")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 5
Current and emerging treatment options for spinal muscular atrophy
Authors Farooq F, MacKenzie A
Received 26 April 2015
Accepted for publication 2 June 2015
Published 17 July 2015 Volume 2015:5 Pages 75—81
DOI https://doi.org/10.2147/DNND.S48420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Thomas Müller
Faraz Farooq,1,2 Alex E MacKenzie2,3
1Science Education Division, Emirates College for Advanced Education, Abu Dhabi, United Arab Emirates; 2Children's Hospital of Eastern Ontario (CHEO) Research Institute, Ottawa, ON, Canada; 3University of Ottawa, Ottawa, ON, Canada
Abstract: Spinal muscular atrophy is one of the most common inherited neuromuscular conditions; our understanding of the genetic pathology and translational research coming from this insight has made significant progress over the past decade. This short review provides the background of the disease along with the bench to bedside progress of some promising treatment options to develop better understanding of the present state of the disease.
Keywords: SMN protein, neurodegenerative disease, orphan disease, therapeutics
Introduction
Spinal muscle atrophy (SMA) is an autosomal recessive neurodegenerative disease and a leading global genetic cause of infant death.1 It is characterized by the loss of α motor neurons from the anterior horn of the spinal cord resulting in muscle weakness, trunk paralysis, and muscle atrophy.2 With an estimated prevalence of 1 in 6,000–11,000 live births and a carrier frequency of 1 in 40 to 1 in 60 among different ethnic groups and geographical location,3–7 SMA is the most common monogenic disease fatal to infants and one of the most common forms of neuromuscular disorder in childhood.
SMA is classified into three major groups based on the age of onset and severity of the disease;8–11 Type I SMA (Werdnig–Hoffmann disease) is the most prevalent and severe form of the disease with postnatal onset within the first six months. These patients are never able to sit and usually succumb within 2–5 years of age due to respiratory failure.11–13 Patients with the intermediary form or type II SMA develop muscle weakness within 6–18 months of age. Although these patients can sit, due to progressive muscle weakness, they can never stand or walk. Type III SMA (Kugelberg–Welander disease) has an onset between 18 months and 30 years of age with patients able to walk on their own (with some assistance).11,12,14,15 The diagnosis of SMA is typically established through physical examination, patient history, electromyography followed by confirmatory genetic testing.3 Muscle biopsy may also be done in some cases16 although less frequently since the advent of genetic testing.
The lack of functional survival motor neuron (SMN) protein due to deletion or mutation in the SMN1 gene is the cause of SMA.17 SMN is an evolutionary conserved RNA-associated protein required for cellular viability; complete loss of functional full-length SMN protein is therefore embryonically lethal.18–20 Humans due to an evolutionary recent duplication event at chromosome 5q17,21 possess a nearly identical SMN paralog, SMN2 (Figure 1) and thus uniquely among all species can survive loss of SMN1.
 | Figure 1 Human SMN locus on chromosome 5q showing inverted duplication of SMN1 and NAIP genes. |
Although, SMN2 has only few translationally insignificant nucleotide differences compared with SMN1, the C to T transition at position 6 of exon 7 results in the production of an alternative splice variant. Consequently, SMN2 produces only ~10% of the full-length functional SMN protein produced by SMN1; the remaining mRNA lacks exon 7 and is translated into a truncated, unstable non-functional protein called SMNΔ7 which, failing to oligomerize, is quickly degraded (Figure 2).22,23
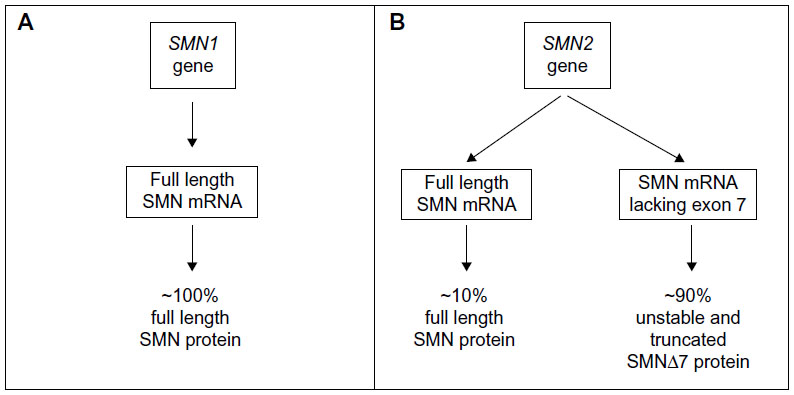 | Figure 2 Schematic diagram showing SMN1 (A) and SMN2 (B) genes along with their corresponding mRNA and protein products. |
Greater than 95% of all SMA patients have homozygous deletions of SMN1 gene.17 All SMA patients have at least one copy of SMN2, which produces low levels of functional SMN protein and acts as a disease modifier. There is an inverse correlation between SMN2 gene copy number and disease severity, ie, an increase in the SMN2 gene copy number decreases the SMA severity.24,25 Typically, SMA type I patients have one or two copies of SMN2 gene compared with two to three copies in type II, and three to four copies in types III and IV. Individuals with more than four copies of the SMN2 gene are completely asymptomatic notwithstanding the deletion of SMN1.
A central function of 294 amino acid long SMN protein is the assembly of small nuclear ribonucleic proteins (snRNPs) which are essential for splicing.26–35 With mutations in the ubiquitously expressed SMN shown to cause SMA, we are left with a question posed in this disease gene cloning era; how do mutations in a gene which is expressed everywhere (ie, SMN) mainly impact neurons (despite several recent studies suggesting a role for SMN in other tissue types as well36–44). Although the precise pathogenic molecular mechanism of SMA is not known, it is believed that the lack of motor neuron SMN protein may lead to a synaptopathy resulting in apoptotic death of motor neurons.20,45
Presently, only multidisciplinary supportive care including, critically, respiratory support is available for most children with SMA.10 Although these interventions have improved both life expectancy and quality of the life, an effective therapy for SMA is eagerly awaited by patients, their families, researchers, clinicians, and support care staff alike. Several approaches are currently being pursued as the following section outlines.
Therapeutic strategies for SMA
Gene therapy
Gene therapy is one of the most promising therapeutic advances for SMA. In the past 6 years, several groups have used self-complementary adeno-associated virus serotypes 8 and 9 carrying human SMN1 cDNA to treat mouse models of SMA. The most encouraging results (amelioration of disease phenotype and dramatic extension in the life span) were observed with pre-symptomatic treatment of SMA mice.46–49
These promising results helped scientists at Nationwide Children’s Hospital to receive a fast track designation from the US Food and Drug Administration (FDA) for ChariSMA™ (gene therapy product) to be tested as a SMA therapeutic. At the time of writing, a phase I clinical trial at Nationwide Children’s Hospital in collaboration with AveXis Inc. and The Sophia’s Cure Foundation is underway to evaluate safety and efficacy of gene transfer (systemic AAV9-delivered SMN1 gene) in SMA type I patients. The major challenges to bring this treatment into clinics are clinical safety, the cost of virus and the possibility of an immune response neutralizing AAV.50
SMN2-dependent therapies
Given the role of SMN2 as a modifier gene for SMA, the inverse correlation between the SMN2 gene copy number and disease severity, the gene itself has become a drug target. Strategies includes a) inducing the expression of SMN2, b) modulating splicing of SMN2-derived transcript, and c) stabilizing the full-length SMN2-derived mRNA and/or protein (Figure 3).51,52
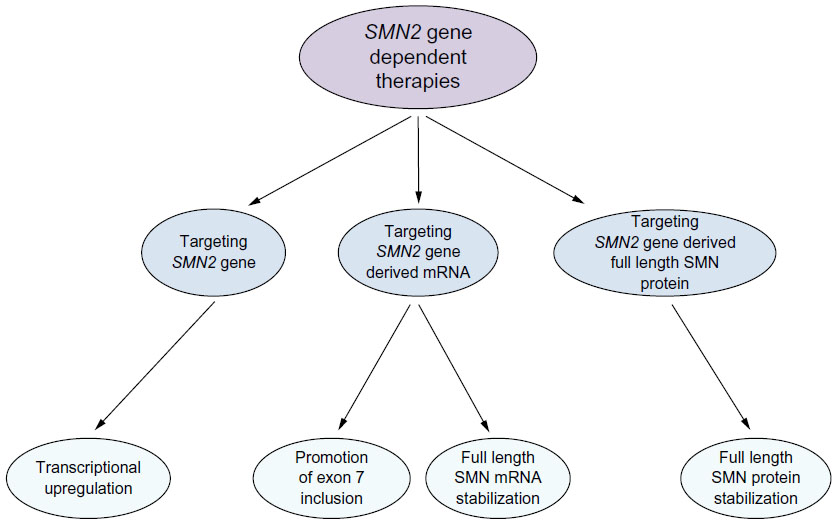 | Figure 3 Current SMN2 gene-derived therapeutic approaches to SMA. |
Activation of SMN2 promoter
Histone deacetylases (HDACs) through chromatin condensation are known to repress transcription of genes such as SMN2. Several HDAC inhibitors including sodium butyrate, valproic acid (VPA), and phenyl butyrate have been assessed for SMN2 induction in cellular and animal models of the disease as well as in clinical trials as potential therapeutic for SMA.53–57 Although they showed promise in cell culture and SMA mouse models, no significant clinical improvement has been observed in SMA patients with HDAC inhibitors.2,58,59
The STAT5 pathway has been implicated in the activation of SMN2 promoter.60–62 The human lactation hormone prolactin (PRL) and human growth hormone (HGH) have been shown to activate the STAT5 pathway which results in an increase in both SMN2 gene transcription and full-length SMN protein both in vitro and in vivo, resulting in attenuation of the SMA mouse model severity. PRL treatment resulted in significant survival and attenuation of disease phenotype in the SMA mouse model, a possible reflection of the significant SMN induction observed with PRL treatment in vivo.61 Although, PRL has been proven safe and was successfully tested in humans for the treatment of lactation-deficient mothers,63 the absence of clinical grade PRL is delaying its further assessment as a potential SMA therapeutic in the patient population. A Phase II trial in SMA type II/III patients showed no improvement in muscle function or strength after 3 months of treatment with HGH.64 However, it should be noted that only a single low dose of HGH was used and peripheral white blood cell SMN levels were not assessed in the patient population. Before crossing HGH off the list of SMA therapeutics, it may be beneficial to do a HGH dose escalation study especially in younger SMA patient population assessing muscle strength and monitoring changes in lymphocyte SMN levels.
The deCODE project initiated by Families of SMA (now cureSMA) identified C5-substituted quinazoline activity in increasing SMN2 promoter activity and thus SMN protein in cell-based assays. The DcpS inhibitor RG3039, a derivative of quinazolines, has since been tested and reported to mildly improve both survival and motor function in two mouse models of SMA through an increase in full-length SMN levels.65,66 The compound was initially out-licensed to Repligen Corporation (Waltham, MA, USA) which in 2012 out-licensed to Pfizer Pharmaceuticals (New York, NY, USA); however, the program has now been halted.
Correction of splicing
The significant majority of transcripts arising from SMN2 gene lack exon 7; agents that suppress exon 7 skipping have thus become the goal of a number of laboratories. Several small compounds including HDAC inhibitors such as VPA, TSA, and sodium butyrate have been assessed for their capacity to increase full-length SMN transcript by altering the splicing process in vitro.53–57,67 However, regardless of any putative effect observed in vitro, no beneficial effect has carried over to clinical trials.
Recently, RO6885247 from PTC Therapeutics (South Plainfield, NJ, USA) and Hoffmann-LaRoche (Nutley, NJ, USA) has been shown to profoundly affect the splicing of SMN2 gene to include more exon 7 showing a dramatic positive impact on SMA model mice.68 A Phase I clinical trial is currently underway to assess the safety profile along with tolerability of this compound.
Antisense oligos (ASOs) are increasingly being used for therapeutic/experimental purposes to treat a number of diseases including SMA, duchenne muscular dystrophy, and myotonic dystrophy.41,69–72 A bifunctional ASO complementary to SMN2 exon 7 pre-mRNA sequences has been designed and tested to promote the inclusion of exon 7 and at the same time inhibit binding of negative splicing factors which ultimately results into increased production of full-length SMN mRNA.41,71
ASOs are not blood brain barrier (BBB) penetrant, which initially posed a hurdle in their use as a SMA therapeutics.73 Several reports have shown that intrathecal injections of ASOs results into improvement in survival and disease phenotype in SMA mice.71 However, in 2011, Hua et al reported an increase in SMN levels along with attenuation of SMA phenotype in SMA mice with systemic delivery of ASO.41 ISIS pharmaceutical has successfully completed early phase trials to show safety, tolerability, and pharmacokinetics of intrathecal ISIS-SMN Rx (ASOs) in SMA patients. A Phase III has been initiated to test the safety and efficacy of intrathecal administration of ISIS-SMN Rx in patients with infantile-onset and later onset of SMA. This represents a significant breakthrough in the field of SMA as this is the first compound which has reached Phase III clinical trial. The results from these trials are eagerly awaited by the SMA community.
Full-length SMN transcript and protein stabilization
In addition to the upregulation of SMN2 transcription and the modulation of splicing, the stabilization of either mRNA or protein is an often overlooked but nonetheless viable potential therapeutic approach. In this regard, SMN mRNA has a specific AU-rich element region in its 3′-untranslated region which marks the mRNA for degradation.74,75 In silico mining of gene expression data sets was used to identify the p38 pathway as a means of enhancing SMN2 levels. In the initial study, it was shown that treatment of neuronal cells with the p38 activator anisomycin results in the translocation of protein HuR to the cytoplasm where it binds the 3′-untranslated region of SMN mRNA and stabilizes the transcript inhibiting its degradation which in turn increases the SMN protein level.74 Since then several p38-activating compounds have been identified and reported to increase the SMN protein level in vitro as well as in vivo.76,77
Celecoxib, a safe, well-tolerated prescription medication used widely for arthritis and in some pediatric rheumatologic diseases, was identified as a BBB penetrant, p38-activating compound.78,79 Treatment with low dose of celecoxib increased SMN protein levels in both human and mouse nerve cell cultures, as well as in patient fibroblasts. In a severe SMA mouse model, mice treated with celecoxib showed an increase in the SMN protein in central nervous system (CNS) tissues. In addition, SMA mice also showed an improved motor function and a statistically significant 40% extension of survival as compared to mice treated with placebo.76 A Phase I/II clinical trial is planned for this FDA approved agent in 2015; it may be with the hope that celecoxib may serve as an adjunctive therapy for SMA, particularly given the low safe doses are required for SMN induction.
Aminoglycoside antibiotics, such as tobramycin and amikacin which have been shown to mask premature stop codon mutations in some genes, have been used to increase SMN protein levels in patient fibroblasts. However, there efficacy and safety has yet to be tested successfully in animal models of the disease.80–82 An alternative potential therapeutic approach involves targeting the ubiquitin–proteasome pathway which targets many proteins including SMN for degradation. The FDA-approved proteasome inhibitor bortezomib has been shown to increase SMN levels both in vitro and in vivo.83 However, the major obstacle to the use of bortezomib as a therapeutic is its inability to cross the BBB.
Finally, through the experience gained from treatments in animal models of the disease, it is the general consensus that, as with most disorders, early timing of the treatment is critical for maximum benefit; optimally, this would involve pre-symptomatic identification of infants with SMA. Newborn screening is therefore an important step in the most effective use of novel therapies of SMA allowing intervention before the clinical course is set.51,84,85 Hopefully through early intervention along with the promising therapeutic candidates described in Table 1, there will be an effective therapy and possibly a cure for SMA in the near future.
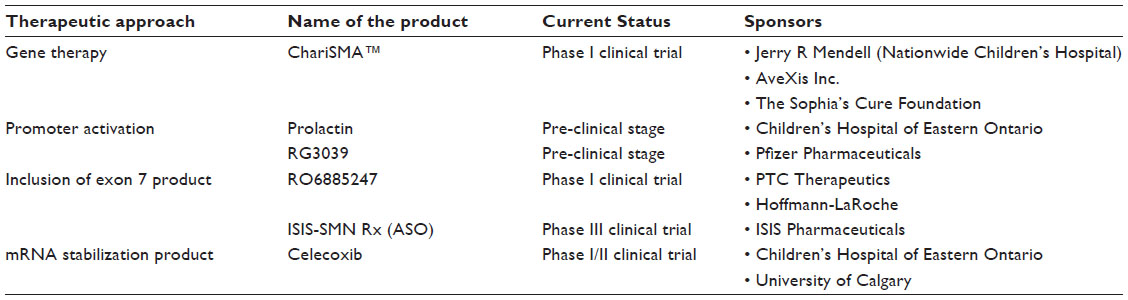 | Table 1 Current therapeutic pipeline for SMA |
Disclosure
The authors report no conflicts of interest in this work.
References
Roberts DF, Chavez J, Court SD. The genetic component in child mortality. Arch Dis Child. 1970;45(239):33–38. | |
Mercuri E, Bertini E, Messina S, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007;68(1):51–55. | |
Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–368. | |
Ogino S, Leonard DG, Rennert H, Ewens WJ, Wilson RB. Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. 2002;110(4):301–307. | |
Ogino S, Wilson RB, Gold B. New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. Eur J Hum Genet. 2004;12(12):1015–1023. | |
Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;15(6):409–413. | |
Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Human Genet. 2012;20(1):27–32. | |
Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromusc Disord. 1995;5(1):3–5. | |
Iannaccone ST, Russman BS, Browne RH, Buncher CR, White M, Samaha FJ. Prospective analysis of strength in spinal muscular atrophy. DCN/Spinal Muscular Atrophy Group. J Child Neurol. 2000;15(2):97–101. | |
Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007; 22(8):1027–1049. | |
Zerres K, Davies KE. 59th ENMC international workshop: spinal muscular atrophies: recent progress and revised diagnostic criteria 17–19 April 1998, Soestduinen, The Netherlands. Neuromusc Disord. 1999; 9(4):272–278. | |
O’Hagen JM, Glanzman AM, McDermott MP, et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromusc Disord. 2007;17(9–10):693–697. | |
Thomas NH, Dubowitz V. The natural history of type I (severe) spinal muscular atrophy. Neuromusc Disord. 1994;4(5–6):497–502. | |
Zerres K, Rudnik-Schoneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67–72. | |
Zerres K, Wirth B, Rudnik-Schoneborn S. Spinal muscular atrophy – clinical and genetic correlations. Neuromusc Disord. 1997; 7(3):202–207. | |
Prior TW, Russman BS. Spinal muscular atrophy. In: Pagon RA, AdamMP, Ardinger HH, et al, editors. GeneReviews®. Seattle, WA: University of Washington; 1993. | |
Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995; 80(1):155–165. | |
Kariya S, Mauricio R, Dai Y, Monani UR. The neuroprotective factor Wld(s) fails to mitigate distal axonal and neuromuscular junction (NMJ) defects in mouse models of spinal muscular atrophy. Neurosci Lett. 2009;449(3):246–251. | |
Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94(18):9920–9925. | |
Simic G. Pathogenesis of proximal autosomal recessive spinal muscular atrophy. Acta Neuropathol. 2008;116(3):223–234. | |
Mahadevan MS, Korneluk RG, Roy N, MacKenzie A, Ikeda J. SMA genes: deleted and duplicated. Nature Genet. 1995;9(2):112–113. | |
Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–6311. | |
Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–1183. | |
Campbell L, Potter A, Ignatius J, Dubowitz V, Davies K. Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am J Hum Genet. 1997;61(1):40–50. | |
Harada Y, Sutomo R, Sadewa AH, et al. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol. 2002;249(9):1211–1219. | |
Buhler D, Raker V, Luhrmann R, Fischer U. Essential role for the tudor domain of SMN in spliceosomal U snRNP assembly: implications for spinal muscular atrophy. Hum Mol Genet. 1999;8(13):2351–2357. | |
Fischer U, Liu Q, Dreyfuss G. The SMN–SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90(6):1023–1029. | |
Friesen WJ, Dreyfuss G. Specific sequences of the Sm and Sm-like (Lsm) proteins mediate their interaction with the spinal muscular atrophy disease gene product (SMN). J Biol Chem. 2000;275(34):26370–26375. | |
Jones KW, Gorzynski K, Hales CM, et al. Direct interaction of the spinal muscular atrophy disease protein SMN with the small nucleolar RNA-associated protein fibrillarin. J Biol Chem. 2001;276(42):38645–38651. | |
Meister G, Buhler D, Laggerbauer B, Zobawa M, Lottspeich F, Fischer U. Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum Mol Genet. 2000;9(13):1977–1986. | |
Mourelatos Z, Abel L, Yong J, Kataoka N, Dreyfuss G. SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J. 2001;20(19):5443–5452. | |
Narayanan U, Achsel T, Luhrmann R, Matera AG. Coupled in vitro import of U snRNPs and SMN, the spinal muscular atrophy protein. Mol Cell. 2004;16(2):223–234. | |
Pellizzoni L, Baccon J, Charroux B, Dreyfuss G. The survival of motor neurons (SMN) protein interacts with the snoRNP proteins fibrillarin and GAR1. Curr Biol. 2001;11(14):1079–1088. | |
Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci U S A. 1999;96(20):11167–11172. | |
Pellizzoni L, Charroux B, Rappsilber J, Mann M, Dreyfuss G. A functional interaction between the survival motor neuron complex and RNA polymerase II. J Cell Biol. 2001;152(1):75–85. | |
Araujo Ade Q, Araujo M, Swoboda KJ. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J Pediat. 2009;155(2):292–294. | |
Bevan AK, Hutchinson KR, Foust KD, et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19(20):3895–3905. | |
Bowerman M, Swoboda KJ, Michalski JP, et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol. 2012;72(2):256–268. | |
Gogliotti RG, Quinlan KA, Barlow CB, Heier CR, Heckman CJ, Didonato CJ. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci. 2012;32(11):3818–3829. | |
Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19(20):3906–3918. | |
Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478(7367):123–126. | |
Martinez TL, Kong L, Wang X, et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J Neurosci. 2012;32(25):8703–8715. | |
Rudnik-Schoneborn S, Vogelgesang S, Armbrust S, Graul-Neumann L, Fusch C, Zerres K. Digital necroses and vascular thrombosis in severe spinal muscular atrophy. Muscle Nerve. 2010;42(1):144–147. | |
Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19(20):4059–4071. | |
Kariya S, Park GH, Maeno-Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17(16):2552–2569. | |
Dominguez E, Marais T, Chatauret N, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2010;20(4):681–693. | |
Foust KD, Wang X, McGovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nature Biotechnol. 2010;28(3):271–274. | |
Passini MA, Bu J, Roskelley EM, et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120(4):1253–1264. | |
Valori CF, Ning K, Wyles M, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2(35):35ra42. | |
MacKenzie A. Genetic therapy for spinal muscular atrophy. Nature Biotechnol. 2010;28(3):235–237. | |
Farooq FT, Holcik M, MacKenzie A. Spinal muscular atrophy: classification, diagnosis, background, molecular mechanism and development of therapeutics. In: Kishore DU, editor. Neurodegenerative Diseases. Rijeka: InTech; 2013:561–579. | |
Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19(R1):R111–R118. | |
Andreassi C, Angelozzi C, Tiziano FD, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12(1):59–65. | |
Brichta L, Hofmann Y, Hahnen E, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12(19):2481–2489. | |
Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci U S A. 2001;98(17):9808–9813. | |
Sumner CJ, Huynh TN, Markowitz JA, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003; 54(5):647–654. | |
Tsai LK, Tsai MS, Ting CH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med (Berlin, Germany). 2008;86(11):1243–1254. | |
Garbes L, Riessland M, Holker I, et al. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum Mol Genet. 2009;18(19):3645–3658. | |
Swoboda KJ, Scott CB, Reyna SP, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PloS one. 2009;4(5):e5268. | |
MacKenzi D,Shamim F, Mongeon K, Trivedi A, MacKenzie A, Farooq F. Human growth hormone increases SMN expression and survival in severe spinal muscular atrophy mouse model. J Neuromusc Dis. 2014;1(1):65–74. | |
Farooq F, Molina FA, Hadwen J, et al. Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway. J Clin Invest. 2011;121(8):3042–3050. | |
Ting CH, Lin CW, Wen SL, Hsieh-Li HM, Li H. Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum Mol Genet. 2007;16(5):499–514. | |
Powe CE, Allen M, Puopolo KM, et al. Recombinant human prolactin for the treatment of lactation insufficiency. Clin Endocrinol (Oxford). 2010;73(5):645–653. | |
Kirschner J, Schorling D, Hauschke D, et al. Somatropin treatment of spinal muscular atrophy: a placebo-controlled, double-blind crossover pilot study. Neuromuscul Disord. 2014;24(2):134–142. | |
Gogliotti RG, Cardona H, Singh J, et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum Mol Genet. 2013;22(20):4084–4101. | |
Van Meerbeke JP, Gibbs RM, Plasterer HL, et al. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum Mol Genet. 2013;22(20):4074–4083. | |
Avila AM, Burnett BG, Taye AA, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117(3):659–671. | |
Naryshkin NA, Weetall M, Dakka A, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345(6197):688–693. | |
Wheeler TM, Leger AJ, Pandey SK, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488(7409):111–115. | |
Aoki Y, Yokota T, Wood MJ. Development of multiexon skipping antisense oligonucleotide therapy for Duchenne muscular dystrophy. Biomed Res Int. 2013;2013:402369. | |
Mitrpant C, Porensky P, Zhou H, et al. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: towards a treatment for spinal muscular atrophy. PLoS One. 2013;8(4):e62114. | |
Porensky PN, Burghes AH. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Hum Gene Ther. 2013;24(5):489–498. | |
MacKenzie A. Sense in antisense therapy for spinal muscular atrophy. New Engl J Med. 2012;366(8):761–763. | |
Farooq F, Balabanian S, Liu X, Holcik M, MacKenzie A. p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum Mol Genet. 2009;18(21):4035–4045. | |
Frevel MA, Bakheet T, Silva AM, Hissong JG, Khabar KS, Williams BR. p38 Mitogen-activated protein kinase-dependent and -independent signaling of mRNA stability of AU-rich element-containing transcripts. Mol Cell Biol. 2003;23(2):425–436. | |
Farooq F, Abadia-Molina F, MacKenzie D, et al. Celecoxib increases SMN and survival in a severe spinal muscular atrophy mouse model via p38 pathway activation. Hum Mol Genet. 2013;22(17):3415–3424. | |
Hadwen J, MacKenzie D, Shamim F, et al. VPAC2 receptor agonist BAY 55-9837 increases SMN protein levels and moderates disease phenotype in severe spinal muscular atrophy mouse models. Orphanet J Rare Dis. 2014;9:4. | |
Hsiao PW, Chang CC, Liu HF, Tsai CM, Chiu TH, Chao JI. Activation of p38 mitogen-activated protein kinase by celecoxib oppositely regulates survivin and gamma-H2AX in human colorectal cancer cells. Toxicol Appl Pharmacol. 2007;222(1):97–104. | |
Steffel J, Akhmedov A, Fahndrich C, Ruschitzka F, Luscher TF, Tanner FC. Differential effect of celecoxib on tissue factor expression in human endothelial and vascular smooth muscle cells. Biochem Biophys Res Commun. 2006;349(2):597–603. | |
Heier CR, DiDonato CJ. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor functio in SMA mice in vivo. Hum Mol Genet. 2009; 18(7):1310–1322. | |
Mattis VB, Rai R, Wang J, Chang CW, Coady T, Lorson CL. Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum Genet. 2006;120(4):589–601. | |
Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL. A non-sequence-specific requirement for SMN protein activity: the role of aminoglycosides in inducing elevated SMN protein levels. Hum Mol Genet. 2005;14(9):1199–1210. | |
Kwon DY, Motley WW, Fischbeck KH, Burnett BG. Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum Mol Genet. 2011; 20(18):3667–3677. | |
Arnold WD, Burghes AH. Spinal muscular atrophy: development and implementation of potential treatments. Ann Neurol. 2013;74(3):348–362. | |
Kayadjanian N, Burghes A, Finkel RS, et al. SMA-EUROPE workshop report: opportunities and challenges in developing clinical trials for spinal muscular atrophy in Europe. Orphanet J Rare Dis. 2013;8:44. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.