and EAU recommendations. Eur Urol. 2005;48(1):1–4. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.