")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Curcumin could reduce the monomer of TTR with Tyr114Cys mutation via autophagy in cell model of familial amyloid polyneuropathy
Authors Li H, Zhang Y, Cao L, Xiong R, Zhang B , Wu L, Zhao Z, Chen S
Received 9 July 2014
Accepted for publication 16 August 2014
Published 31 October 2014 Volume 2014:8 Pages 2121—2128
DOI https://doi.org/10.2147/DDDT.S70866
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Hui Li,1,* Yu Zhang,1,* Li Cao,1 Ran Xiong,1 Bei Zhang,1 Li Wu,1 Zongbo Zhao,1 Sheng-Di Chen1,2
1Department of Neurology and Institute of Neurology, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, 2Key Laboratory of Stem Cell Biology and Laboratory of Neurodegenerative Diseases, Institute of Health Science, Shanghai Institutes of Biological Sciences, Chinese Academy of Science, and Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Transthyretin (TTR) familial amyloid polyneuropathy (FAP) is an autosomal dominant inherited neurodegenerative disorder caused by various mutations in the transthyretin gene. We aimed to identify the mechanisms underlying TTR FAP with Tyr114Cys (Y114C) mutation. Our study showed that TTR Y114C mutation led to an increase in monomeric TTR and impaired autophagy. Treatment with curcumin resulted in a significant decrease of monomeric TTR by recovering autophagy. Our research suggests that impairment of autophagy might be involved in the pathogenesis of TTR FAP with Y114C mutation, and curcumin might be a potential therapeutic approach for TTR FAP.
Keywords: curcumin, familial amyloid polyneuropathy, transthyretin, autophagy
Introduction
Transthyretin (TTR) familial amyloid polyneuropathy (FAP) is an autosomal dominant inherited disease, characterized clinically by progressive sensory, motor, and autonomic impairment, which typically lead to death around a decade after diagnosis.1 Since the first identification of TTR with Val30Met mutation (TTR V30M), the most common gene mutation in FAP patients, more than 100 TTR mutations have been found to cause FAP.2 However, the detailed pathogenesis underlying TTR FAP remains undefined. Previous studies of the TTR V30M mutant have shown that misfolding and self-aggregation of TTR are implicated in the pathogenesis of TTR FAP involving abnormal endoplasmic reticulum (ER) stress.3
Corresponding to the various TTR gene mutations and a wide range of geographical distributions, FAP presents diverse characteristics in genotype-phenotype in different regions. We have recently published the first report of a TTR Tyr114Cys (TTR Y114C) mutation in a Chinese family with TTR FAP.4 Compared with TTR V30M, the TTR Y114C mutation showed different clinical manifestations, and was also observed in a Japanese family.5,6 This suggests that the pathogenesis of the TTR Y114C and TTR V30M mutations might be different. Studies focused on monomer generation and tetramer depolymerization have been performed.1,2 However, the mechanisms underlying the clearing of the abnormally increased monomer are unknown.
Autophagy is the major lysosomal pathway via which cells degrade intracytoplasmic protein. It is widely accepted that autophagy plays a key role in the process of amyloid deposition in certain neurodegenerative diseases, including alpha-synuclein, beta peptides, tau oligomers, and misfolded prion protein.7 Therefore, autophagy may be involved in degradation of the TTR monomer in TTR FAP.
Curcumin and its analogs have demonstrated a protective effect in many diseases involving antimicrobial, antitubercular,8 and anticancer mechanisms,9 and they can also modulate innate immunity.10 Of note, curcumin has been shown to promote autophagy.11 Therefore, we hypothesized that autophagy might be involved in the pathogenetic mechanism of the TTR Y114C mutation in TTR FAP and curcumin might have potential therapeutic role in this disease. In this study, we aimed to identify the role of autophagy in the pathogenetic mechanism of TTR FAP and to assess the therapeutic effect of curcumin in the disease.
Materials and methods
Plasmid construction, cell culture, and stable cell line generation
Human TTR complementary DNA was amplified from the human fetal brain complementary DNA library (Invitrogen, Carlsbad, CA, USA) and ligated to the pcDNA3.0 vector with a C terminal Myc tag. TTR Y114C and TTR V30M were generated using a site-directed mutagenesis kit (Qiagen, Hilden, Germany). Human embryonic kidney (HEK) 293T cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin. TTR Y114C and TTR V30M were transfected into HEK293T cells and stable clones were selected with 500 μg/mL G418.
Cell viability assay
The cytotoxic effect of curcumin was determined using the cell proliferation reagent 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich). In brief, HEK293T cells were seeded at 2×104 cells/well in 96-well flat-bottomed plates and incubated at 37°C overnight. The cells were then treated with 0, 2.5, 5.0, 7.5, 10, 15, 20, or 25 μM curcumin for 24 hours. Next, they were exposed to 10 μL of the MTT (0.5 mg/mL) reagent for 4 hours at 37°C. Absorbance was measured at 570 nm using a microplate reader. All experiments were performed in triplicate.
Antibodies and reagents
The following antibodies were used: anti-LC3 rabbit (Novus Biologicals, San Diego, CA, USA), Myc-Tag (19C2) mouse monoclonal antibody (Abmart, Shanghai, People’s Republic of China), BiP antibody (Cell Signaling Technology, Danvers, MA, USA), eukaryotic initiation factor 2 alpha (eIF2α) antibody (Cell Signaling Technology), phospho-eIF2α (Ser51) antibody (p-eIF2α) (Cell Signaling Technology), and mouse monoclonal anti-beta-actin antibody (Sigma-Aldrich). Curcumin with purity greater than 95% was obtained from Sigma and dissolved in dimethyl sulfoxide. Fresh stock solutions of curcumin were prepared in dimethyl sulfoxide and diluted to stock concentrations with 10 mM and used immediately. All the curcumin solutions were kept in the dark to avoid exposure to light. 3-Methyladenine (3-MA), a phosphatidylinositol 3-phosphate kinase inhibitor, was purchased from Sigma and dissolved in dimethyl sulfoxide to produce a 300 mM stock solution.
Western blotting
Cells were plated in six-well plates at a density of 2×106 cells per well for 36 hours prior to treatment with 5 μM curcumin or 1 mM 3-MA. Equal amounts of the supernatant from each sample were then mixed with a native sample buffer (0.05 MTris-HCl at pH 6.8, 10% glycerol) using the method described by Tojo et al.12 The supernatant was mixed with non-natured buffer (50 mM Tris⋅HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate), with protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA) and 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich). The cells were lysed in RIPA buffer (50 mM Tris⋅HClpH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) with protease inhibitor cocktail and 1 mM phenylmethylsulfonyl fluoride for 30 minutes on ice. After centrifugation at 14,000× g for 30 minutes, the protein concentration was assayed using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). The samples were detected by immunoblotting. Band intensities were quantified by densitometric analyses using NIH ImageJ software. For all the Western blot results, at least three independent experiments were done and the most representative result was shown.
Immunocytochemistry assay
Cells were seeded on poly-L-lysine-coated glass coverslips, fixed with 4% paraformaldehyde for 20 minutes, permeabilized with 0.2% Triton X-100 for 15 minutes, blocked with 3% bovine serum albumin in phosphate-buffered saline and 0.2% Tween 20 for 1 hour, and then incubated with primary antibodies against LC3-II overnight at 4°C, followed by washing in phosphate-buffered saline and 0.2% Tween 20 three times for 10 minutes each. An Alexa Fluor 488 anti-rabbit immunoglobulin G tagged secondary antibody (Invitrogen) was added. After washing three times using phosphate-buffered saline, anti-fade mounting medium (Vector Laboratories Inc., Burlingame, CA, USA) was added and the stained cells were analyzed using a phase contrast fluorescence microscope (Eclipse E400; Nikon, Tokyo, Japan).
Statistical analysis
The statistical analysis was carried out using Statistical Package for the Social Sciences version 16.0 software (SPSS Inc., Chicago, IL, USA). Quantitative data were expressed as the mean ± standard deviation and compared using one-way analysis of variance. Statistical significance was set at a P-value less than 0.05. All analyses were performed blind to the experimental conditions.
Results
TTR Y114C mutation led to increased monomeric TTR and impaired autophagy in vitro
To investigate the alteration of monomeric TTR with different mutations, we generated HEK293T cell lines with wild-type TTR, TTR Y114C, and stable overexpression of TTR V30M. Wild-type TTR represented the normal control and TTR V30M represented the positive control. Western blotting analysis of the TTR level in the cells when cultured for 24 hours showed that the monomer of TTR Y114C and TTR V30M was increased by approximately 2.3 times and 2.78 times, respectively, compared with wild-type TTR (Figure 1A and B). Mutation of TTR Y114C was related to the increase in monomeric TTR, as well as the mutation of TTR V30M.
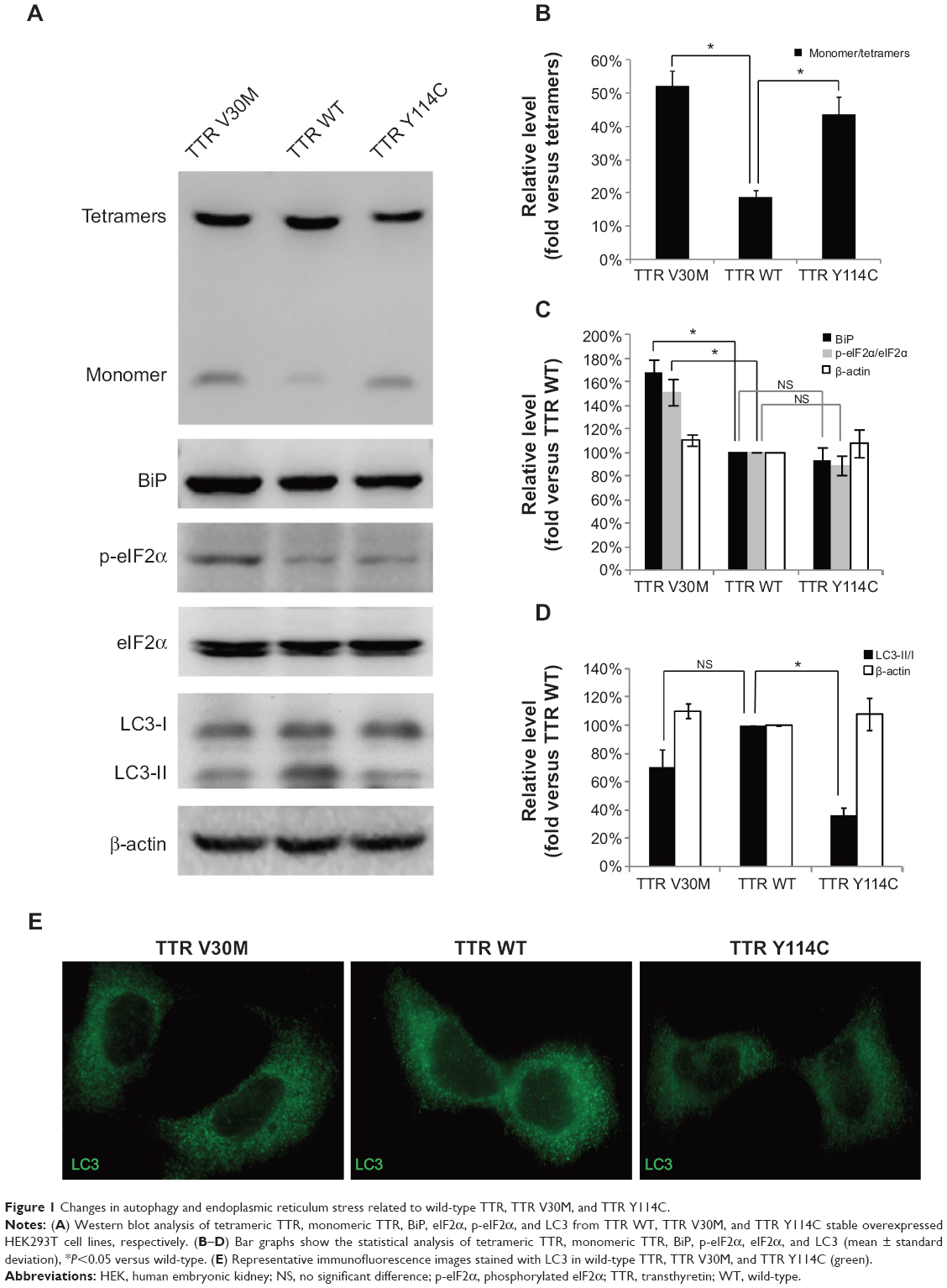 | Figure 1 Changes in autophagy and endoplasmic reticulum stress related to wild-type TTR, TTR V30M, and TTR Y114C. |
Next we investigated the activation of several markers associated with ER stress, including ER-resident chaperone BiP and p-eIF2α. Our results showed the levels of BiP and p-eIF2α is higher in TTR V30M than those in wild-type TTR. In contrast, BiP and p-eIF2α levels in TTR Y114C were similar to those in wild-type TTR (Figure 1A and C), indicating ER stress might not be the main pathogenetic mechanism for the TTR Y114C mutation. We then investigated whether autophagy plays a role in the mechanism of TTR Y114C mutation. LC3-II is well known to be a robust marker of autophagosomes, and immunofluorescent staining of LC3-II can be used to assay for autophagosome formation. A high ratio of LC3-II to LC3-I would indicate induction of autophagy. Our results revealed that the ratio of LC3-II/I was markedly decreased for TTR Y114C, but less suppressed for TTR V30M (Figure 1A and D).Likewise, a significant decrease in LC3-II immunoreactivity was detected in TTR Y114C (Figure 1E). The results of Western blotting and immunofluorescence indicated that autophagy in TTR Y114C was significantly downregulated. Therefore, impaired autophagy might be responsible for the pathogenesis of TTR Y114C mutation.
Curcumin decreased monomeric TTR by promoting autophagy
The effects of curcumin were investigated in TTR Y114C and wild-type TTR stable overexpressed HEK293T cells. Curcumin did not show toxic effects in the stable overexpressed cell lines at curcumin concentrations below 10 μM (Figure 2A and B). We chose 5 μM as the experimental concentration, because it is the minimal effective concentration of curcumin in these cell lines. Further, we wanted to determine whether curcumin could decrease monomeric TTR by promoting autophagy at the minimal effective concentration. Therefore, we used curcumin (2.5 μM and 5 μM) as a protective agent to assess whether it could decrease monomeric TTR with mutation by promoting autophagy. Quantification of LC3-II and LC3-I indicated markedly higher activation of LC3 in TTR Y114C treated with curcumin 5 μM for 24 hours (Figure 2D). In contrast, treatment with curcumin at different concentrations could not activate LC3 in wild-type TTR (Figure 2C, E). We next examined the ratio of monomers to tetramers in TTR Y114C, which was significantly decreased after 24 hours of treatment with 5 μM curcumin compared with no treatment with curcumin (Figure 2D and F). However, for wild-type TTR, the ratio of monomers to tetramers was unchanged after treatment with curcumin (Figure 2C and E). These results indicate that treatment with curcumin 5 μM for 24 hours was able to decrease the monomer in the TTR Y114C mutation by promoting autophagy.
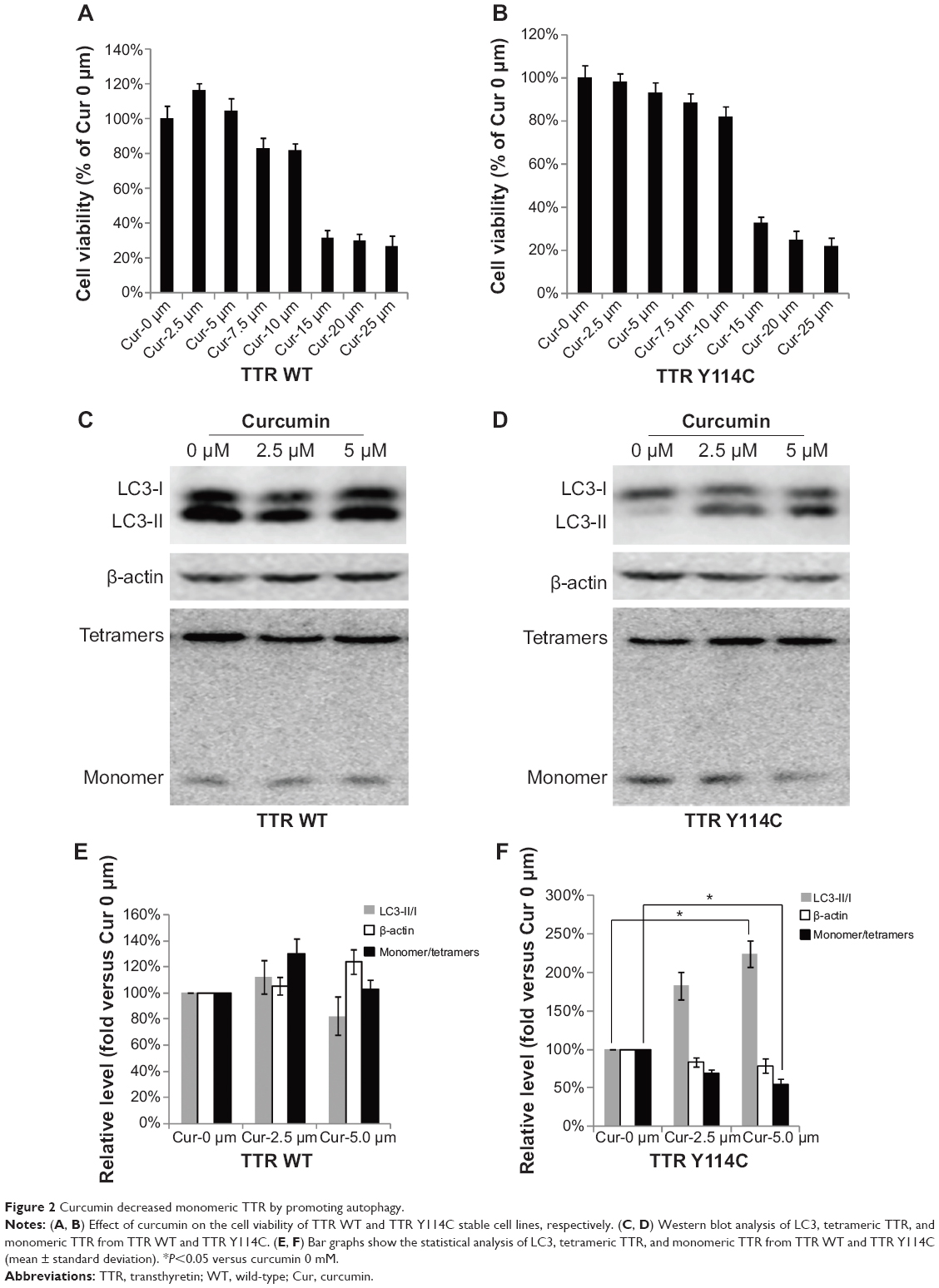 | Figure 2 Curcumin decreased monomeric TTR by promoting autophagy. |
Protective effect of curcumin on TTR Y114C could be partially blocked by 3-MA
To further validate whether the decrease in monomer by curcumin in our experiments was mediated by autophagy, 3-MA, an inhibitor of autophagosome formation, was implied to negatively regulate autophagy. 3-MA (1 mM) was added to the cell culture medium 2 hours before curcumin and incubated for 24 hours. Analysis of LC3, tetrameric TTR, and monomeric TTR from TTR Y114C revealed that 3-MA partly reversed the LC3 II activation induced by curcumin and increased the monomer of TTR Y114C (Figure 3). These results confirm that curcumin induced the decrease in the TTR Y114C monomer by promoting the autophagy pathway.
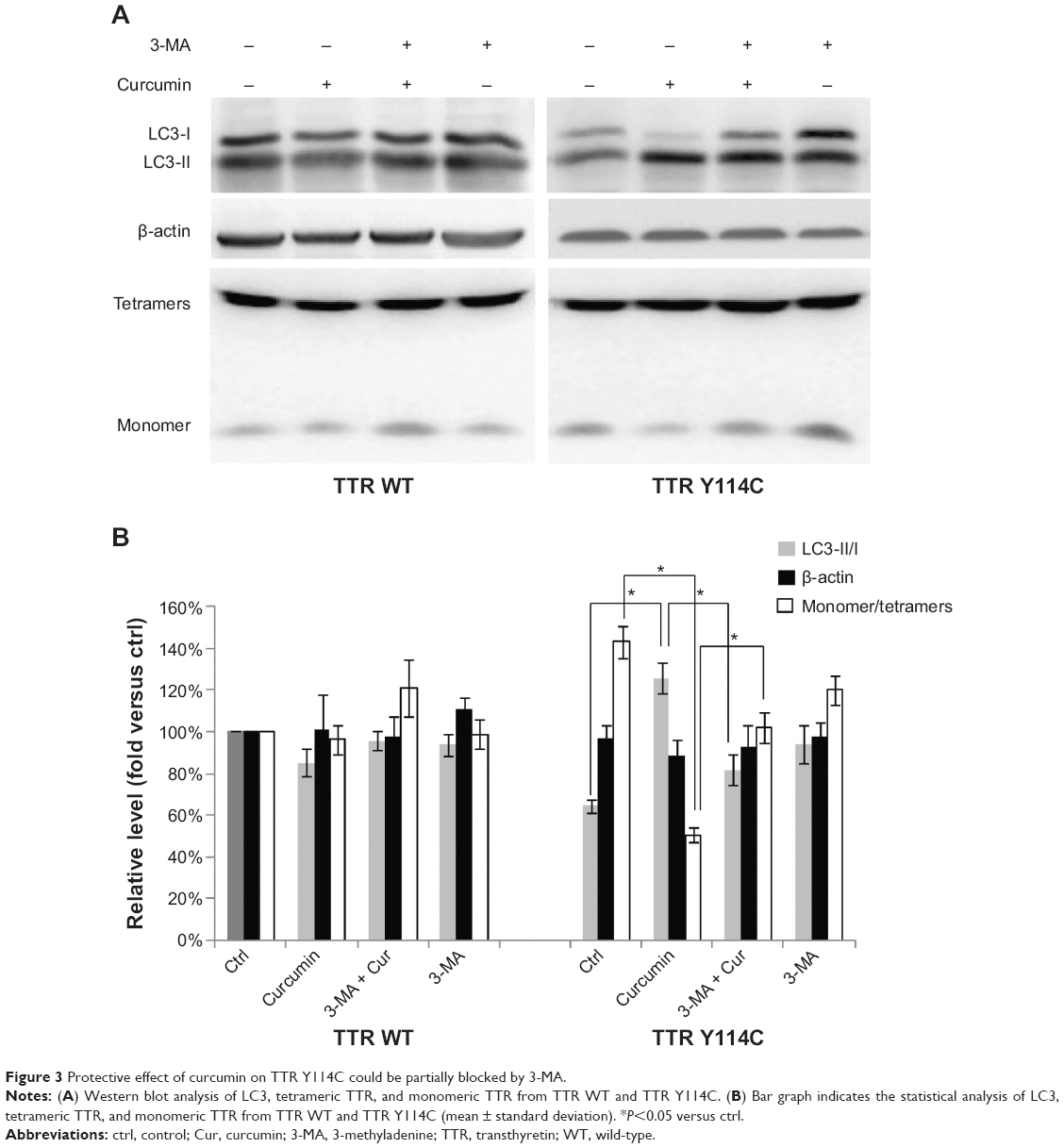 | Figure 3 Protective effect of curcumin on TTR Y114C could be partially blocked by 3-MA. |
Discussion
TTR FAP is a severe autosomal dominant inherited disease, for which the treatment options are limited. Liver transplantation performed early in the course of the disease is the only therapeutic strategy known to stabilize this neuropathy.1,13 More recently, tafamidis meglumine, a potent inhibitor of misfolding and deposition of mutated TTR, has completed an 18-month, placebo-controlled Phase II/III clinical trial for the treatment of FAP.14 However, in June 2012, the US Food and Drug Administration Peripheral and Central Nervous System Drugs Advisory Committee rejected this drug, stating a lack of convincing data supporting its efficacy.15 Hence, it is important to identify the pathogenetic mechanism of FAP to find an alternative effective treatment strategy.
Accumulating studies focused on the TTR mutation gene and protein have provided insights into the pathogenesis of TTR FAP, including decreased stability of TTR tetramers, conformational change in the crystal structure of variant TTR, altered kinetics of denaturation, and disturbing endoplasmic ER quality control system.1,16–18 Previous studies have demonstrated that increased levels of ER stress are correlated with extracellular TTR deposition. Two ER stress markers, BiP and p-eIF2α, have been observed to be present and upregulated in the salivary gland tissue of FAP patients.3 However, the precise molecular mechanisms underlying TTR FAP and its phenotypic heterogeneity are not yet fully understood.
Our current study investigated whether the two mutations, TTR Y114C and TTR V30M, share the same pathogenesis and evaluated the effect of pathogenic mutations on the clearance of the monomer. Our results show that the ratio of LC3-II/I was markedly decreased, while BiP and p-eIF2α levels remained constant in TTR Y114C when compared with wild-type TTR and TTR 30M. The results of our research indicate the impaired autophagy contributed to the TTR Y114C mutation, but not ER stress. This observation indicates that abnormal accumulation of TTR caused by a different mutation might be cleared by different pathways, and more studies are necessary to confirm whether this difference applies to other TTR mutations.
Curcumin is known to have neuroprotective properties through a variety of mechanisms.8–11 Our research indicates that curcumin decreased the monomeric TTR by promoting autophagy, and without toxic effects. Moreover, this protective effect of curcumin on TTR Y114C could be partially blocked by 3-MA. Pullakhandam et al showed that curcumin binds to wild-type TTR and prevents urea-induced perturbations in the tertiary structure of TTR in vitro.19 Recently, Ferreira et al reported that dietary curcumin modulated TTR amyloidogenicity.20 Therefore, curcumin might be an effective therapy for FAP involving multiple molecular pathways.
Overall, our findings show that abnormal accumulation of TTR caused by different mutations might be cleared in different ways, and curcumin might be an effective therapy for FAP by promoting autophagy. Further studies are necessary to determine whether this phenomenon exists in other TTR mutations.
Acknowledgments
This study was supported by grants from the National Program of Basic Research of China (2010CB945200, 2011CB504104), The National Natural Science Foundation of China (number 81301081), the Shanghai Key Project of Basic Science Research (10411954500), and the Program for Outstanding Medical Academic Leader (LJ 06003).
Disclosure
The authors report no conflicts of interest in this work.
References
Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086–1097. | ||
Saraiva MJ, Birken S, Costa PP, Goodman DS. Amyloid fibril protein in familial amyloidotic polyneuropathy, Portuguese type. Definition of molecular abnormality in transthyretin (prealbumin). J Clin Invest. 1984;74(1):104–119. | ||
Teixeira PF, Cerca F, Santos SD, Saraiva MJ. Endoplasmic reticulum stress associated with extracellular aggregates. Evidence from transthyretin deposition in familial amyloid polyneuropathy. J Biol Chem. 2006;281(31):21998–22003. | ||
Zhang Y, Deng YL, Ma JF, et al. Transthyretin-related hereditary amyloidosis in a Chinese family with TTR Y114C mutation. Neurodegener Dis. 2011;8(4):187–193. | ||
Ando Y, Almeida M, Ohlsson PI, et al. Unusual self-association properties of transthyretin Y114C related to familial amyloidotic polyneuropathy: effects on detection and quantification. Biochem Biophys Res Commun. 1999;261(2):264–269. | ||
Nakamura M, Yamashita T, Ueda M, et al. Neuroradiologic and clinicopathologic features of oculoleptomeningeal type amyloidosis. Neurology. 2005;65(7):1051–1056. | ||
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10 Suppl:S10–S17. | ||
Bukhari SN, Franzblau SG, Jantan I, et al. Current prospects of synthetic curcumin analogs and chalcone derivatives against mycobacterium tuberculosis. Med Chem. 2013;9(7):897–903. | ||
Bukhari SN, Jantan I, Unsal Tan O, et al. Biological activity and molecular docking studies of curcumin-related α,β-unsaturated carbonyl-based synthetic compounds as anticancer agents and mushroom tyrosinase inhibitors. J Agric Food Chem. 2014;62(24):5538–5547. | ||
Jantan I, Bukhari SN, Lajis NH, et al. Effects of diarylpentanoid analogues of curcumin on chemiluminescence and chemotactic activities of phagocytes. J Pharm Pharmacol. 2012;64(3):404–412. | ||
Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72(1):29–39. | ||
Tojo K, Sekijima Y, Kelly JW, et al. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56(4):441–449. | ||
Stangou AJ, Hawkins PN. Liver transplantation in transthyretin-related familial amyloid polyneuropathy. Curr Opin Neurol. 2004;17(5):615–620. | ||
Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. | ||
de Carvalho M. Is it better than it seems or just good enough? The tafamidis saga. Muscle Nerve. 2012;46(6):839–840. | ||
Benson MD. Pathogenesis of transthyretin amyloidosis. Amyloid. 2012;19 Suppl 1:14–15. | ||
Lei M, Yang M, Huo S. Intrinsic versus mutation dependent instability/flexibility: a comparative analysis of the structure and dynamics of wild-type transthyretin and its pathogenic variants. J Struct Biol. 2004;148(2):153–168. | ||
Jiang X, Smith CS, Petrassi HM, et al. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40(38):11442–11452. | ||
Pullakhandam R, Srinivas PN, Nair MK, Reddy GB. Binding and stabilization of transthyretin by curcumin. Arch Biochem Biophys. 2009;485(2):115–119. | ||
Ferreira N, Santos SA, Domingues MR, Saraiva MJ, Almeida MR. Dietary curcumin counteracts extracellular transthyretin deposition: insights on the mechanism of amyloid inhibition. Biochim Biophys Acta. 2013;1832(1):39–45. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.