")
Back to Journals » Cancer Management and Research » Volume 11
CSNK2A1 Promotes Gastric Cancer Invasion Through the PI3K-Akt-mTOR Signaling Pathway
Authors Jiang C , Ma Z, Zhang G, Yang X, Du Q, Wang W
Received 13 July 2019
Accepted for publication 12 November 2019
Published 2 December 2019 Volume 2019:11 Pages 10135—10143
DOI https://doi.org/10.2147/CMAR.S222620
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Chao Jiang,1,2 Zhenghong Ma,2 Guoan Zhang,3,4 Xigui Yang,2 Qin Du,5 Weibo Wang1
1Department of Oncology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong 250021, People’s Republic of China; 2Departments of Oncology, Affiliated Hospital of Shandong Academy of Medical Sciences, Shandong First Medical University, Jinan, Shandong 250031, People’s Republic of China; 3Forensic Science Center, Jining Medical University, Jining, Shandong 272067, People’s Republic of China; 4Cancer Pathology Institute, Jining Medical University, Jining, Shandong 272000, People’s Republic of China; 5Departments of Oncology, Affiliated Hospital of Jining Medical University, Jining, Shandong 272000, People’s Republic of China
Correspondence: Weibo Wang Tel +86 151 6888 8787
Fax +86 531 5862 8751
Email [email protected]
Objective: Casein kinase 2 a1 (CSNK2A1) has been shown to be involved in tumorigenesis by enhancing several oncogenic signaling pathways in various cancers. However, the function and mechanism of CSNK2A1 in gastric cancer remain unclear, and this study aimed to elucidate the role of CSNK2A1 in gastric cancer.
Methods: CSNK2A1 expression was assessed by Western blot and qPCR in four gastric cancer (GC) cell lines and one normal gastric epithelial cell line. Stable cancer cell lines with CSNK2A1 gene overexpression or knockdown were established to investigate the function and mechanism of CSNK2A1 in GC cells.
Results: CSNK2A1 expression was higher in GC cells than in normal gastric epithelial cells. Stable overexpression of CSNK2A1 in SNU216 cells significantly increased cellular proliferation, invasion, and migration. Silencing CSNK2A1 expression in SGC-790 cells effectively inhibited its oncogenic function. We further verified that epithelial-mesenchymal transition (EMT) was affected by CSNK2A1 and that CSNK2A1 promotes GC cell invasion through the PI3K-Akt-mTOR signaling pathway.
Conclusion: Our findings suggested that CSNK2A1 plays important oncogenic roles in GC invasion via EMT and the PI3K-Akt-mTOR signaling pathway and that CSNK2A1 may serve as a novel prognostic and/or therapeutic target in GC.
Keywords: gastric cancer, CSNK2A1, function, mechanism
Introduction
Gastric cancer (GC) is the fifth most common malignant tumor, and its mortality rate ranks third worldwide. As a result of national screening plans and improvements in chemotherapy, mortality rates have significantly decreased. However, the mortality rate of GC is relatively higher in developing countries than in developed countries.1 GC displays significant global variations, and its highest incidence is observed in East Asia.2 In recent years, traditional chemotherapy and molecular target therapy have made great progress, but most GC cases are diagnosed at an advanced stage, and a number of these patients have distance metastases,3 so treatment efficacy remains limited. The occurrence and development of GC are part of a complex process involving multiple mechanisms and multiple factors. However, the mechanisms underlying the development of GC are not fully understood.
Casein kinase II (CSNK2) is a pleiotropic serine/threonine kinase that phosphorylates hundreds of substrates and participates in diverse cellular processes, including cell cycle regulation,4,5 DNA replication and repair,6 development and differentiation,7 transcription,8 cell signaling,9 carcinogenesis,10 and apoptosis.11 With its two catalytic subunits (α) and two regulatory subunits (β) that form a stable heterotetramer, CSNK2 is a highly conserved serine/threonine kinase involved in various aspects of cell biology, and >300 targets of CSNK2 are known. CSNK2 has two types of catalytic subunits, CK2α and CK2α’, which are encoded by CSNK2A1 and CSNK2A2, respectively.12–15 The expression and kinase activity of CSNK2A1 are higher in malignant tumor cells than in normal counterpart cells, and it has been suggested that CSNK2A1 plays a tumorigenic role in breast,16 lung, 17,18 kidney,19 colorectal20,21 and prostate22 cancers. Positive expression of CSNK2A1 predicts a shorter overall survival and relapse-free survival for several cancers.16–22 To confirm the expression level of CSNK2A1 in GC tissues, we retrieved and analyzed TCGA data using the online database UALCAN (http://ualcan.path.uab.edu/analysis.html).23 The expression level of CSNK2A1 was significantly higher in GC tissues than in normal tissues (P<0.001, Figure S1). However, there was no correlation between the CSNK2A1 expression level and survival time in GC patients (p = 0.56, Figure S1).
CSNK2A1 has been found to be related to the invasion and migration of various cancer cells through epithelial-mesenchymal transition (EMT)24 and nuclear factor-kappa B (NF-κB)16,20 signaling pathways, but the function of CSNK2A1 in GC remains unclear. Previous research has only shown that CSNK2A1 is a gene related to the PI3K-Akt signaling pathway in CRC tumoral tissue.21 However, the relationship and function of CSNK2A1 in the PI3K-Akt-mTOR signaling pathway in GC remains unclear.
In this study, we investigated the expression pattern of CSNK2A1 in the tumorigenesis of GC cells. Furthermore, we studied the involvement of CSNK2A1 in GC cell proliferation, tumor formation, invasion, and migration. We also identified and illustrated its potential mechanism.
Materials and Methods
Cell Culture and Chemical Agents
GC cell lines (SGC-790, SNU216, BGC823, and HGC27) and a gastric epithelial cell line (GES-1) were purchased from the American Type Culture Collection (Manassas, VA, USA) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Basal Media, Shanghai, China) and penicillin-streptomycin (BasalMedia, Shanghai, China). All of the cells were incubated in a humidified air atmosphere with 5% CO2 at 37°C. LY294002, a PI3-kinase inhibitor (ab120243), was used to treat cells for 2 hrs at 30 µM.
siRNA, shRNA and Transfection
CSNK2A1-specific siRNAs and shRNA were designed based on GeneBank data combined with primer design principles. The siRNA target sense and antisense sequences were as follows: CSNK2A1, 5ʹ-CAUUUAGUUACUGGGCAUA-3ʹ and 5ʹ-UAUGCCCAGUAACUAAAUG-3ʹ; negative control, 5ʹ-CCUACGCCACCAAUUUCGU-3ʹ and 5ʹ-ACGAAAUUGGUGGCGUAGG-3ʹ. CSNK2A1 knockdown lentiviruses were obtained from GeneChem (Shanghai, China) and were used to establish a CSNK2A1-knockdown SGC790 cell line. CSNK2A1-overexpressing lentiviruses were also obtained from GeneChem Company (Shanghai, China) and were used to generate a CSNK2A1-overexpressing SNU216 cell line. Negative control cell lines were also established with SGC790 and SNU216 cells. According to the manufacturer’s instructions, Lipofectamine 2000 (Invitrogen, USA) was used to transfect oligonucleotides or plasmids into GC cells. Forty-eight hours after transfection, the cells were collected for further experiments.
Western Blot Analysis
Total proteins were extracted from cells using RIPA lysate buffer (1% NP40, 0.1% SDS, 5 mM EDTA, 0.5% deoxycholate sodium, and 1 mM pervanadate) containing protease and phosphatase inhibitors. Equal amounts of protein were separated by 12% SDS-PAGE and transferred to PVDF membranes (Millipore, USA), which were subjected to Western blotting according to standard methods. The membranes were incubated with primary antibodies at 4°C overnight after blocking with 5% skim milk and then incubated with horseradish peroxidase-conjugated secondary antibodies. GAPDH was used as a loading control. An ECL system was used to detect the signals. The following antibodies were used in our experiments: anti-CSNK2A1 (#ab76040, Abcam, Cambridge, UK; 1:100), anti-E-cadherin (#3195, Cell Signaling Technology, Shanghai, China; 1:1000), anti-N-cadherin (#13,116; Cell Signaling Technology, Shanghai, China; 1:1000), anti-vimentin (#5741; Cell Signaling Technology, Shanghai, China; 1:1000), anti-ALK (#ab16670, Abcam, Cambridge, UK; 1:100;), anti-AKT1 (phospho S473) (#ab81283, Abcam, Cambridge, UK; 1:6000), anti-pan-Akt (phospho T308) (#ab38449, Abcam, Cambridge, UK; 1:500), anti-mTOR (#ab134903, Abcam, Cambridge, UK; 1:10,000), and anti-mTOR (phospho S2448) (#ab109268, Abcam, Cambridge, UK; 1:5000).
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. qRT-PCR was performed using SYBR Premix Ex Taq (Takara, Japan) to assess gene expression. The specific primers used for RT-PCR were as follows: CSNK2A1 F: 5`-GAACGCTTTGTCCACAGTGA-3`, R: 5`-TATCGCAGCAGTTTGTCCAG-3`; GAPDH F: 5`-AACAGCGACACCCACTCCTC-3`, R: 5`-GGAGGGGAGATTCAGTGTGGT-3`. The data were normalized to the geometric mean of the expression of the housekeeping gene GAPDH, and relative expression levels were calculated using the 2-ΔΔCT method.
Cell Counting Kit 8 (CCK-8) Assay
CCK-8 assays were used to determine the proliferation ability of GC cells. Cells were seeded into 96-well plates at a density of 3,000 cells per well and cultured overnight. Cells were quantified using CCK-8 assays (Dojindo, Tokyo, Japan) according to the manufacturer’s instructions. The absorbance data (OD value) were measured at 450 nm, and the experiment was repeated 3 times.
Plate Clone Formation Assay
Cells were seeded at a density of 3000 cells/6-cm dish and incubated at 37°C and 5% CO2 for 2 weeks; fresh medium was added every three days. The cell clones were washed with PBS, fixed for 30 mins and stained with Giemsa for 30 mins at room temperature. The number of stained clones was then counted and analyzed.
Wound Healing Assay
A total of 1.2 × 106 cells were plated into each well of a 6-well plate and cultured overnight before being scraped with a 10-μl pipette tip. After scratching (recorded as 0 h), the cells were incubated with serum-free medium for 24 hrs. ImageJ software was used to measure the area of the scratches at 0 and 24 hrs.
Transwell Migration and Invasion Assays
Cell migration/invasion assays were performed in a 24-well Boyden chamber with an 8-mm pore size polycarbonate membrane (Corning, Union City, CA, USA). An appropriate amount of Matrigel (1:8) was added to the upper chamber of transwell plates for the invasion assay, while plates without Matrigel in the upper chamber were used for the migration assay. Approximately 5× 104 cells suspended in 200 µL of serum-free medium were added to the upper chamber, and 700 µl of medium containing 10% FBS was added to the lower chamber. After 24 hrs, the cells were fixed with 4% paraformaldehyde and stained with a 0.1% crystal violet solution for 30 mins each at room temperature. Subsequently, at least six randomly selected fields were counted under a microscope (magnification, X200), and the average number was calculated.
Confocal Laser Scanning Microscopy (CLSM)
Cells were fixed with 4% paraformaldehyde for 20 mins and treated with 0.1% Triton X-100 for 5 mins. The cells were blocked with 5% normal goat serum diluted in 0.2% Triton X-100 for 1 hr. After that, the cells were incubated with E-cadherin (Cell Signaling Technology, #3195) and N-cadherin (Cell Signaling Technology, #13,116) antibodies overnight at 4°C, followed by incubation with secondary antibodies conjugated with fluorescent Alexa Fluor 594 (Cell Signaling Technology) at 37°C for 60 mins. The nuclei were stained with DAPI. Immunofluorescence was visualized under a fluorescence microscope (LSM 5, Carl Zeiss, Germany).
Statistical Analysis
Experimental groups were compared using a t-test for pairwise comparisons or ANOVA (followed by Tukey’s HSD test). Two-sided P values <0.05 were considered statistically significant. SPSS 22.0 (SPSS, Chicago, IL, USA) software was used for statistical analyses. For all experiments, statistical significance is indicated as follows: N.S., nonsignificant; *p<0.05; **p<0.01; and ***p<0.001.
Results
CSNK2A1 Is Upregulated in GC Cells
To determine the expression of CSNK2A1 in GC, we performed a Western blot analysis of four GC cell lines (SGC-790, SNU216, BGC823, and HGC27) and one gastric epithelial cell line (GES-1). The results showed that CSNK2A1 was higher in the four GC cell lines than in the gastric epithelial cells. The expression of CSNK2A1 was highest in SGC790 cells and lowest in SNU216 cells (Figure 1A). Quantitative real-time PCR (qRT-PCR) results showed that CSNK2A1 mRNA and protein levels were consistent in GC cells and gastric epithelial cells (Figure 1B).
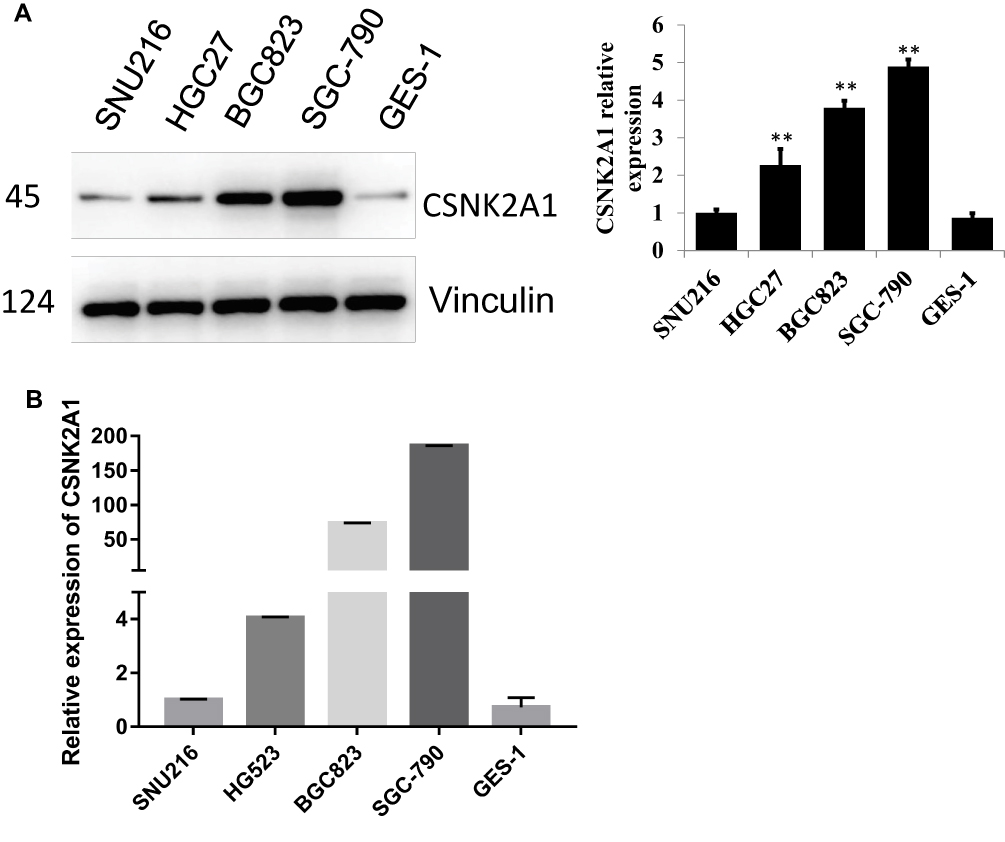 |
Figure 1 Expression of CSNK2A1 in GC. (A) Western blot analysis of CSNK2A1 expression in four human GC cell lines (SGC-790, SNU216, BGC823, and HGC27) and one gastric epithelial cell line (GES-1). (B) qRT-PCR analysis of CSNK2A1 in four human GC cell lines and one gastric epithelial cell line. (**P<0.01). Anti-CSNK2A1 antibody [EP1963Y] (ab76040) (dilution, 1:100; Abcam, Cambridge, UK). |
CSNK2A1 Enhances Cellular Proliferation, Invasion, and Migration
To explore the oncogenic function of CSNK2A1 in GC, SGC790 cells were used to establish a CSNK2A1-knockdown cell line. SNU216 cells were used to generate a CSNK2A1-overexpressing cell line. CSNK2A1 expression was evaluated by Western blot analysis (Figure 2A). In vitro functional assays were performed to investigate the role of CSNK2A1 in the proliferation, migration, and invasion of GC cells.
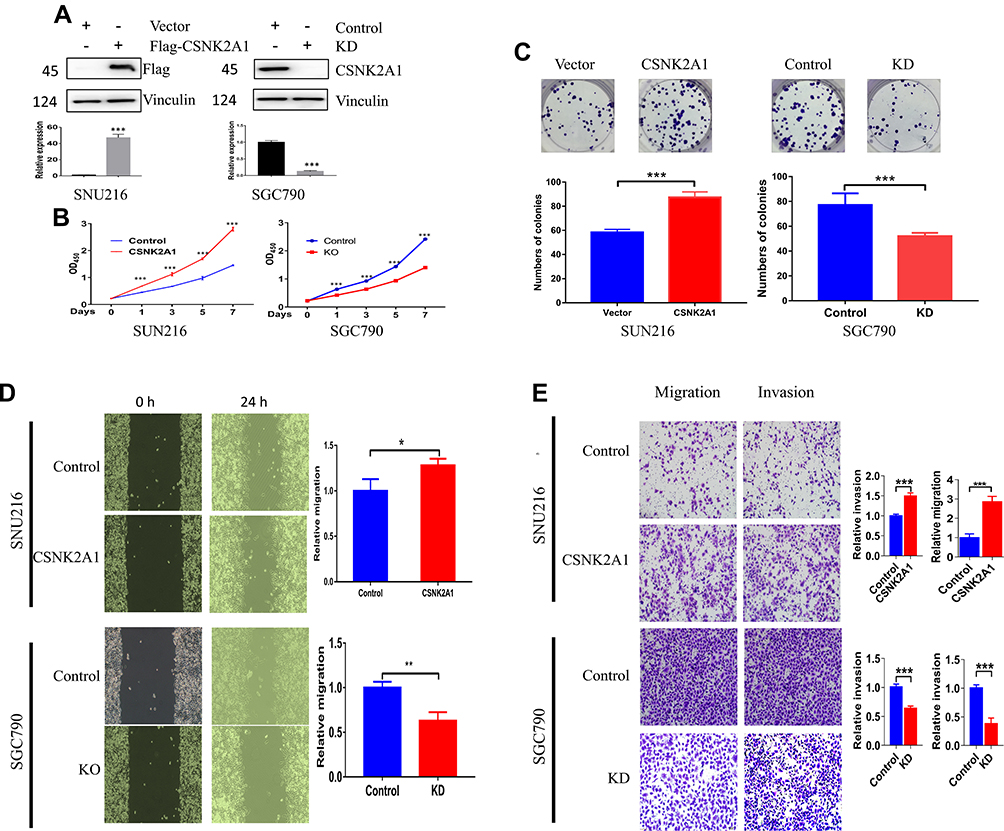 |
Figure 2 The function of CSNK2A1 in GC. (A) CSNK2A1 knockdown and overexpression effects were determined by Western blot analysis, and the results showed that the CSNK2A1-overexpressing cell line and CSNK2A1-knockdown cell line were established. (B) The CCK-8 assay results revealed that CSNK2A1 overexpression effectively increased the rate of cell proliferation and that CSNK2A1 silencing effectively reduced the rate of cell proliferation. (C) The plate clone formation assay showed that CSNK2A1 overexpression effectively increased the rate of clonal formation, and CSNK2A1 silencing effectively reduced the rate of clonal formation. (D) The wound healing assay showed that CSNK2A1-overexpressing cells had a greater migration ability than control cells, while CSNK2A1 knockdown reduced gastric cell migration. (E) Transwell invasion and migration assays showed that CSNK2A1 overexpression effectively increased invasive and migratory abilities and that CSNK2A1 silencing effectively reduced invasive and migratory abilities. (*P<0.05; **P<0.01; ***P<0.001). |
The results of the cell counting kit 8 (CCK-8) assay showed that CSNK2A1 substantially increased the rate of cell proliferation (Figure 2B). The plate clone formation assay showed that the clonal formation rate was higher in CSNK2A1-overexpressing cells than in control cells (p<0.05), while CSNK2A1-knockdown cells displayed reduced clonal formation capability (Figure 2C). To explore the migration function of CSNK2A1 in GC cells, a wound healing assay was applied. The results showed that CSNK2A1 promotes GC cell migration (p<0.05) (Figure 2D). To further confirm the invasion and migration function of CSNK2A1, transwell invasion and migration assays were performed in SGC790 and SNU216 cells. The results of the transwell invasion and migration assay showed that the invasive and migratory abilities were significantly higher in CSNK2A1-overexpressing cells than in the corresponding control cells (p<0.05) (Figure 2E).
CSNK2A1 Promotes the EMT Phenotype in GC Cells
EMT is a process by which epithelial cells lose their cell polarity and cell-cell adhesion and gain mesenchymal cell-like migratory and invasive properties. Therefore, we examined whether CSNK2A1 enhanced the EMT phenotype. The Western blot and CLSM results showed that CSNK2A1 increased the expression of the mesenchymal markers vimentin and N-cadherin and reduced the expression of E-cadherin, an epithelial marker (Figure 3).
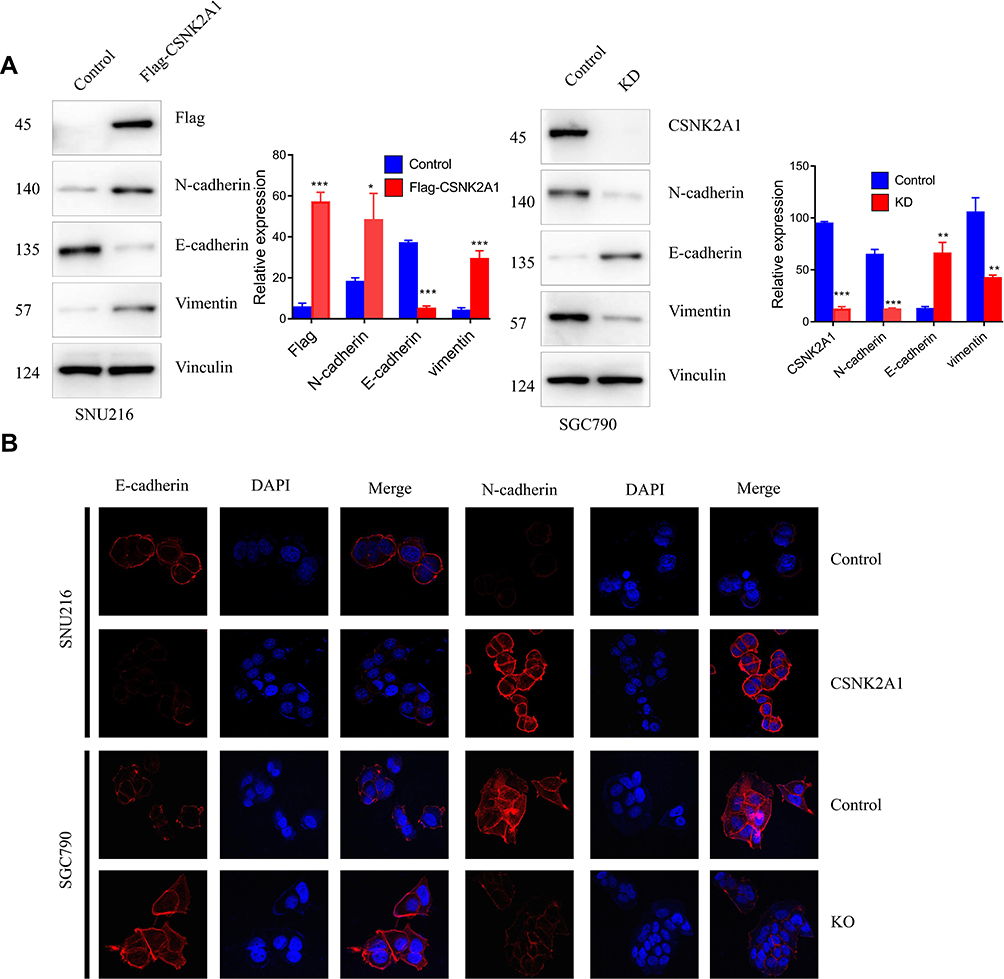 |
Figure 3 CSNK2A1 promotes the EMT phenotype. (A) Western blot results showed that N-cadherin/vimentin protein levels were increased in CSNK2A1-overexpressing cells and decreased in CSNK2A1-knockdown cells. E-cadherin protein levels were decreased in CSNK2A1-overexpressing cells and increased in CSNK2A1-knockdown cells. (B) CLSM results showed that E-cadherin was decreased and that N-cadherin was increased in CSNK2A1-overexpressing cells. Conversely, E-cadherin was increased, and N-cadherin was decreased in CSNK2A1-knockdown cells. (*P<0.05; ** P<0.01; ***P<0.001). |
PI3K-Akt-mTOR Signaling Pathway Exploration
PI3K signaling is one of the most commonly overactivated pathways in cancer. Thus, we explored whether it is involved in the CSNK2A1-enhanced invasive phenotype in GC cells. Western blotting showed that Akt and mTOR phosphorylation levels were increased in response to CSNK2A1 overexpression and decreased when CSNK2A1 was decreased (Figure 4A). To further investigate the relationship and function of CSNK2A1 in the PI3K-Akt-mTOR signaling pathway in GC, we used a PI3K inhibitor (ly294002) to treat CSNK2A1-overexpressing SNU216 cells. The Western blot results showed that ly294002 decreased the levels of the downstream molecules of the signaling pathway (p-Akt S473/T308, p-mTOR) (Figure 4B). Transwell invasion and migration assays showed that the migratory and invasive abilities of CSNK2A1-overexpressing GC cells were markedly reversed by ly294002 (Figure 4C). To exclude the possible effect of proliferation on migration and invasion, a CCK-8 assay showed that ly294002 suppressed cell proliferation by approximately 1.5 times; this reduction (approximately 1.5 times lower) cannot explain the large difference in migration and invasion (approximately 3~4 times lower) (Figure 4D). These findings indicate that CSNK2A1 promotes the proliferation, invasion and migration of GC through the PI3K-Akt-mTOR pathway.
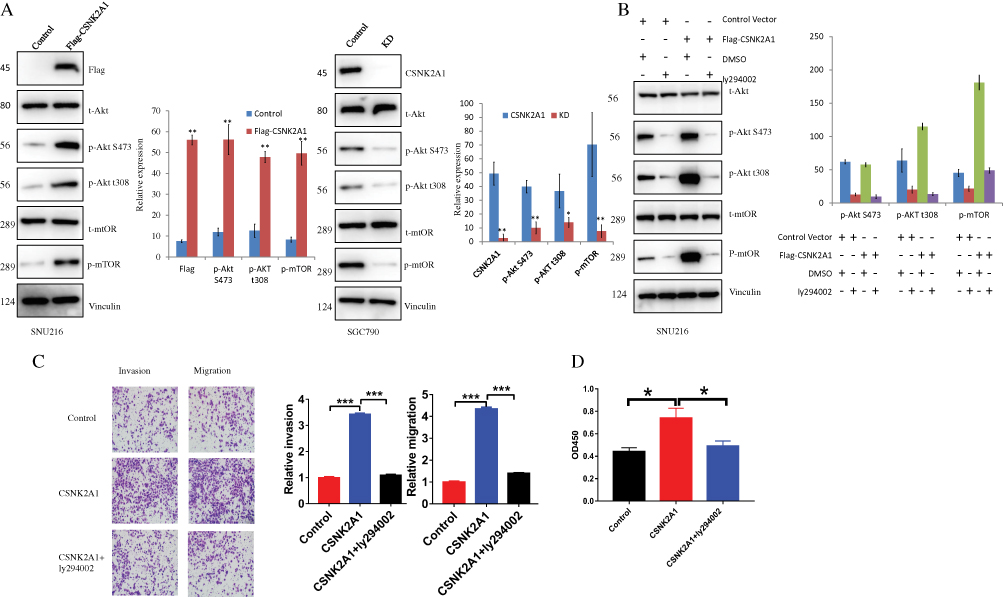 |
Figure 4 CSNK2A1 modulates the PI3K-Akt-mTOR signaling pathway. (A) The Western blot results show that the levels of p-Akt S473/T308 and p-mTOR were increased in CSNK2A1-overexpressing cells and decreased in CSNK2A1-knockdown cells. (B) The levels of p-Akt S473/T308 and p-mTOR were decreased after treatment with a PI3K inhibitor (ly294002) in the CSNK2A1-overexpressing GC cell line. (C) The migratory and invasive abilities were markedly reduced after PI3K inhibitor ly294002 treatment in the CSNK2A1-overexpressing GC cell line. (D) A CCK-8 assay was used to detect the viability of cells treated with empty vector, CSNK2A1 and CSNK2A1+ ly294002. (*P<0.05; ** P<0.01; ***P<0.001). |
Discussion
In this study, we validated that CSNK2A1 was more highly expressed in GC cell lines (SGC-790, SNU216, BGC823, and HGC27) than in a normal gastric epithelial cell line (GES-1). In agreement with our results, the expression of CSNK2A1 was found to be higher in breast cancer,16 lung cancer,17,18 kidney cancer,19 colorectal cancer,20,21 and prostate cancer.22 In the present study, CSNK2A1 was shown to play a tumorigenic role. CSNK2A1-mediated phosphorylation of SIRT6 regulates proliferation and invasion in breast carcinoma, and knockout of CSNK2A1 decreased the proliferation and invasion of breast cancer cells. Overexpression of CSNK2A1 was found to increase proliferation and invasion.16 CSNK2A1 modulates cell proliferation and invasion by regulating EMT-related genes in colorectal cancer. In our study, CSNK2A1 was downregulated and upregulated in SGC790 and SNU216 cells, respectively, and cell function studies revealed that CSNK2A1 expression was associated with migration, invasion, and proliferation in GC cells. The results showed that CSNK2A1 overexpression significantly promoted the migration, invasion and proliferation of GC cells. In addition, CSNK2A1 knockdown led to the opposite results. These results are consistent with the results of other studies.25
Several indicator molecules were found to regulate GC cell invasion and metastasis via the EMT signaling pathway.26–28 CSNK2A1 was overexpressed in colorectal cancer and modulated cell proliferation and invasion via regulating EMT-related genes. That study found that CSNK2A1 knockdown decreased vimentin, snail1, and smad2/3 expression levels, increased E-cadherin expression levels, and thus suppressed cell motility and invasion.16 In this study, the Western blot and CLSM results showed that N-cadherin/vimentin protein levels were increased in CSNK2A1-overexpressing cells and decreased in CSNK2A1-knockdown cells. E-cadherin protein levels were decreased in CSNK2A1-overexpressing cells and increased in CSNK2A1-knockdown cells. We demonstrated that CSNK2A1 can affect the expression of EMT-related indicator molecules to regulate oncogenic function. Therefore, the EMT signaling pathway is one of the carcinogenic mechanisms of CSNK2A1 in GC.
PI3K was first discovered more than two decades ago, and its critical role in oncogenesis and cancer progression is well described.29,30 A high frequency of PI3K mutations is among the most common changes found in human cancers.31 The PI3K-Akt-mTOR signaling pathway regulates various cellular processes, such as proliferation, growth, apoptosis and cytoskeletal rearrangement in various cancers. The identification of downstream kinases of the pathways provides potential targets for therapies.32 Several genes regulate proliferation, metastasis, aggressiveness and angiogenesis in GC through the PI3K-Akt-mTOR signaling pathway.33,34 CSNK2A1 was confirmed to be a gene related to the PI3K-Akt signaling pathway in CRC tumoral tissue in a previous study.21 However, the role of CSNK2A1 in the PI3K-Akt-mTOR signaling pathway in GC remains elusive. The results of this study showed that CSNK2A1 increased PI3K-Akt-mTOR signaling pathway activity by increasing p-Akt S473/T308 and p-mTOR levels. The PI3K inhibitor ly294002 decreased the levels of p-Akt S473/T308 and p-mTOR and suppressed the migration and invasion induced by CSNK2A1 overexpression. Therefore, CSNK2A1 promotes the migration and invasion of GC through the PI3K-Akt-mTOR pathway.
In conclusion, we found that CSNK2A1 upregulation in human GC may indicate an acquired malignant phenotype in tumor cells. Silencing CSNK2A1 in GC cells suppressed cellular proliferation, migration, invasion and tumor formation, suggesting that CSNK2A1 inhibition could be exploited as a therapeutic strategy for GC.
Acknowledgment
The study was funded by Shandong Medical and Health Science and Technology Development Plan Project (2016WS0522).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.v68.6
2. Markar SR, Karthikesalingam A, Jackson D, Hanna GB. Long-term survival after gastrectomy for cancer in randomized, controlled oncological trials: comparison between West and East. Ann Surg Oncol. 2013;20(7):2328–2338. doi:10.1245/s10434-012-2862-9
3. Digklia A, Wagner AD. Advanced gastric cancer: current treatment landscape and future perspectives. World J Gastroenterol. 2016;22(8):2403–2414. doi:10.3748/wjg.v22.i8.2403
4. St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: from birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66(11–12):1817–1829. doi:10.1007/s00018-009-9150-2
5. Guerra B, Issinger OG. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis. 1999;20(2):391–408. doi:10.1002/(SICI)1522-2683(19990201)20:2<391:AID-ELPS391>3.0.CO;2-N
6. Loizou JI, El-Khamisy SF, Zlatanou A, et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117(1):17–28. doi:10.1016/S0092-8674(04)00206-5
7. Gotz C, Montenarh M. Protein kinase CK2 in development and differentiation. Biomed Rep. 2017;6(2):127–133. doi:10.3892/br.2016.829
8. Ghavidel A, Schultz MC. TATA binding protein-associated CK2 transduces DNA damage signals to the RNA polymerase III transcriptional machinery. Cell. 2001;106(5):575–584. doi:10.1016/S0092-8674(01)00473-1
9. Seldin DC, Landesman-Bollag E, Farago M, Currier N, Lou D, Dominguez I. CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol Cell Biochem. 2005;274(1–2):63–67. doi:10.1007/s11010-005-3078-0
10. Trembley JH, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell Mol Life Sci. 2009;66(11–12):1858–1867. doi:10.1007/s00018-009-9154-y
11. Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2–a key suppressor of apoptosis. Adv Enzyme Regul. 2008;48:179–187. doi:10.1016/j.advenzreg.2008.04.002
12. Duncan JS, Litchfield DW. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta. 2008;1784(1):33–47. doi:10.1016/j.bbapap.2007.08.017
13. Bodenbach L, Fauss J, Robitzki A, et al. Recombinant human casein kinase II. A study with the complete set of subunits (alpha, alpha’ and beta), site-directed autophosphorylation mutants and a bicistronically expressed holoenzyme. Eur J Biochem. 1994;220(1):263–273. doi:10.1111/j.1432-1033.1994.tb18622.x
14. Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17(3):349–368. doi:10.1096/fj.02-0473rev
15. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi:10.1038/nature19057
16. Bae JS, Park SH, Jamiyandorj U, et al. CK2alpha/CSNK2A1 phosphorylates SIRT6 and is involved in the progression of breast carcinoma and predicts shorter survival of diagnosed patients. Am J Pathol. 2016;186(12):3297–3315. doi:10.1016/j.ajpath.2016.08.007
17. Wang Z, Liu H, Liu B, et al. Gene expression levels of CSNK1A1 and AAC-11, but not NME1, in tumor tissues as prognostic factors in NSCLC patients. Med Sci Monit. 2010;16(8):CR357–CR364.
18. Xie ZC, Tang RX, Gao X, et al. A meta-analysis and bioinformatics exploration of the diagnostic value and molecular mechanism of miR-193a-5p in lung cancer. Oncol Lett. 2018;16(4):4114–4128. doi:10.3892/ol.2018.9174
19. Rabjerg M. Identification and validation of novel prognostic markers in renal cell carcinoma. Dan Med J. 2017;64(10):
20. Slattery ML, Mullany LE, Sakoda L, et al. The NF-kappaB signalling pathway in colorectal cancer: associations between dysregulated gene and miRNA expression. J Cancer Res Clin Oncol. 2018;144(2):269–283. doi:10.1007/s00432-017-2548-6
21. Zhang T, Ma Y, Fang J, Liu C, Chen L. A deregulated PI3K-AKT signaling pathway in patients with colorectal cancer. J Gastrointest Cancer. 2019;50(1):35–41. doi:10.1007/s12029-017-0024-9
22. Laramas M, Pasquier D, Filhol O, Ringeisen F, Descotes JL, Cochet C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2alpha) is associated with poor prognostic factors in human prostate cancer. Eur J Cancer. 2007;43(5):928–934. doi:10.1016/j.ejca.2006.11.021
23. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN:a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
24. Kim S, Ham S, Yang K, Kim K. Protein kinase CK2 activation is required for transforming growth factor beta-induced epithelial-mesenchymal transition. Mol Oncol. 2018;12(10):1811–1826. doi:10.1002/1878-0261.12378
25. Zou J, Luo H, Zeng Q, Dong Z, Wu D, Liu L. Protein kinase CK2alpha is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J Transl Med. 2011;9:97. doi:10.1186/1479-5876-9-97
26. Sohn SH, Kim B, Sul HJ, et al. INC280 inhibits Wnt/beta-catenin and EMT signaling pathways and its induce apoptosis in diffuse gastric cancer positive for c-MET amplification. BMC Res Notes. 2019;12(1):125. doi:10.1186/s13104-019-4163-x
27. You J, Zhao Q, Fan X, Wang J. SOX5 promotes cell invasion and metastasis via activation of Twist-mediated epithelial-mesenchymal transition in gastric cancer. Onco Targets Ther. 2019;12:2465–2476. doi:10.2147/OTT.S197087
28. Cao QH, Liu F, Li CZ, et al. Testes-specific protease 50 (TSP50) promotes invasion and metastasis by inducing EMT in gastric cancer. BMC Cancer. 2018;18(1):94. doi:10.1186/s12885-018-4000-y
29. Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985;315(6016):239–242. doi:10.1038/315239a0
30. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. doi:10.1038/nrg1879
31. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi:10.1126/science.1096502
32. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi:10.1038/nrc839
33. Xing X, Zhang L, Wen X, et al. PP242 suppresses cell proliferation, metastasis, and angiogenesis of gastric cancer through inhibition of the PI3K/AKT/mTOR pathway. Anticancer Drugs. 2014;25(10):1129–1140. doi:10.1097/CAD.0000000000000148
34. Huang YK, Kang WM, Ma ZQ, Liu YQ, Zhou L, Yu JC. NUCKS1 promotes gastric cancer cell aggressiveness by upregulating IGF-1R and subsequently activating the PI3K/Akt/mTOR signaling pathway. Carcinogenesis. 2019;40(2):370–379. doi:10.1093/carcin/bgy142
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.