")
Back to Journals » Journal of Inflammation Research » Volume 14
CTRP9 Enhances Efferocytosis in Macrophages via MAPK/Drp1-Mediated Mitochondrial Fission and AdipoR1-Induced Immunometabolism
Authors Song CX, Chen JY, Li N, Guo Y
Received 20 January 2021
Accepted for publication 10 March 2021
Published 23 March 2021 Volume 2021:14 Pages 1007—1017
DOI https://doi.org/10.2147/JIR.S302944
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Cheng-Xiang Song,1,2 Ji-Ying Chen,2,3 Na Li,1,2 Yuan Guo3
1Department of Cardiology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 2The Key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministry of Education, Chinese National Health Commission and Chinese Academy of Medical Sciences, The State and Shandong Province Joint Key Laboratory of Translational Cardiovascular Medicine, Qilu Hospital of Shandong University, Jinan, 250012, People’s Republic of China; 3Department of General Practice, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China
Correspondence: Yuan Guo
Department of General Practice, Qilu Hospital, Cheeloo College of Medicine, Shandong University, 107 Wenhuaxi Road, Jinan, 250012, People’s Republic of China
Tel +86-531-82169092
Email [email protected]
Background: Clearance of apoptotic cells (ACs) by phagocytes (efferocytosis) suppresses post-apoptotic necrosis and alleviates inflammation. Defective efferocytosis induces diseases that include atherosclerosis and autoimmune diseases. C1q/TNF-related protein 9 (CTRP9), a novel adipokine, has been reported to protect against various cardiovascular disease; however, the effect of CTRP9 on efferocytosis has not been elucidated.
Methods: 1. The efferocytosis of macrophages incubated with ACs with or without CTRP9 treatment was detected by flow cytometry (FCM) and immunostaining. The unengulfed ACs of CTRP9-KO and wild-type (WT) mice after dexamethasone injection were detected by TUNEL assay. 2. As mitochondrial fission is important for promoting efferocytosis, the effect of CTRP9 on mitochondrial fission was measured by fission/fusion-related proteins (MFN2, DRP1, MFF, and OPA1) and visualized by staining with MitoTracker. 3. On account of metabolism insults in engulfed macrophages, we conducted a two-stage efferocytosis assay, and the protective effects of CTRP9 on metabolism were investigated by Western blot.
Results: CTRP9 significantly facilitated macrophage efferocytosis, and it promoted mitochondrial fission by increasing the expression of p-DRP1 (s616) and the translocation of DRP1 from the cytoplasm to the mitochondria. The p38/Jnk-MAPK pathway was activated after treatment with 1 μg/mL CTRP9. When we blocked the activation of MAPK signaling by SB203580 and SP600125, the mediated effect on p-DRP1 (s616) was reduced. Moreover, CTRP9 increased the levels of ABCA1, PPAR-y, HIF-1a and GLUT1, as well as the release of lactate in basal and engulfed macrophages, which revealed that the metabolism of macrophages was advanced. Apoptotic cell-conditioned media (ACCM) and ACs increased the expression of adiponectin receptor 1 (AdipoR1). Down-regulation of AdipoR1 by siRNA could abrogate the immunometabolism effects of CTRP9.
Conclusion: CTRP9 promoted efferocytosis in macrophages via MAPK/drp1-mediated mitochondrial fission and AdipoR1-induced immunometabolism.
Keywords: CTRP9, macrophage, efferocytosis, mitochondrial fission, immunometabolism
Introduction
The effective clearance of dying cells by phagocytes, a process known as efferocytosis, plays an essential role in tissue homeostasis. While defective efferocytosis underlies a growing list of chronic inflammatory diseases, such as autoimmune disease and atherosclerosis, where it promotes disease progression.1 Therefore, identification of mechanisms required for efficient efferocytosis may provide new molecular targets for treatment or prevention of autoimmune/inflammatory diseases.
Mitochondrial fission plays an important role in promoting the continued clearance of apoptotic cells by macrophage, and its inhibition was reported to reduce efferocytosis.2 Moreover, dynamin-related protein 1 (Drp1) is a key regulator of mitochondrial fission, and the Mitogen-activated protein kinase (MAPK)/Drp1 pathway is reported to have association with mitochondrial fission during melanoma lesions.3 Indeed, AICAR enhances the phagocytic ability of macrophages towards apoptotic cells through p38-MAPK activation,4 while blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly.5
An interesting and important feature of efferocytosis is the ability of phagocytes to ingest multiple apoptotic cells (ACs) over a relatively short period, referred to as continual efferocytosis. This capability of efferocytes is essential when the AC-to-phagocyte ratio is high.6 Efficient clearance depends on the capacity of a single phagocyte to ingest multiple apoptotic cells successively, and to process the corpse-derived cellular material.7 Phagolysosomal degradation of ACs results in large amounts of metabolic cargo, including amino acids, lipids and nucleic acids, that must be processed in a safe and productive manner. For example, LXR-ABCA1 pathway is mediated by LDL-related protein1 (LRP1)-mediated efferocytosis, which activates peroxisome proliferator-activated receptor-y (PPAR-y), an inducer of LXR- and ABCA1-mediated cholesterol efflux.8 In addition, one study found that soon after phagocytes degrade ACs, the mitochondrial membrane potential (MMP) increases and this is related to the delivery of AC cargo, as the uptake of beads does not affect the MMP.9 Indeed, Ucp2-MMP axis enhanced engulfment.9 Expect that, glycolysis is generally considered to be proinflammatory, whereas blocking aerobic glycolysis suppresses efferocytosis, blocking oxidative phosphorylation does not. Experimental inhibition of GLUT1 induction results in the suppression of both initial and continual efferocytosis in vitro and in vivo.10
The C1qTNF‐related proteins (CTRPs) are a recently discovered and highly conserved family of adiponectin (APN) paralogs. Among CTRPs, CTRP9 is highly expressed in heart and adipose tissue. The globular domain isoform of CTRP9 (gCTRP9) is generated via proteolytic cleavage of full‐length CTRP9 (fCTRP9) and is the main active isoform.11 It has been demonstrated that the CTRP9 has protective roles in the regulation of obesity, diabetes, and hyperlipidemia, which are vital risk factors for atherosclerosis.11 But little is known about whether the anti-atherosclerotic effect of CTRP9 is mediated by efferocytosis. CTRP9 specifically stimulates MAPK phosphorylation and activation in macrophages,12 and increases mitochondrial biogenesis.13,14 Moreover, CTRP9 induces mitochondrial biogenesis via AdipoR1-SIRT1-PGC-1α activation.15 CTRP9 decreases LOX-1 expression in cholesterol-loaded VSMCs with a significant increase in the levels of ABCA1 and ABCG1.16 CTRP9 also affects glucose and insulin metabolism, and overexpression of CTRP9 in mice significantly reduces the serum glucose levels and lowers serum insulin levels in ob/ob mice.17
Adiponectin receptor 1 (AdipoR1) serves as the receptor of CTRP9 besides adiponectin. Accumulating evidence indicates that CTRP9 signaling is transduced via the AdipoR1 in RAW 264.7 macrophages,18 endothelial cells (ECs),15 and cardiac myocytes.19 Upon binding to AdipoR1, CTRP9 can activate a variety of signaling pathways to regulate glucose and lipid metabolism, and also suppress the inflammation.17 Therefore, the present study aimed to investigate whether CTRP9 promotes efferocytosis by facilitating mitochondrial fission via the MAPK/drp1 signaling pathway and immunometabolism via AdipoR1.
Materials and Methods
Chemicals and Reagents
Recombinant mouse gCTRP9 protein was purchased from Aviscera Bioscience (Santa Clara, CA). Antibodies for Glut1, ABCA1, PPAR-y, HIF-1a, AdipoR1, actin, tubulin was obtained from Abcam, Drp1, p-Drp1 (Ser616), Erk1/2, p-Erk1/2(Thr202/Tyr204), Jnk, p-Jnk (Thr183/Tyr185), p38, p-p38 (Thr180/Tyr182), Opa1, Mff, Mfn-2, Ucp2 were bought from CST. U0126, SP600125 and SB203580 were obtained from MCE. Cell culture medium was purchased from HyClone Laboratories (Utah, USA).
Preparation and Culture of Mouse Peritoneal Macrophages
The study was approved by the Medical Ethics Committee of Qilu Hospital, Shandong University, China (Ethics Reference Number: DWLL-2017-015), and was conducted according to the Regulations of Experimental Animal Administration issued by the State Committee of Science and Technology of People’s Republic of China. Mouse peritoneal macrophages were elicited by 6% starch. Briefly, eight-week-old wild-type male C57BL/6 mice were injected with 6% starch into the peritoneal cavity. Peritoneal exudates were obtained 72 h after injection by flushing the peritoneal cavity with ice-cold PBS. Peritoneal lavage was pooled and centrifuged in a 50-mL conical centrifuge tube at 800 rpm for 5 min at 4°C, and the pellet was resuspended in DMEM medium supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. Macrophages were allowed to adhere to tissue culture wells for 1 h prior to removal of other cells by washing with serum-free DMEM.
Generation of Apoptotic Cells and Their Conditioned Media
Apoptotic thymocytes were induced by 1 mM dexamethasone (Sigma-Aldrich) for 6 h at 37 °C. To get conditioned media, we incubated the generated apoptotic for another 4 h in RPMI with 10% FBS following washing. Afterward, the cell medium was obtained by centrifugation at 13,000 g for 10 min and then filtered through 0.2 mm pore filters to remove apoptotic bodies.
Mitochondrial Morphology Analysis
To view mitochondria, macrophages were labeled with 150 nM MitoTracker Green for 15 min in serum-free DMEM. Cells were viewed by confocal fluorescence microscopy (NIKON A1 Confocal microscope, 63X oil objective). The number of individuals was quantified using ImageJ software.
In vitro Efferocytosis Assay
Peritoneal macrophages harvested 3 days after intraperitoneal injection of 6% starch in mice had been previously immunized with 6% starch were plated in 12-well dishes at a density of 4x105 cells per well. Fluorescent ACs were added to macrophages (10:1) for 45 min unless noted otherwise, followed by vigorous washing 3 times with 1X PBS. Macrophages were dissociated from the wells and centrifuged at 800 rpm for 5min. After resuspended with 100 ul PBS, cells were incubated with FITC-F4/80 antibody (1:100 dilution, biolegend) for 30 min at 37°C and analyzed by flow cytometry (BD Canto II). For two-stage efferocytosis experiments, unlabeled ACs were incubated with macrophages for 45 min followed by vigorous rinsing 3 times with 1X PBS. Macrophages were then incubated for another 12 h, followed by addition of labeled ACs. After 45 min, unbound ACs were removed by rinsing, and then the macrophages were analyzed by flow cytometry.
Analysis of Cell Clearance in the Thymus
Five-to-six-week-old wild-type and CTRP9-deficient mice were intraperitoneally injected with 300 ul of PBS containing 250 ug dexamethasone dissolved in ethanol (10 mg/mL). Twelve hours after injection, the mice were killed and thymuses were extracted. Apoptotic cells were detected with TUNEL staining and analyzed by microscopy. Over 200 thymocytes were analyzed per sample.
Immunofluorescence Staining
Peritoneal macrophages seeded in non-tissue culture plates were stimulated with 1 µg/mL CTRP9 for 2 h. Cells were washed with ice-cooled PBS and blocked the Fc receptors. Cells were then fixed and stained with anti-drp1 (s616). Cells were washed with 1X PBS and analyzed by fluorescence microscopy (NIKON microscope, 40X objective).
Western Blotting
Cell lysates were prepared from peritoneal macrophages (5 × 106 cells) in lysis buffer according to the manufacturer’s protocol and boiled for 10 min. The samples were diluted with 1×lysis buffer and were separated by electrophoresis and transferred onto polyvinylidene fluoride membranes (PVDF). After blocking with 5% skimmed milk. The membranes were reacted with primary antibodies against DRP1 (1:1000 dilution), p-DRP1 (Ser616) (1:1000 dilution), ERK1/2 (1:1000 dilution), p-ERK1/2 (1:1000 dilution), GLUT1 (1:1000 dilution), and β-actin (1:1000 dilution) at 4 °C overnight, followed by incubation with peroxidase-conjugated secondary antibodies at room temperature for 2 h. The blots were visualized with enhanced chemiluminescence detection reagent. β-Actin was used as internal controls for total protein. Bands were quantified by densitometry using Quantity One software (Bio-Rad, Hercules, CA).
RNA Interference
Cells cultured with Opti-MEM reduced serum media were transfected with AdipoR1 siRNA or negative control siRNA (GenePharma) along with RNAiMAX according to the manufacturer’s instructions. After transfection for 48 h, cells were collected and the efficiency of gene silencing was determined by Western blot.
Statistical Analysis
SPSS software 22.0 was used for statistical analysis. Data were presented as mean ± SD of at least three independent experiments. The differences were determined by one-way ANOVA with Tukey’s post hoc test. P<0.05 was considered to be statistically significant.
Results
CTRP9 Enhances Efferocytosis in Macrophages
To determine whether CTRP9 contributes to the clearance of apoptotic cells in vitro, FITC-labeled macrophages were incubated with apoptotic cells labeled with TAMRA-SE in the presence or absence of CTRP9, as determined by flow cytometric analysis of dual-labeled and total macrophages. Results showed that stimulation of apoptotic cell uptake was dependent on the dose of CTRP9 in the phagocytosis assay (Figure 1A), based on this result, we used 1 µg/mL CTRP9 for the subsequent experiments. In agreement with these data, microscopic analyses also revealed that CTRP9 stimulated the ingestion of CFSE–labeled apoptotic cells by mouse peritoneal macrophages (Figure 1B).
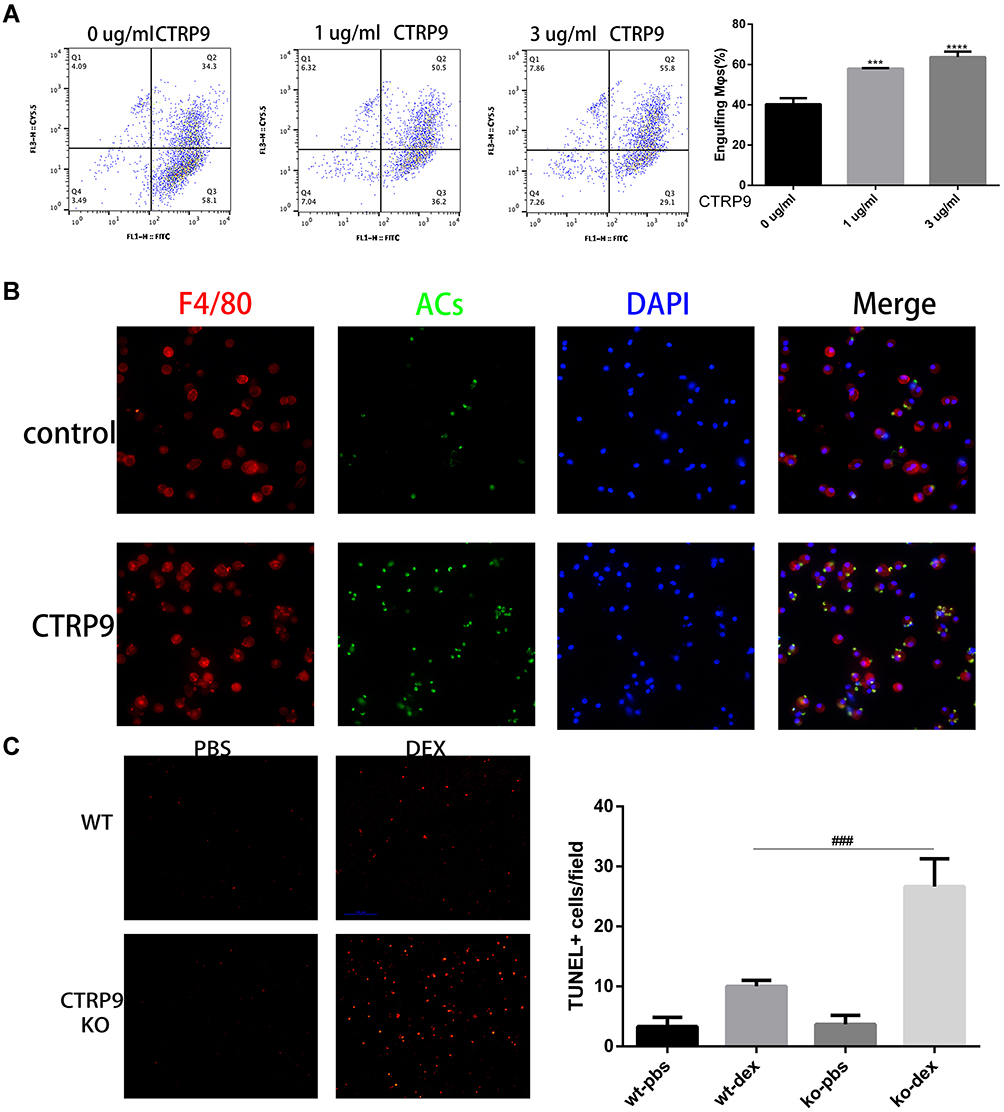 |
Figure 1 CTRP9 treatment promotes macrophages efferocytosis in vitro and in vivo. (A) TAMRA-labeled apoptotic cells were incubated with FITC-labeled macrophages at 10:1 ratio for 45 min. The unengulfed ACs were removed by rinsing, and the macrophages were detached from the plate for flow cytometric analysis of TAMRA+ FITC+ cells, which reflects efferocytosis. (n=3). (B) Macrophages (red) were incubated with CFSE-labeled apoptotic cells (green) for 45 min at a 10:1 AC: macrophage ratio. Macrophages that had or had not engulfed an AC (AC+, AC–) were visualized by microscopy. (C) WT and CTRP9-KO mice were treated with dexamethasone (DEX), and thymi were stained for apoptotic cells with TUNEL. The number of TUNEL-positive cells per microscopic field for the different experimental conditions is reported. (n=5). All data are expressed as mean ± SD. ***p<0.001, ****p<0.0001; versus control. ###p<0.001; versus WT-DEX. |
Next, to determine whether CTRP9 contributes to the clearance of apoptotic cells in vivo, CTRP9-deficient (CTRP9-KO) and WT mice were injected with dexamethasone to induce massive thymocyte apoptosis.20 At 24 hours after dexamethasone treatment, the thymi of CTRP9-KO and WT mice were assessed for apoptotic cells in histological sections by TUNEL staining. Few TUNEL-positive cells were observed with untreated CTRP-KO and WT mice in the thymi. However, the thymi of dexamethasone-treated CTRP-KO mice showed 2.6-fold greater remnant apoptotic cells than those of similarly treated WT mice (Figure 1C).
CTRP9 Facilitates Mitochondrial Fission via MAPK Signaling
As mitochondrial fission plays a role in promoting the continued clearance of apoptotic cells by macrophage, and its inhibition was reported to reduce efferocytosis, we explored whether CTRP9 promoted macrophage efferocytosis via mitochondrial fission. We found that CTRP9 treatment significantly increased the phosphorylation of drp1 at S616, the expression of Mff and the ratio of short-chain (S-chain) to long-chain (L-chain) of OPA1, while decreased the expression of mfn-2, resulting in mitochondrial fission (Figure 2B and C). Then, we verified the p-drp1 (s616) expression by immunostaining, indicating that CTRP9 promoted the expression of p-drp1 (s616) (Figure 2A). Immunoblot of mitochondrial fraction of macrophages showed that treatment with CTRP9 increased drp1 translocation to mitochondria (Figure 2D). Furthermore, we observed the mitochondrial morphology by confocal microscope, and the mitochondria were shorter and more fragmented in CTRP9 treatment versus control macrophages (Figure 2E). Above all, these data clearly showed that CTRP9 facilitated mitochondrial fission.
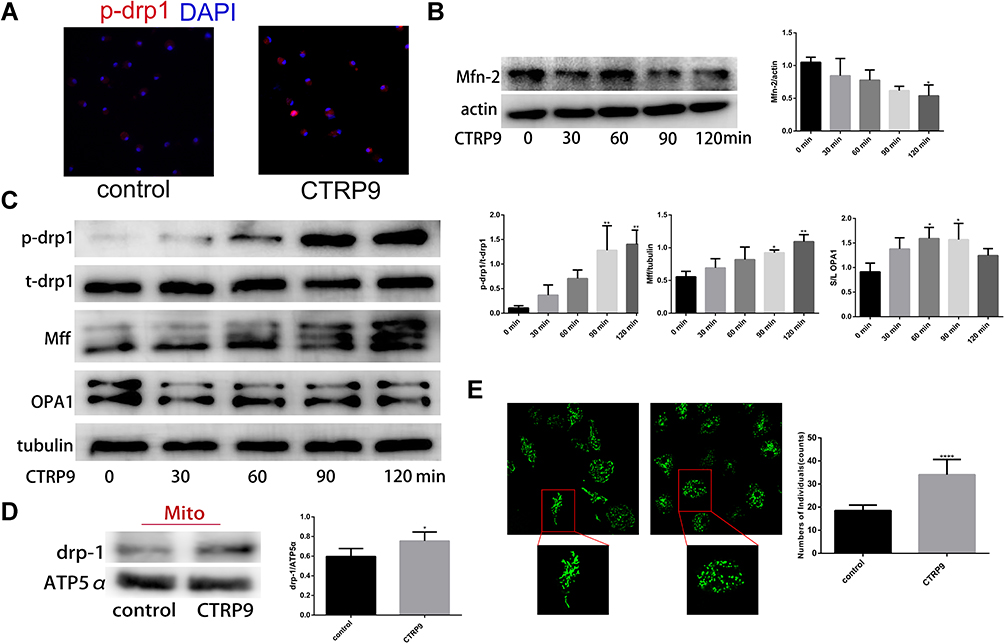 |
Figure 2 CTRP9 induces mitochondrial fission in mouse peritoneal macrophages. (A) Representative immunofluorescence staining for p-drp1 s616 (red) and DAPI (blue) obtained at 2 h 1 µg/mL CTRP9 incubation. (B and C) Protein expression were assessed using Western blot in macrophages after 1 µg/mL CTRP9 treatments for 2 h. (D) Immunoblot of the indicated proteins in mitochondrial fractions of macrophages incubated without or with 1 µg/mL CTRP9 for 2 h. (E) Mitochondrial morphologies of macrophages with or without 1 µg/mL CTRP9 treatment for 2 h. Green, MitoTracker Green. All data are expressed as mean ± SD (n=3). *p< 0.05; **p<0.01; ****p<0.0001; versus control. |
To elucidate the effect of CTRP9 on MAPK activation, peritoneal macrophages were incubated for 0–2 h with 1 µg/mL CTRP9. We tested phosphorylation of Erk, p-38, Jnk and the results showed increased phosphorylation of Erk, p-38, Jnk in peritoneal macrophages (Figure 3A––C). Moreover, U0126 (inhibitor of Erk), SB203580 (inhibitor of p38), SP600125 (inhibitor of Jnk) individually restrained the phosphorylation of Erk, p-38, Jnk after CTRP9 treatment for 2h (Figure 3D–F).
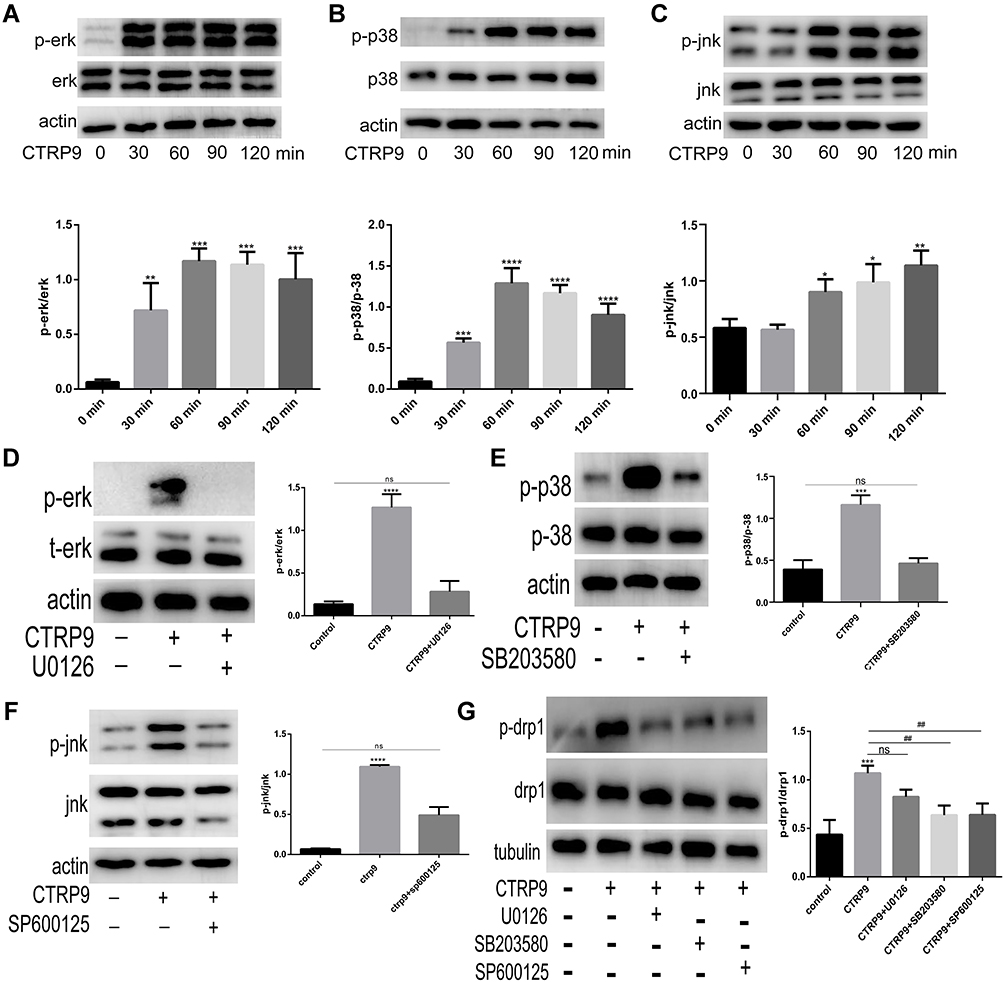 |
Figure 3 CTRP9 promotes macrophage mitochondrial fission through the activation of p38/Jnk MAPK signaling. (A–C) The protein levels of JNK, ERK, P-38 induced by 1 µg/mL CTRP9 for 0–2 h. (D–F) Immunoblot analysis of Erk, p38 and Jnk after interrupted by CTRP9 and U0126, SB203580, SP600125 (10 µM 1h). (G) Immunoblot analysis of p-drp1 s616 after interrupted by 1 µg/mL CTRP9 and U0126, SB203580, SP600125. All data are expressed as mean ± SD (n=3). “ns” means no significance. ns, p >0.05; *p< 0.05; **p<0.01; ***p<0.001, ****p<0.0001; versus control. ##p<0.01; versus CTRP9 group. |
We further investigated the role of CTRP9 in the promotion of mitochondrial fission effects via MAPK pathway. Given that the drp1 plays a leading role in mitochondrial fission, we mainly examined the changes of drp1 in the interruption of CTRP9. Results showed that the phosphorylation of drp1 at S616 was decreased when MAPK was inhibited by SB203580 and SP600125 (Figure 3G). These results indicated that mitochondrial fission effect of CTRP9 was partly dependent on the activation of p38/Jnk MAPK in macrophages.
CTRP9 Improves Macrophage Immunometabolism After the Ingestion of Apoptotic Cells
Mitochondrial fission facilitated the phagocytosis of ACs into macrophages, but the ingestion of ACs by phagocytic cells results in accumulation of extra material in these cells, and, indeed, the mass of their intracellular content initially doubles.21 Efficient metabolism to cope with the dramatic increase in lipid, protein, nucleotide, and carbohydrate content is necessary to continued clearance of apoptotic cells by macrophage. Based on this data, we hypothesized that CTRP9 enabled macrophages to internalize ACs after the first efferocytosis. To test this hypothesis, we conducted a two-stage efferocytosis experiment (Figure 4A) in which macrophages were first incubated for 120 min with ACs, after AC removal and a 12-hour interval with or without CTRP9 treatment, then incubated with a second round of ACs labeled with TAMRA and CypHer5E. Consistent with the hypothesis, the percentage of macrophages that had internalized both labels was more in CTRP9-treated macrophages than in control cells (Figure 4B). To verify that the above data were due to effects on improved immunometabolism, macrophages were incubated for 0–24 h with 1 µg/mL CTRP9. Western blot was used to assess the protein expression levels of PPAR-y, HIF-1a, ABCA1, GLUT1 and UCP2, a time-dependent rise in PPAR-y, HIF-1a, ABCA1, GLUT1 expression was observed, while there is no effect on UCP2 expression (Figure 4C). Next, we incubated macrophages with ACs (AC+) and some do not (AC_), after AC removal with or without CTRP9 treatment, then we tested the level of ABCA1, GLUT1 and lactate. Only treatment with ACs or CTRP9 could increase the expression of ABCA1 and GLUT1 and the release of lactate (Figure 4D and E). After treatment with ACs, CTRP9 further enhanced the effect of improving immunometabolism.
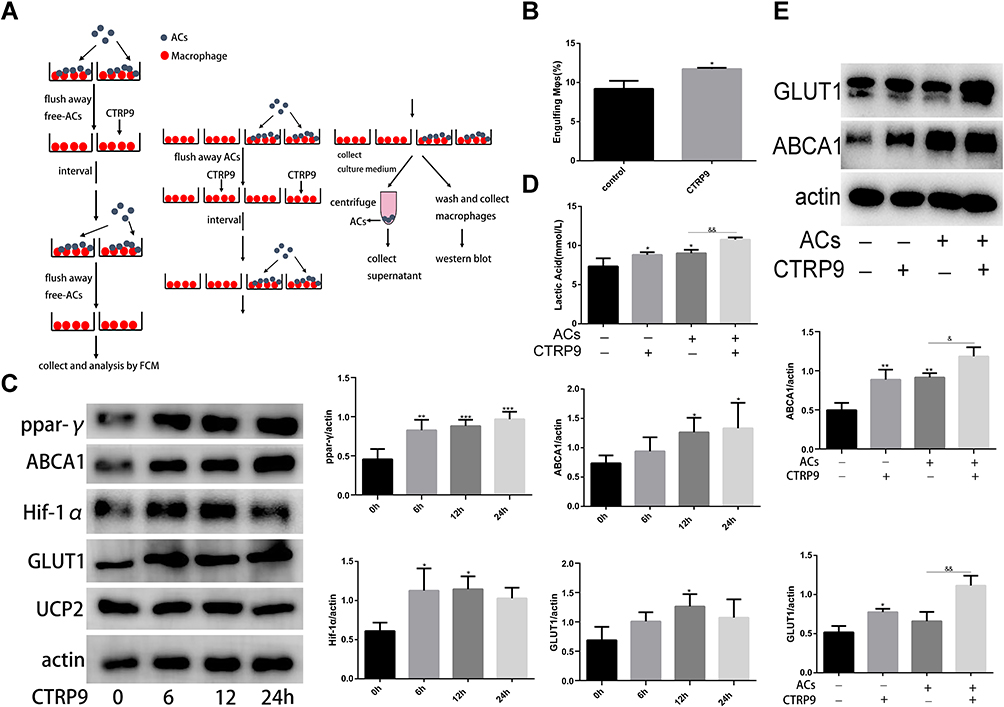 |
Figure 4 CTRP9 improved macrophage immunometabolism (A) The detailed procedure of two-stage efferocytosis assay. (B) Macrophages were incubated with ACs at a ratio of 10:1 ACs: macrophages for 120 min. The ACs were removed, and then, after a 12h interval with or without CTRP9 treatment, the macrophages were incubated with TAMRA-CypHer5E-labeled ACs at a ratio of 10:1 for 45 min. The unengulfed ACs were removed, and efferocytosis was quantified as the total percentage of macrophages that were positive for TAMRA-CypHer5E-labeled ACs by flow cytometric analysis. (C) Macrophages were stimulated with 1 μg/mL of CTRP9 for 0–24 h, and cell lysates were analyzed for PPAR-y, ABCA1, HIF-1a, GLUT1 and Ucp2 by Western blot. (D and E) Macrophages were incubated with ACs at a ratio of 10:1 ACs: macrophages for 120 min. The ACs were removed, and then, after a 12 h interval with or without CTRP9 treatment. Lactate was measured in the medium. Representative Western blot analyses and quantitative data are shown. All data are expressed as mean ± SD (n=3). *p< 0.05; **p<0.01; ***p<0.001; versus control. &p<0.05; &&p<0.01; versus ACs group. |
AdipoR1 Mediates the Immunometabolism Effects of CTRP9
Several studies reported that the AdipoR1 contributed to the regulation of glucose and lipid metabolism of CTRP9. To analyze the involvement of AdipoR1 on macrophage efferocytosis, we found that apoptotic cell-conditioned media (ACCM), time-dependently upregulated AdipoR1 in macrophages in vitro (Figure 5A). We further showed that ACs also contributed to AdipoR1 induction in macrophages in vitro (Figure 5B). Thus, we explored the role of AdipoR1 in the macrophage immunometabolism reactions of CTRP9. With AdipoR1 siRNA transfected in cells, Western blot demonstrated that the AdipoR1-homo-666 showed the high inhibition efficiency (Figure 5C) and was chosen for the subsequent experiments. Macrophages pretreated with negative control siRNA or AdipoR1 siRNA were exposed to CTRP9 or not for the indicated time. Compared to NC-group, AdipoR1 knockdown itself had no effect on the expression of HIF-1a, ABCA1, GLUT1, but decreased the levels of PPAR-y, which indicates that the effect of CTRP9 on PPAR-y was partly induced by AdipoR1. Knockdown of AdipoR1 reversed the promotion effects of CTRP9 on the expression of PPAR-y, HIF-1a, ABCA1, GLUT1 (Figure 5C). It is demonstrated that CTRP9 activated immunometabolism through AdipoR1 in macrophages.
 |
Figure 5 AdipoR1 mediated the immunometabolism effects of CTRP9 (A and B) Macrophages were incubated with apoptotic cell-conditioned media (ACCM) (A) or apoptotic cells (B), and the expression of AdipoR1 was detected. (C) Macrophages were treated with control or AdipoR1 siRNA prior to stimulation with 1 μg/mL of CTRP9 for 12 h and the expression of related molecules were analyzed Western blot. All data are expressed as mean ± SD (n=3). “ns” means no significance. ns, p>0.05; *p< 0.05; **p<0.01; versus control. #p< 0.05; ##p<0.01; versus NC+CTRP9 group. |
Discussion
In the present study, we investigated the effects of CTRP9 on macrophage efferocytosis and the underlying molecular mechanism. Our results suggest that the observed increase of macrophage efferocytosis induced by CTRP9 was mediated, at least in part, by MAPK/Drp1 signaling and improving immunometabolism.
Pulanco et al found that complement protein C1q enhances macrophage foam cell efferocytosis; moreover,22 Galvanl et al showed that complement protein C1q and adiponectin stimulate Mer tyrosine kinase-dependent engulfment of apoptotic cells through a shared pathway,23 Takemura et al demonstrated that adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies.20 Although the effect of CTRP9 on clearance of apoptotic cells has not been investigated, CTRP9, C1q and adiponectin share a short N-terminal domain, a collagen-like repeating sequence (glycine-X-Y) and a C-terminal globular domain with homology to the TNF family.24 It is possible that other members of the CTRP family may also promote efferocytosis. In the present study, our results demonstrated that CTRP9 enhanced macrophages efferocytosis in vivo and vitro experiments.
Mitochondrial fission is involved in many physiological and pathological processes. It participates in the development and progression of cardiovascular pathologies, including diabetic cardiomyopathy, atherosclerosis, cardiac hypertrophy and decompensated heart failure.25 Drp1 also promoted vascular calcification by regulating osteogenic differentiation.26 A study by Wang et al showed that Drp1 expression was increased in the macrophage exposed to ACs, while Drp1 inhibition reduced the continued efferocytosis.2 These observations were consistent with our results in Figures 1 and 2 showing that CTRP9 enhanced efferocytosis in macrophages via mitochondrial fission.
Mitochondrial fission is associated with MAPK pathway. Previous study showed that Erk2-mediated phosphorylation of drp1 on Serine 616 promotes mitochondrial fission and MAPK-driven tumor growth.27 Gui et al demonstrated that drp1 is a substrate for p38 MAPK, and p38 MAPK-mediated phosphorylation drp1 at serine 616 to activate drp1 and is associated with increased mitochondrial fission.28 Quan et al indicated that AICAR increased the phagocytic ability of macrophages toward apoptotic cells through activation of p38 MAPK in macrophages.4 Our experiments demonstrated that CTRP9 activated MAPK protein expression, enhanced Drp1-essential mitochondrial fission. Furthermore, we found that SP600125 and SB203580 reduced the effects of CTRP9 on MAPK/Drp1 expression and inhibited macrophage mitochondrial fission. Taken together, these findings indicated that CTRP9 activates MAPK expression, which in turn increased phosphorylation drp1 at serine 616 and, subsequently, increased mitochondrial fission.
After apoptotic cells are degraded in phagolysosomes, macrophages become overloaded with macromolecular constituents and therefore have evolved elegant mechanisms to either use or efflux this cargo. Some of the material engulfed by phagocytic cells can be completely destroyed, while the indigestible cholesterol is effluxed via the ATP-binding cassette (ABC) transporters to extracellular cholesterol acceptors, notably the various classes of apolipoprotein A1-containing high-density lipoproteins. Toward this end, PtdSer and lipid-sensing nuclear receptors can upregulate the level of ABCA1 transporters.21 Moreover, previous study showed that the burden of cholesterol from degraded apoptotic cells activates members of the PPAR and liver X receptor (LXR) families of nuclear receptors and drive ABCA1 and ABCG1 expression, which mediate cholesterol efflux from the cells.8 In addition, the digested macromolecules provide the extra energy and induce cellular changes required for a successful continuation of phagocytosis. The changes include increased expression of the factors involved in the phagocytosis such as liver X receptor (LXR) α/β and peroxisome proliferator-activated receptor (PPAR) δ/γ, which are sensitive to changes in the lipid content of the macrophage engulfing ACs.29,30 Park et al demonstrated that the influx of AC-derived metabolic cargo can increase the mitochondrial membrane potential, but macrophages have evolved a compensatory mechanism, whereby upregulation of the UCP2, renders the mitochondrial membrane potential increase transient.9 UCP2 also reduces the formation of ROS and reduces the risk of atherosclerosis. Sho et al proposed that the addition of ACs to macrophages increased glycolysis, which was mediated by induction of both GLUT1 (SLC2A1) and a kinase that activates GLUT1 called SGK1. When these molecules were targeted both in vitro and in vivo, including deletion of GLUT1 in fat-fed Ldlr−/- mice, lesional efferocytosis was impaired and plaque necrosis was worsened. To evaluate the extent of CTRP9 in terms of macrophage immunometabolism, we incubated the macrophages with CTRP9 to detect the variation of metabolic molecules expression. The result suggested that CTRP9 treatment promoted the expression of ABCA1 PPAR-y HIF-1a GLUT1; however, there is no obvious influence on the expression of UCP2. Furthermore, we established a two-stage efferocytosis experiment to determine the effect of CTRP9 on improving metabolism. The flow cytometry (FCM) result verified that CTRP9 could enhance macrophage efferocytosis after engulfed apoptotic cells and the Western blot also demonstrated that CTRP9 could relieve the metabolic pressure of macrophages that have engulfed apoptotic cells.
As mentioned above, AdipoR1 serves as the receptor of CTRP9 besides adiponectin. Mice studies have demonstrated that AdipoR1 mediated metabolic actions of adiponectin, indicating that AdipoR1 helped to regulate normal glucose metabolism and insulin sensitivity.31 Matsunami et al demonstrated that downregulation of AdipoR1 increased synthesis and decreased oxidation of fatty acids, contributing to the progression of nonalcoholic steatohepatitis (NASH).32 Accumulating evidence indicates that CTRP9 signaling is transduced via the AdipoR1 in RAW 264.7 macrophages, endothelial cells (ECs), and cardiac myocytes. Upon binding to AdipoR1, CTRP9 can activate a variety of signaling pathways to regulate glucose and lipid metabolism, vascular relaxation and cell differentiation. Based on the above findings, we decided to explore whether CTRP9 promoted immunometabolism through AdipoR1. In this study, we examined the expression level of AdipoR1 after incubated with ACCM or ACs, and our results demonstrated that in macrophages, AdipoR1 expression was increased when cultured with ACCM or ACs. The up-regulation of AdiopR1expression seemed to be a compensatory mechanism to better transduce CTRP9 signals to meet the metabolic needs of macrophages that have phagocytosed apoptotic cells. We found that AdipoR1 knockdown itself, compared with NC-group, could significantly decreased the expression of PPAR-y. It seems that AdipoR1 is an important upstream signal molecular, which maintains the basic expression of PPAR-y in unexcited status. Further study should be carried out to explore this phenomenon.
In summary, we elucidated that AdipoR1 was partly participated in the improving immunometabolism process of CTRP9 in macrophages.
Conclusion
In conclusion, we demonstrated that CTRP9 promoted efferocytosis by facilitating mitochondrial fission via the MAPK/drp1 signaling pathway and immunometabolism via AdipoR1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81671950).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16(9):907–917. doi:10.1038/ni.3253
2. Wang Y, Subramanian M, Yurdagul A
3. Serasinghe MN, Wieder SY, Renault TT, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. 2015;57(3):521–536. doi:10.1016/j.molcel.2015.01.003
4. Quan H, Kim JM, Lee HJ, Lee SH, Choi JI, Bae HB. AICAR enhances the phagocytic ability of macrophages towards apoptotic cells through P38 mitogen activated protein kinase activation independent of AMP-activated protein kinase. PLoS One. 2015;10(5):e0127885. doi:10.1371/journal.pone.0127885
5. De Maeyer RPH, van de Merwe RC, Louie R, et al. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol. 2020;21(6):615–625. doi:10.1038/s41590-020-0646-0
6. Han CZ, Ravichandran KS. Metabolic connections during apoptotic cell engulfment. Cell. 2011;147(7):1442–1445. doi:10.1016/j.cell.2011.12.006
7. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. doi:10.1038/nature06307
8. Xian X, Ding Y, Dieckmann M, et al. LRP1 integrates murine macrophage cholesterol homeostasis and inflammatory responses in atherosclerosis. Elife. 2017;6.
9. Park D, Han CZ, Elliott MR, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. 2011;477(7363):220–224. doi:10.1038/nature10340
10. Morioka S, Perry JSA, Raymond MH, et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature. 2018;563(7733):714–718. doi:10.1038/s41586-018-0735-5
11. Yang Y, Li Y, Ma Z, et al. A brief glimpse at CTRP3 and CTRP9 in lipid metabolism and cardiovascular protection. Prog Lipid Res. 2016;64:170–177. doi:10.1016/j.plipres.2016.10.001
12. Yan W, Guo Y, Tao L, et al. C1q/tumor necrosis factor-related protein-9 regulates the fate of implanted mesenchymal stem cells and mobilizes their protective effects against ischemic heart injury via multiple novel signaling pathways. Circulation. 2017;136(22):2162–2177. doi:10.1161/CIRCULATIONAHA.117.029557
13. Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–1295. doi:10.1038/nm788
14. Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99(25):15983–15987. doi:10.1073/pnas.252625599
15. Cheng L, Li B, Chen X, et al. CTRP9 induces mitochondrial biogenesis and protects high glucose-induced endothelial oxidative damage via AdipoR1 -SIRT1- PGC-1alpha activation. Biochem Biophys Res Commun. 2016;477(4):685–691. doi:10.1016/j.bbrc.2016.06.120
16. Liu Q, Zhang H, Lin J, et al. C1q/TNF-related protein 9 inhibits the cholesterol-induced vascular smooth muscle cell phenotype switch and cell dysfunction by activating AMP-dependent kinase. J Cell Mol Med. 2017;21(11):2823–2836. doi:10.1111/jcmm.13196
17. Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, et al. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 2009;23(1):241–258. doi:10.1096/fj.08-114991
18. Zhang P, Huang C, Li J, et al. Globular CTRP9 inhibits oxLDL-induced inflammatory response in RAW 264.7 macrophages via AMPK activation. Mol Cell Biochem. 2016;417(1–2):67–74. doi:10.1007/s11010-016-2714-1
19. Kambara T, Shibata R, Ohashi K, et al. C1q/tumor necrosis factor-related protein 9 protects against acute myocardial injury through an adiponectin receptor I-AMPK-dependent mechanism. Mol Cell Biol. 2015;35(12):2173–2185. doi:10.1128/MCB.01518-14
20. Takemura Y, Ouchi N, Shibata R, et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. 2007;117(2):375–386. doi:10.1172/JCI29709
21. Kiss RS, Elliott MR, Ma Z, Marcel YL, Ravichandran KS. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol. 2006;16(22):2252–2258. doi:10.1016/j.cub.2006.09.043
22. Pulanco MC, Cosman J, Ho -M-M, et al. Complement protein C1q enhances macrophage foam cell survival and efferocytosis. J Immunol. 2017;198(1):472–480. doi:10.4049/jimmunol.1601445
23. Galvan MD, Hulsebus H, Heitker T, Zeng E, Bohlson SS. Complement protein C1q and adiponectin stimulate Mer tyrosine kinase-dependent engulfment of apoptotic cells through a shared pathway. J Innate Immun. 2014;6(6):780–792. doi:10.1159/000363295
24. Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96(5):1723–1732.
25. Vásquez-Trincado C, García-Carvajal I, Pennanen C, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016;594(3):509–525. doi:10.1113/JP271301
26. Rogers MA, Maldonado N, Hutcheson JD, et al. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res. 2017;121(3):220–233. doi:10.1161/CIRCRESAHA.116.310293
27. Kashatus JA, Nascimento A, Myers LJ, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57(3):537–551. doi:10.1016/j.molcel.2015.01.002
28. Gui C, Ren Y, Chen J, et al. p38 MAPK-DRP1 signaling is involved in mitochondrial dysfunction and cell death in mutant A53T alpha-synuclein model of Parkinson’s disease. Toxicol Appl Pharmacol. 2020;388:114874. doi:10.1016/j.taap.2019.114874
29. Mukundan L, Odegaard JI, Morel CR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15(11):1266–1272. doi:10.1038/nm.2048
30. Rőszer T, Menéndez-Gutiérrez MP, Lefterova MI, et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor γ or retinoid X receptor α deficiency. J Immunol. 2011;186(1):621–631. doi:10.4049/jimmunol.1002230
31. Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13(3):332–339. doi:10.1038/nm1557
32. Matsunami T, Sato Y, Ariga S, et al. Regulation of synthesis and oxidation of fatty acids by adiponectin receptors (AdipoR1/R2) and insulin receptor substrate isoforms (IRS-1/-2) of the liver in a nonalcoholic steatohepatitis animal model. Metabolism. 2011;60(6):805–814. doi:10.1016/j.metabol.2010.07.032
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.