")
Back to Journals » OncoTargets and Therapy » Volume 8
Crosstalk with cancer-associated fibroblasts induces resistance of non-small cell lung cancer cells to epidermal growth factor receptor tyrosine kinase inhibition
Authors Choe C , Shin Y, Kim C, Choi S, Lee J, Kim S, Cho YB , Kim J
Received 31 May 2015
Accepted for publication 19 September 2015
Published 7 December 2015 Volume 2015:8 Pages 3665—3678
DOI https://doi.org/10.2147/OTT.S89659
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr William C. Cho
Chungyoul Choe,1,* Yong-Sung Shin,1,* Changhoon Kim,2 So-Jung Choi,1 Jinseon Lee,1 So Young Kim,1 Yong Beom Cho,3 Jhingook Kim1,4
1Samsung Biomedical Research Institute, Samsung Medical Center Sungkyunkwan University, School of Medicine, 2Department of Biomedical Science, Graduate School of Biomedical & Engineering, Hanyang University, 3Department of Surgery, 4Department of Thoracic Surgery, Samsung Medical Center, Sungkyunkwan University, School of Medicine, Seoul, Republic of Korea
*These authors contributed equally to this work
Abstract: Although lung cancers with activating mutations in the epidermal growth factor receptor (EGFR) are highly sensitive to selective EGFR tyrosine kinase inhibitors (TKIs), these tumors invariably develop acquired drug resistance. Host stromal cells have been found to have a considerable effect on the sensitivity of cancer cells to EGFR TKIs. Little is known, however, about the signaling mechanisms through which stromal cells contribute to the response to EGFR TKI in non-small cell lung cancer. This work examined the role of hedgehog signaling in cancer-associated fibroblast (CAF)-mediated resistance of lung cancer cells to the EGFR TKI erlotinib. PC9 cells, non-small cell lung cancer cells with EGFR-activating mutations, became resistant to the EGFR TKI erlotinib when cocultured in vitro with CAFs. Polymerase chain reaction and immunocytochemical assays showed that CAFs induced epithelial to mesenchymal transition phenotype in PC9 cells, with an associated change in the expression of epithelial to mesenchymal transition marker proteins including vimentin. Importantly, CAFs induce upregulation of the 7-transmembrane protein smoothened, the central signal transducer of hedgehog, suggesting that the hedgehog signaling pathway is active in CAF-mediated drug resistance. Indeed, downregulation of smoothened activity with the smoothened antagonist cyclopamine induces remodeling of the actin cytoskeleton independently of Gli-mediated transcriptional activity in PC9 cells. These findings indicate that crosstalk with CAFs plays a critical role in resistance of lung cancer to EGFR TKIs through induction of the epithelial to mesenchymal transition and may be an ideal therapeutic target in lung cancer.
Keywords: CAFs, lung cancer, NSCLC, direct coculture, hedgehog signaling, EMT, actin, EGFR TKIs
Introduction
Lung cancer is the leading cause of cancer-related death worldwide, and ~85% of all lung cancers are non-small cell lung cancer (NSCLC).1,2 NSCLC is defined by the accumulation of multiple genotypic alterations and comprises diverse histologic subtypes.3,4 These alterations have been intensely pursued as therapeutic targets, and the development of a series of promising molecular inhibitors for cancers harboring them has revolutionized the treatment of NSCLC.5 However, as is common in cancer therapy, acquired resistance to these targeted drugs invariably occurs and there is no effective therapy for individuals who develop such resistance. Therefore, it is imperative to understand the mechanism of drug resistance in the clinic.
Epidermal growth factor receptor (EGFR), a member of a family of closely related growth factor receptor tyrosine kinases, is overexpressed or amplified in 62% of cases of NSCLC and its expression correlates with poor prognosis.6–8 Furthermore, in unselected NSCLC samples, mutations in EGFR are present in ~10% of cases in North American and Western Europe but ~30%–50% of cases in individuals of East Asian descent, and are associated with over 50% of adenocarcinomas with bronchioalveolar features that arise in nonsmokers.9 Of various small-molecule tyrosine kinase inhibitors (TKIs) directed against EGFR that have been developed over the past 30 years, first-generation EGFR TKIs such as erlotinib and gefitinib are currently used in the clinic as first-line drugs for patients with NSCLC.10–13 Erlotinib and gefitinib are reversible inhibitors that compete with endogenous adenosine triphosphate (ATP) for binding to the kinase domain, thus preventing its tyrosine-phosphorylating activity and blocking downstream signaling.13,14 Intriguingly, with respect to efficacy and potency, retrospective studies have revealed that lung cancers that express mutated EGFRs (eg, exon 19 deletion) respond better to treatment with EGFR TKIs than lung cancers with wild-type EGFR, although some NSCLC patients with wild-type EGFR have been reported to benefit from treatment with EGFR TKIs.15,16 It has been proposed that lung cancers with mutated EGFR are dependent on the EGFR pathway and these inhibitors have a stronger binding affinity for mutant than wild-type EGFR, leading to good results in many patients.17 Invariably, however, most patients develop resistance to EGFR TKI therapy and lung cancer with heterogeneity in resistance mechanisms starts to regrow.18
Studies performed over the last several years have identified several mechanisms of acquired resistance to EGFR TKI (reviewed in Janne et al,19 Pao and Chmielecki,20 Sharma et al,21 Wheeler et al,22 and Holohan et al23). Among these, one common mechanism is a secondary point mutation of the kinase domain (eg, T790M, L747S, D761Y, and T854A in exon 2). Among those mutations, T790M mutation is found in 50%–65% of EGFR mutant resistance cases.24,25 The T790M mutation changes proper binding of the drug to the ATP binding pocket of EGFR and/or restores the affinity for ATP versus drug back to the level of wild-type EGFR, leading to more intense and sustained activation of EGFR signaling.26,27 The second well-known mechanism of gefitinib/erlotinib resistance is an oncogene kinase switch system (eg, MET amplification, AXL activation, HER2 upregulation, or KRAS activation) (reviewed in Niederst and Engelman28). For example, MET creates a bypass signaling track that activates AKT through HER3-mediated activation of PI3K in the presence of EGFR TKIs, conferring resistance to EGFR TKIs.29–31
Although the principal resistance mechanisms mentioned above have been studied for a long time, the tumor microenvironment has only recently been recognized to play a crucial role in drug resistance (reviewed in Kong and Mooney32 and McMillin et al33). In solid tumors, the microenvironment consists of tumor-associated normal epithelial and stromal cells, immune cells, and vascular cells. Indeed, NSCLC is typically characterized by a prominent desmoplastic stroma with abundant inflammation.34,35 The desmoplastic stroma creates a tumor microenvironment conducive to tumorigenesis, angiogenesis, metastatic spread of tumor cells, and drug resistance. Several signaling pathways have been reported to induce desmoplasia, including sonic hedgehog (SHh), notch, and transforming growth factor-beta (TGFβ).36,37 However, which aspects of the desmoplastic microenvironment contribute to drug resistance is still unknown.
Among desmoplastic stromal cells, cancer-associated fibroblasts (CAFs) play a crucial role in the development of desmoplasia and therapeutic resistance through signaling molecules and cell–cell interaction.38,39 Of the well-studied signaling molecules, Wnt16B and hepatocyte growth factor secreted by CAFs have shown to promote the epithelial to mesenchymal transition (EMT) program, a form of cell plasticity in which epithelial cells acquire mesenchymal phenotypes as defined by the combined loss of epithelial cell junction proteins such as E-cadherin and the gain of mesenchymal markers such as vimentin or fibronectin.23,33,40,41 The EMT phenotype was first recognized as an important feature of embryogenesis and morphogenesis during embryonic development. Further studies reported that the EMT program not only plays a crucial role in tumor cell progression but also induces drug resistance.42,43 In support of this notion, a recent study involving gene expression profiling of a large panel of NSCLC cell lines that were obtained from therapy-naïve patients showed that the EMT gene expression signature could be used as a predictive factor of resistance to the EGFR TKI erlotinib and inhibitors of PI3K-mTOR signaling.42 Moreover, resistance to EGFR TKIs was observed in cell lines undergoing EMT.44,45 The signaling that regulates the EMT is diverse and incredibly complex, involving activation of a number of different pathways (eg, TGFb, Wnt, notch, hedgehog [Hh]) that converge on several prominent transcription factor families (eg, zinc finger proteins Snail and Slug, Twist, Zeb 1/2, Smads, Gli 1/2/3) and frequently involves crosstalk between the pathways (reviewed in Thiery and Sleeman40). Indeed, the clinic development of EMT was observed in a patient with NSCLC who acquired resistance to erlotinib as a result of adaptation to therapy in the absence of known resistance mechanisms such as the EGFR T790M mutation and MET amplification.37,46–48 Given that cancer cells are surrounded by the stromal microenvironment, these findings strongly highlight the significance of the stromal microenvironment in drug resistance.
In NSCLC tumors, CAFs are a significant component of the tumor microenvironment with respect to its structure and function.49–52 Although numerous studies have reported a very clear link between resistance to chemotherapy and targeted therapy and the EMT phenotype, there are few systematic studies of the molecular and cellular mechanisms by which CAFs influence the resistance to EGFR TKIs in the context of the pathophysiology of the tumor microenvironment in NSCLC. Our previous study using direct coculture with CAFs found that the GLI1 transcriptional targets snail family zinc finger 1 and snail family zinc finger 2 are upregulated in NSCLC H358 cells, suggesting that the Hh signaling pathway is active in the tumor microenvironment.53 In the present study, we tested the hypothesis that direct contact with CAFs isolated from lung cancer patients contributes to a distinctive microenvironment that influences the sensitivity of NSCLC cells to EGFR TKIs through the Hh signaling pathway.
Materials and methods
Cell culture and reagents
Ethical approval was not sought as cell lines were commercially sought. Study protocol followed the principles outlined in the Declaration of Helsinki. Human pulmonary adenocarcinoma PC9 cells (EGFR exon 19 del E-746-A750) and bronchioalveolar carcinoma H358 cells (EGFR wild-type) were obtained from the Korean Cell Line Bank (Seoul, Republic of Korea). Cells were maintained in Roswell Park Memorial Institute (RPMI-1640) medium supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific Inc, Waltham, MA, USA). Erlotinib and cyclopamine were obtained from Cayman Chemical (Ann Arbor, MI, USA) and Sigma-Aldrich Co. (St Louis, MO, USA), respectively, and were dissolved in dimethyl sulfoxide (DMSO) at 30 and 10 mM, respectively.
Primary culture of fibroblasts
CAFs were isolated from lung cancer specimens of patients who underwent surgical resection in 2012 at the Samsung Medical Center, Seoul, Republic of Korea, as described previously (IRB No 2011-03-025-001).54 Briefly, 30 minutes after surgery, fresh tissue samples (3×3×3 mm) were minced with surgical blades in 5 mL of RPMI-1640 without serum to an approximate size of 40–60 μm in diameter or 40–200 cells/clump. Tissue fragments were subjected to enzyme digestion in a solution containing collagenase I (450 U/mL; EMD Millipore, Billerica, MA, USA) and DNase I (60 U/mL; Hoffman-La Roche Ltd., Basel, Switzerland) for 20 minutes and then seeded in a T25 culture flask in 5 mL RPMI-1640 medium containing 10% fetal bovine serum. Fibroblasts were separated from epithelial cells by treatment with Dispase II (14 U/mL; Roche) and subsequent mechanical ablation with a pulled glass pipette tip. The culture medium was changed after 48 hours to remove unattached cells and debris in suspension. Attached cells were cultured for 7–10 days to yield 1×106 cells. All fibroblasts were used after 3–7 passages of the primary culture.
Generation of stable NSCLC-GFP cell lines
Stable cells expressing green fluorescence protein (GFP) were generated by transduction with lentiviral vector carrying the GFP gene (Applied Biological Materials Inc., Richmond, Canada). From 6 days after transfection, stable GFP-expressing H358 (H358-GFP) and PC9 (PC9-GFP) cells were sorted twice by fluorescence-activated cell sorting (FACS) using a FACSAriaIII (Becton Dickinson Biosciences, San Jose, CA, USA) over a period of 3 weeks until more than 95% of the cells were stably GFP-positive.
Coculture of lung cancer cells with fibroblasts
Direct coculture in which two cell types were grown in physical contact was performed. In brief, 30,000 CAFs were plated in 24-well plate and grown for 24 hours to ~80% confluence in growth media. The following day, NSCLC cells were seeded on top of the preincubated CAFs at a ratio of 1:4. To avoid the effects of cell confluence or passage on the interpretation of data, the coculture was maintained for no longer than 7 days. Coculture plates were cultured under normal culture conditions.
Inhibitor treatments of cells
PC9 cells in monoculture and coculture were treated with inhibitors or equal volumes of the vehicle control solvent DMSO and incubated at 37°C in 5% CO2. Following incubation, as specified for each analysis, cells were processed for downstream analyses, including immunocytochemistry, proliferation assay, and gene expression as described below.
Immunocytochemistry
Fibroblasts were seeded on sterile glass coverslips and immunocytochemical staining was performed. Briefly, cells on coverslips were fixed with 4% paraformaldehyde for 10 minutes and then permeabilized with 0.5% Triton-X 100 for 5 minutes. Cells were then blocked for 1 hour with blocking solution (1% bovine serum albumin in phosphate-buffered saline [PBS]), followed by incubation with primary antibody against smoothened (SMO) (SC-166685; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 hours at room temperature. Subsequently, the cells were incubated with Alexa 594-conjugated antimouse IgG (A-11032, Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA) for 60 minutes at room temperature. To assess the subcellular organization of actin microfilaments, cells were incubated with rhodamine-conjugated phalloidin (Molecular Probes, Eugene, OR, USA) at a dilution of 1:200 (1.5 units/mL final concentration). The cells were then washed with PBS, and the coverslips were mounted on a glass slide in 10% Mowiol 4–88, 1 μg/mL 4′,6-diamidino-2-phenylindole dihydrochloride, and 25% glycerol in PBS. The cells were observed with a ZEISS FL Microscope Axiovert 200 (Zeiss, Oberkochen, Germany).
Cell sorting
After 48 hours of coculture, cells expressing GFP were trypsinized and centrifuged at 300× g for 5 minutes. The cell pellet was suspended in 2 mL PBS and the cells were sorted by FACS using a FACSAriaIII and a 488 nm optical filter. Sort gates were set by analyzing 50,000 cells from GFP-expressing and control cells. Cells were collected, centrifuged at 500× g for 10 minutes, and processed for analysis of gene expression.
Quantitative real-time reverse transcription-polymerase chain reaction
Total RNA was extracted from the sorted cocultured cells and first-strand cDNA was synthesized using oligo-dT primers and M-MLV reverse transcriptase (Invitrogen). Real-time quantitative PCR reactions were performed in triplicate in a final volume of 10 μL containing 1× SYBR® Green PCR master mix (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA), 10 ng of cDNA, and 20 pmol of each primer. Real-time quantitative PCR was performed using a 7900HT fast real-time polymerase chain reaction system (Applied Biosystems) with the following conditions: 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control in each reaction. To verify specific amplification, melting curve analysis was performed (55°C–95°C, 0.5°C/s). Quantification of relative expression was performed by the ΔΔCT method. Genes and their primers are shown in Table 1. Expression of each messenger RNA (mRNA) was normalized to that of GAPDH in the same sample.
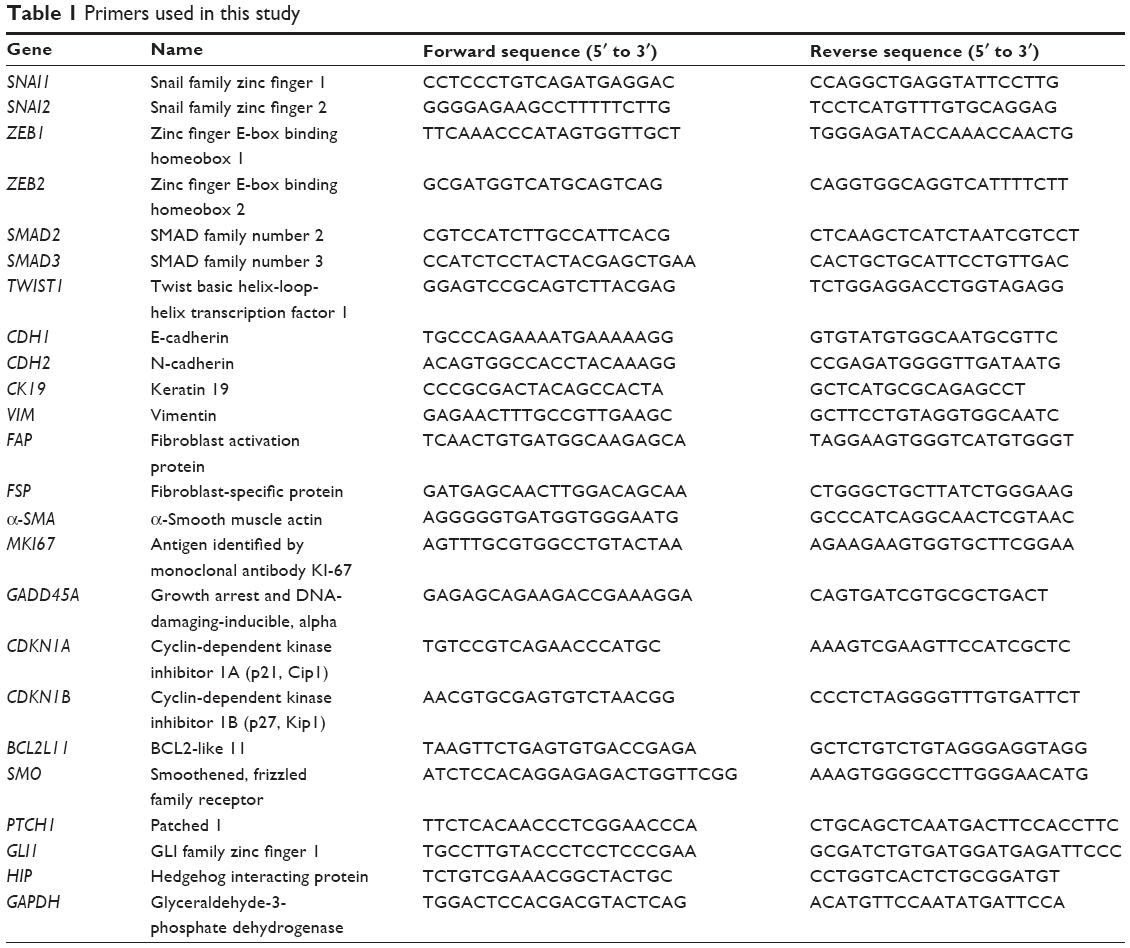 | Table 1 Primers used in this study |
Western blot analysis
Cells were lysed with RIPA buffer (sc-24,948, Santa Cruz Biotechnology) for 30 minutes on ice, and lysates were cleared by centrifugation at 13,000× g for 15 minutes at 4°C. Supernatants were incubated with 4× Laemmli sample buffer (#161-0747; Bio-Rad Laboratories Inc., Hercules, CA, USA) at 95°C for 5 minutes. The samples were then separated with sodium dodecyl sulphate-polyacrylamide gel electrophoresis gel and immunoblotted with the indicated antibodies: Vimentin (ab92547, Abcam, Cambridge, MA, USA), SMA (ab5694, Abcam), E-Cadherin (4065S, Cell Signaling Technology), Snail1 (SC-28199, Santa Cruz Biotechnology), CDK1 (610037, BD Pharmingen), CDK2 (610145, BD Pharmingen), and GAPDH (2118S, Cell Signaling Technology). GAPDH was used to show equal loading of protein.
Cell proliferation analysis
The fluorescence of cells in 24-well plates was quantified in triplicate using a GloMax plate reader (Promega Corporation, Fitchburg, WI, USA) with filter settings of excitation at 460 nm and emission at 550 nm and a bandwidth of 30 nm. Fluorescence intensity was calculated in relative fluorescence units. The nonspecific signals of wells containing cell-free medium (blank value) were subtracted from the results to give the fluorescence signals of the H358-GFP cells. Viability was determined relative to untreated controls. Each experiment was performed at least three times, each with triplicate samples. Cell viability was calculated using the following equation:
|
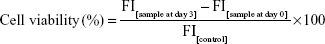
where FI[sample at day 3] is the average fluorescence intensity of erlotinib-treated sample at day 3, FI[sample at day 0] is the average fluorescence intensity of erlotinib-treated sample at day 0, and FI[control] is the average fluorescence intensity of untreated control sample at day 3. The half maximal inhibitory concentration50 (IC50) was determined from the plot of viability versus concentration of the EGFR TKI erlotinib. IC50 values were calculated by fitting the data to a sigmoid dose–response curve using nonlinear regression in Sigma plot (Systat Software, Inc., San Jose, CA, USA). Differences in IC50 were compared using Student’s unpaired t-test with P<0.05 as the limit of statistical significances.
Coculture wound healing assay PC9-GFP
Cells in coculture were allowed to grow to 80% confluence in a 100 mm dish and were then wounded by making a single scratch in the monolayer with an Eppendorf P20 pipette tip. The medium was then replaced to remove floating cells and debris, and cells were subsequently allowed to grow for 3 days to close the wound in the presence of the SMO antagonist cyclopamine (10 μM) or equal volumes of the vehicle control solvent DMSO. Photographs of the same four zones along the scratch were taken at 0, 24, and 48 hours postwound using a Nikon Eclipse inverted phase-contrast microscope.
Results
Coculture with CAFs induces erlotinib resistance in lung cancer cells
To investigate whether the erlotinib sensitivity of lung cancer cells with EGFR-activating mutations was affected by crosstalk with the host microenvironment, we cocultured NSCLC H358 and PC9 cells expressing GFP (PC9-GFP and H358-GFP, respectively) with CAFs in direct contact conditions. The ratio of cell number of NSCLC cells to CAFs is 1:4. In monoculture, the same number of NSCLC cells as in coculture was used for viability assay. PC9 cells, which harbor an exon 19 deletion E746-A750 mutation and contain five copies of the EGFR gene per cells, are purportedly sensitive to erlotinib, whereas H358 cells have been known to be resistant to erlotinib.55 A growth inhibition assay based on the measurement of GFP intensity in cancer cells showed that the EGFR-mutant cell line PC9 was sensitive to erlotinib with an IC50 <0.01 μM, whereas the EGFR wild-type cell line H358 was resistant to erlotinib with an IC50 value close to 2 μM. In contrast, both PC9 and H358 cells became resistant to erlotinib in the presence of CAFs, IC50 =0.05 μM (P<0.05) and IC50 >10 μM (P<0.05), respectively (Figure 1). Indeed, the difference in cell number between monoculture and coculture did not affect the drug response up to 1 μM of erlotinib (data not shown). Therefore, given the fact that the physiological range of erlotinib concentration is less than 1 μM, we conclude that PC9 cells are significantly resistant to erlotinib in the presence of CAFs. Taken together, these results suggest that CAF-derived secreted signaling molecules and direct physical contact, alone or in combination, induced erlotinib resistance in lung cancer cells with EGFR-activating mutations and wild-type EGFR.
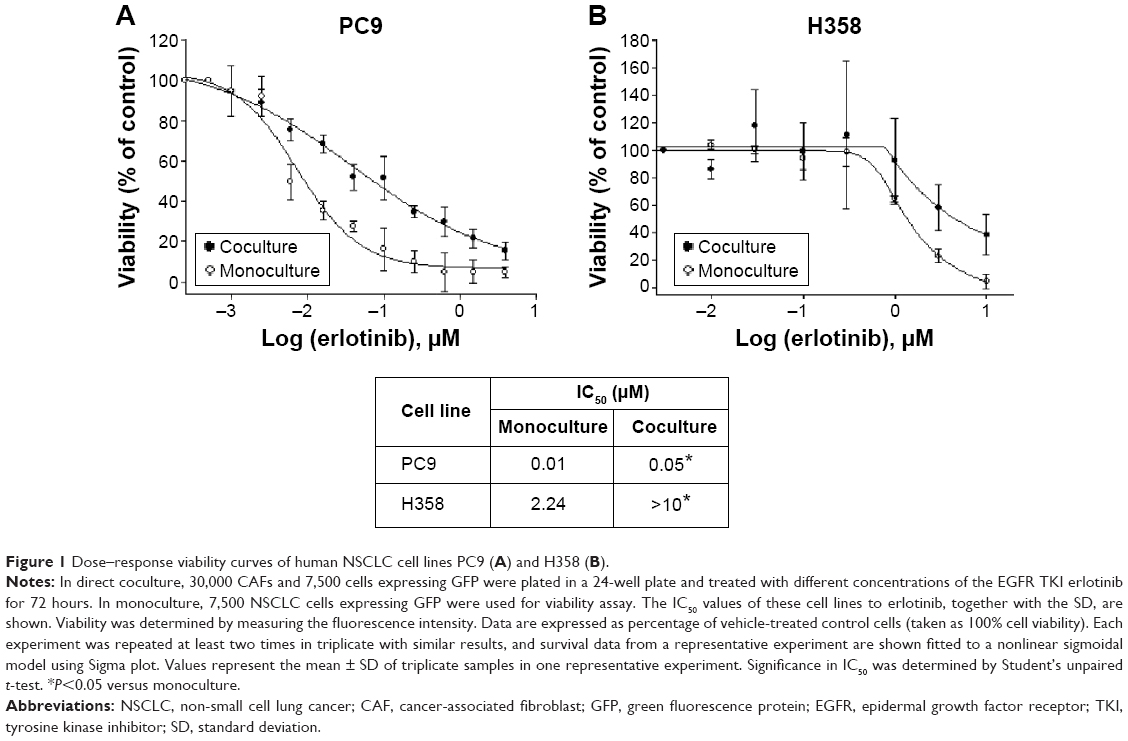 | Figure 1 Dose–response viability curves of human NSCLC cell lines PC9 (A) and H358 (B). |
Induction of EMT in human NSCLC PC9 cells by CAFs in a SMO-dependent manner
To further examine the role of CAFs in EGFR TKI resistance, we investigated the behavior of PC9 cells in direct coculture. Microscopically, PC9 cells expressing GFP alone showed typical epithelial cobblestone morphology with homotypic cell adhesion, but cells with primary cultured CAFs exhibited EMT features, including the gain of a mesenchymal phenotype with elongated and nonpolarized morphology, reorganization of filamentous actin (F-actin) with a significant enhancement of stress fibers, and decrease of cell size in comparison with monoculture (Figure 2A). Consistent with EMT induction, these cells also dispersed in coculture. To further examine the molecular mechanism underlying the induction of EMT in NSCLC PC9 cells in coculture with CAFs, we systematically studied the effect of CAFs on the expression of molecular markers for the EMT phenotype and related upstream and downstream signaling events in NSCLC PC9 cells at the levels of mRNAs (Figure 2B) and proteins (Figure 2C). Quantitative real-time reverse transcription-polymerase chain reaction analysis showed that the mesenchymal cytoskeletal markers FAP and vimentin were highly upregulated in PC9 cells cocultured with CAFs compared with the control monoculture, whereas the differentiated mesenchymal lineage marker α-SMA was activated at a relatively lower level (Figure 2B). Expression of FAP and vimentin is characteristic of mesenchymal cells and these proteins are typically absent or only weakly expressed in epithelial cell lineages.40 Furthermore, the expression level of the most crucial marker of EMT, CDH1 (E-cadherin), was highly downregulated in PC9 cells in coculture with primary cultured fibroblasts compared with PC9 cells cultured alone (Figure 2C). Among cell cycle-related proteins, expression of cyclin-dependent kinase inhibitor 1A (CDKN1A [p21CIP1]) was specifically increased in coculture with the primary cultured fibroblasts (Figure 2B). Interestingly, the seven-pass transmembrane protein SMO, which mediates Hh signaling,56 was highly upregulated in coculture and in coculture treated with erlotinib (Figure 2B). Hh signaling has been shown to be very active in cancer, especially in lung, where it promotes stromal hyperplasia, myoblast differentiation, and production of extracellular matrix, which may promote cancer cells to undergo EMT process to further facilitate the strong propensity of cancer cells for invasion and metastasis.57–65 Key components of Hh signaling include the ligands SHh, Indian Hh, and desert Hh; the negative and positive effector receptors of signaling, patched 1 (PTCH1), and SMO, respectively; and the GLI transcription factors controlling Hh target gene expression.56 Consistent with the mRNA expression levels, immunocytochemical staining using antibody to SMO showed upregulation of SMO protein (Figure 2A). In light of the fact that Hh signaling influences EMT induction, our findings suggest that the effect of CAFs on the EMT is in part mediated by Hh signaling. Furthermore, our observation that the expression of EMT markers changed in response to erlotinib (Figure 2B) indicates that the Hh-mediated EMT could be responsible for the observed drug resistance (Figure 1). Given that Hh signaling influences EMT induction, our findings further suggest that the Hh-mediated effect of CAFs on the EMT renders PC9 cells resistant to erlotinib in combination with other cellular signaling induced by CAFs. However, our hypothesis remains to be tested. Taken together, these results suggest that primary cultured CAFs derived from patients with lung cancer are potent inducers of the EMT phenotype, which contributes to acquired resistance of human NSCLC PC9 cells to the EGFR TKI erlotinib.
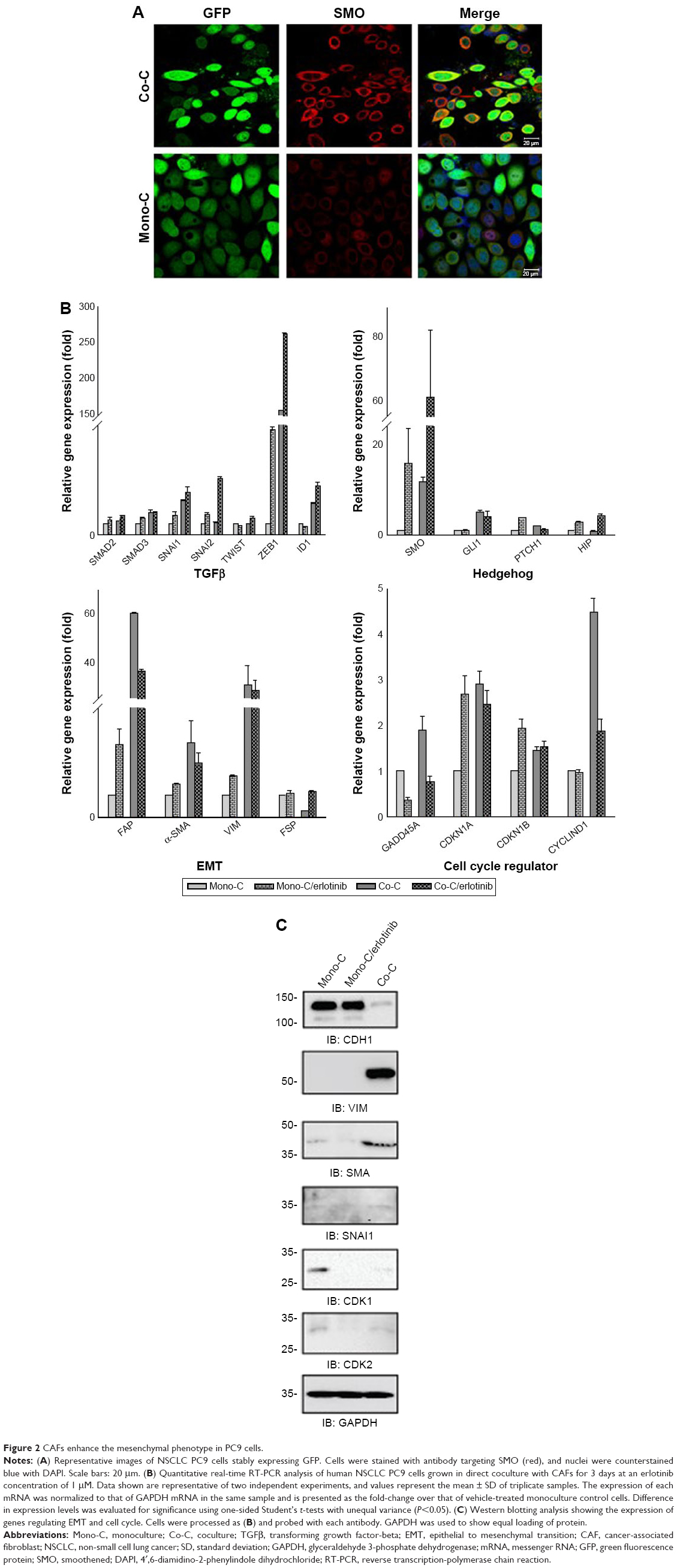 | Figure 2 CAFs enhance the mesenchymal phenotype in PC9 cells. |
SMO inhibitor inhibits the induction of EMT markers in a noncanonical manner
To examine whether the SMO-mediated Hh pathway induced by CAFs contributes to resistance to erlotinib we used cyclopamine, a plant-derived steroidal alkaloid that specifically inhibits the activity of SMO, to block the SHh pathway in NSCLC cells.66,67 Quantitative real-time reverse transcription-polymerase chain reaction analysis showed that cyclopamine downregulated the expression of genes associated with EMT (Figure 3A) but did not affect expression of GLI1 target genes, such as cyclin D1, Bcl2, and GLI itself (Figure 3A). Hh pathway activation occurs not only through activation of GLI transcription factors, but also by mechanisms of remodeling of actin cytoskeleton.68–70 To test the latter, we treated cells with cyclopamine and analyzed changes in the actin cytoskeleton (Figure 3B). Cells showed formation of stress fiber in coculture in the absence of cyclopamine. In contrast, addition of cyclopamine leads to change in actin cytoskeleton, suggesting that Hh signals in coculture stimulated the remodeling of the actin cytoskeleton. Taken together, these findings suggest that SMO-mediated Hh signaling by CAFs is transduced through actin cytoskeleton independently of GLI-mediated transcriptional activity.
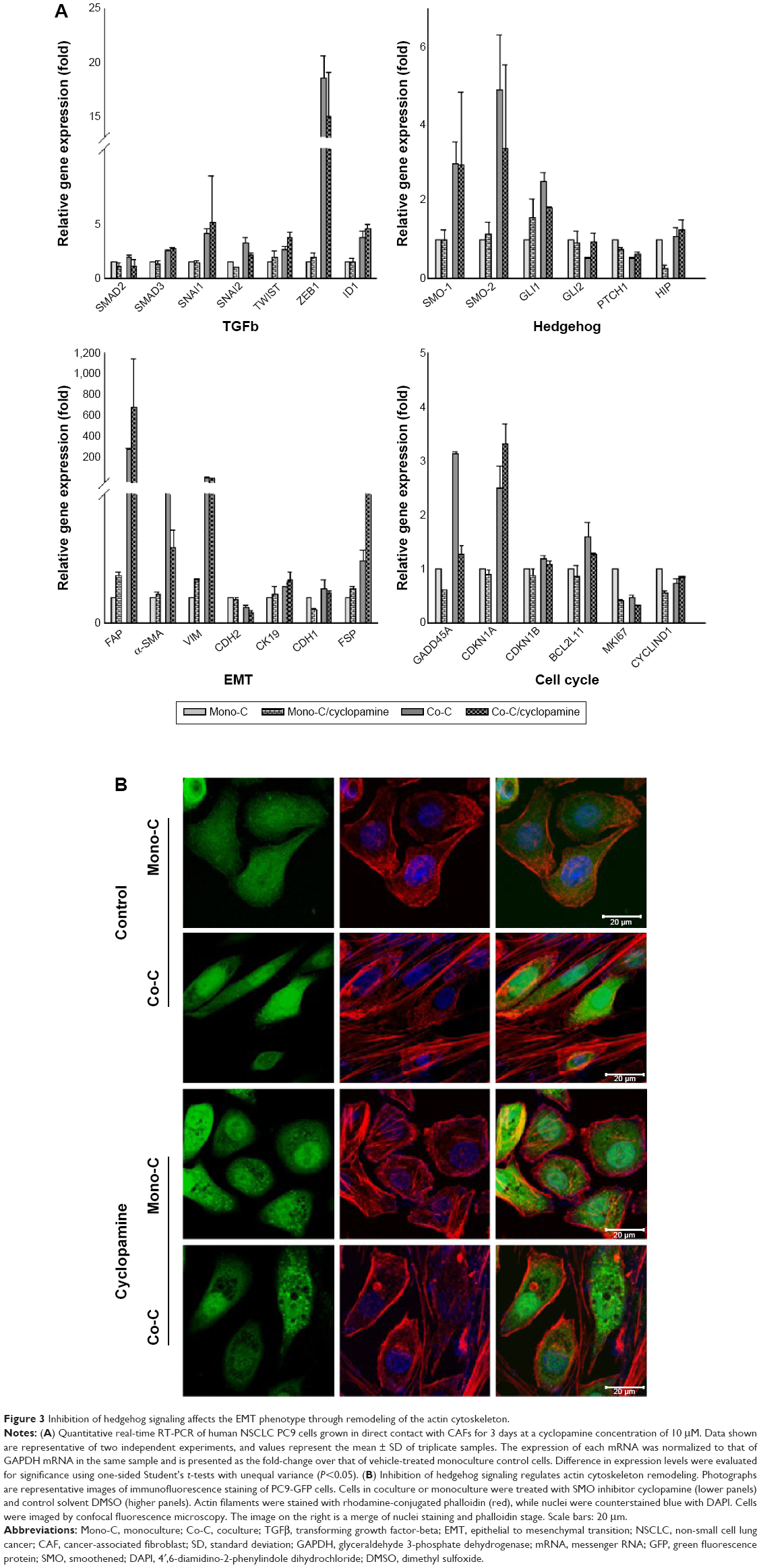 | Figure 3 Inhibition of hedgehog signaling affects the EMT phenotype through remodeling of the actin cytoskeleton. |
Inhibition of Hh signaling suppresses cell motility
To further investigate the effect of the actin-mediated SMO signaling on EMT, we studied the motility of NSCLC PC9 cells in coculture conditions. PC9-GFP cells in coculture with primary cultured fibroblasts were treated with cyclopamine and scratched and the subsequent migration of cells to close the gap was evaluated for 48 hours. Cyclopamine did not make any difference in PC9-GFP cells mobility in control monoculture, suggesting that actin-mediated EMT is not active in monoculture. In contrast, PC9-GFP cells in coculture showed the decrease in mobility in the presence of cyclopamine (Figure 4). Interestingly, cyclopamine obviously increases cell proliferation in monoculture, but not in coculture during 48 hours, indicating that cyclopamine has an opposite role for proliferation and mobility. Taken together, these results lead to the conclusion that CAFs affect drug resistance through induction of the EMT which is mediated by actin cytoskeleton rather than through regulation of proliferation.
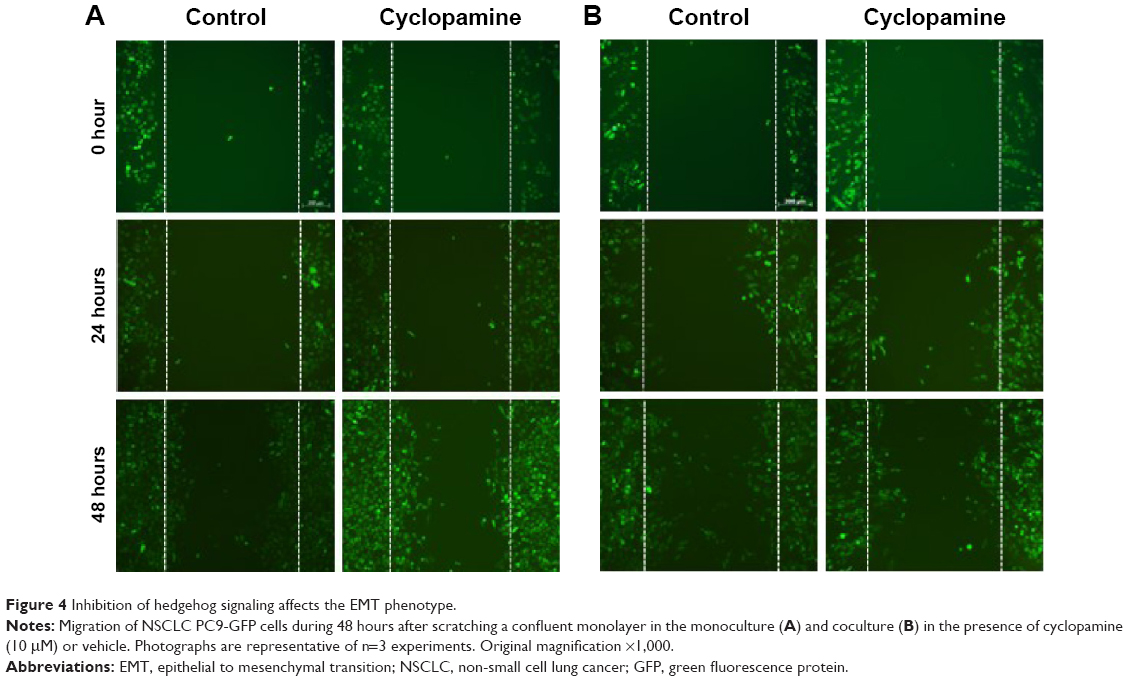 | Figure 4 Inhibition of hedgehog signaling affects the EMT phenotype. |
Discussion
A major shift in the paradigm of drug development targeting the recurrence of primary tumors following treatment with targeted kinase inhibitors is to prevent or delay their acquired resistance through regulation of normal tissue homeostasis and architecture.23 In this study, we have shown that crosstalk between the tumor cells and CAFs is responsible for the induction of resistance to the EGFR TKI erlotinib in NSCLC. CAFs activate Hh signaling, which induces EMT plasticity that renders NSCLCs resistant to erlotinib. We further showed proof-of-concept evidence that EGFR TKI resistance induced by CAFs could be circumvented by targeting the SMO signaling protein. These results indicate that targeting the crosstalk between tumor cells and CAFs may be an important novel strategy for overcoming the EGFR TKI resistance of lung cancers.
A principal question regarding the sensitivity of solid tumors to targeted kinase inhibitors is why some tumors respond and others do not. An ever-increasing list of mechanisms have been reported to mediate the response to EGFR TKIs in models based on lung cancer cells, such as genotypic alterations in EGFR and oncogenic bypass, leading to alterations in the drug target, activation of pro-survival pathways, or ineffective induction of cell death (reviewed in Janne et al,19 Pao and Chmielecki,20 Sharma et al,21 Wheeler et al,22 and Holohan et al23). In addition, studies have shown that histologic changes occur during adaptation to therapy.47,71 More importantly, the tumor microenvironment has recently been recognized to confer resistance to EGFR TKIs such as erlotinib and gefitinib.32,33 The stromal microenvironment of a tumor consists of immune cells, fibroblasts, and lymphatic and vascular endothelial cells. These stromal cells contribute to the creation of desmoplasia that involves the activation of multiple signaling pathways, including SHh that is secreted from cancer cells.36 In turn, activated stromal cells induce multiple signaling pathways in the cancer cells, including extracellular signal-related kinase and AKT pathways. Extracellular matrix proteins were also found to play a role in promoting cancer in several studies.65 For example, the extracellular matrix protein SPARC promotes EMT in cancer cells and cell–cell interactions.72 Taken together, these observations support the conclusion that desmoplastic stroma provides a novel drug target to overcome acquired resistance to EGFR TKIs.
CAFs, the principal cellular component of the tumor microenvironment, have been linked to several activities that regulate tumor progression, including angiogenesis, EMT, progressive genetic instability, deregulation of antitumor immune responses, enhanced metastasis, and enhanced growth (reviewed in Sirica38 and Madar et al39). In this regard, CAFs act as a major source of chemoattractants, which facilitate tumor cell motility and metastasis through EMT. In the present study, we find that erlotinib-sensitive PC9 cells, which harbor an exon 19 deletion E746-A750 mutation and contain five copies of the EGFR gene per cells, transdifferentiate to EMT plasticity in coculture with CAFs, leading to enhanced viability in the presence of erlotinib. How does CAF induce resistance to EGFR TKIs independent of an acquired secondary EGFR mutation? Emerging evidence indicates that CAFs provide a refuge for cancer cells from therapeutic drugs through secreted CAF-derived proteins including Wnt16B and hepatocyte growth factor, thus allowing the cancer cells to evade apoptosis and develop acquired resistance that ultimately leads to disease recurrence.33,36 In support of this idea, Wang et al demonstrated that CAFs might protect tumors through upregulation of hepatocyte growth factor expression until the drug-resistant minor clones become dominant.43 The mechanism underlying the protection provided by secreted CAF-derived proteins has been demonstrated to involve the EMT, a form of cell plasticity in which epithelial cells acquire mesenchymal phenotypes. In addition to the EMT induced by secreted CAF-derived proteins, two recent studies in lung cancer cells have identified the RTK AXL pathway as a mechanism of overcoming EGFR inhibitor resistance that is associated with development of the mesenchymal phenotype.42,73 Moreover, a recent study using a large-scale small interfering RNA screen to identify regulators of ALK-mediated resistance to EGFR TKI identified MED12, which is a component of the mediator transcription complex that is often mutated in cancers.74 MED12 knockdown was shown to induce an EMT-like feature through the activation of TGFβR signaling, and this change was associated with drug resistance. Conversely, inhibition of TGFβR signaling was shown to restore EGFR TKI responsiveness in MED12-depleted cells. These observations suggest that EMT contributes to the development of drug resistance. However, the action mechanism of EMT leading to drug resistance remains poorly understood.
In our previous study, we reported that CAFs upregulate GLI1 expression in NSCLC H358 cells.53 In the present study, we confirmed and further extended these observations in PC9 cells, suggesting that CAFs upregulate GLI1 expression regardless of cell types. However, we hypothesize that the resistance to the erlotinib could be dependent on the induction of a SMO-mediated reorganization of actin cytoskeleton in combination with GLI1 transcriptional activity. This idea is supported by our observation that H358 cells do not change the organization of actin cytoskeleton, in contrast to the PC9 cells (data not shown). SMO and patched proteins mediate the cellular response to the secreted Hh protein.56,75 During normal Hh signaling, Hh protein binds to PTCH1, thereby alleviating PTCH-mediated suppression of SMO, a distant relative of G-protein coupled receptors. SMO activation then triggers a series of intracellular events, culminating in the activation of GLI-dependent transcription.56,75 GLI activates expression of a variety of target genes, such as BMP4, FOXA2, ISL1, and FPXMI. With respect to EMT, Joost et al reported that the inhibition of GLI promotes EMT through directly regulating the transcription of E-cadherin (CDH1), a main determinant of epithelial tissue organization and cell polarity, in pancreatic cancer cells.59 In contrast to this finding, we observed that although GLI is activated through SMO activation, it does not activate any GLI target genes associated with EMT. Instead, SMO activation regulates remodeling of actin cytoskeleton. In support of our result, several studies reported that actin-mediated EMT is important in drug resistance.76,77 However, the mechanism underlying this resistance remains incompletely understood. Here, we hypothesize that cells undergoing actin-mediated EMT proliferate slowly and consequently are less susceptible to cell death induced by EGFR inhibitor.
In summary, we show that CAF-induced EMT could be responsible for resistance of lung cancers to EGFR TKIs. Our results suggest that targeting this crosstalk may be an important new strategy for overcoming resistance to EGFR TKIs in NSCLC. Thus, a more thorough understanding of which aspects of the EMT program contribute to resistance to EGFR TKIs is worthy of investigation and could lead to new therapeutic interventions in drug resistance.
Acknowledgments
This study was supported by grants from the Seoul R&BD program (SS100010) and from Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2013R1A1A2062457).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. | ||
Oxnard GR, Binder A, Janne PA. New targetable oncogenes in non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1097–1104. | ||
Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. | ||
Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet. 2013;382(9893):720–731. | ||
Cox G, Jones JL, O’Byrne KJ. Matrix metalloproteinase 9 and the epidermal growth factor signal pathway in operable non-small cell lung cancer. Clin Cancer Res. 2000;6(6):2349–2355. | ||
Hirsch FR, Varella-Garcia M, Bunn PA Jr, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21(20):3798–3807. | ||
Volm M, Rittgen W, Drings P. Prognostic value of ERBB-1, VEGF, cyclin A, FOS, JUN and MYC in patients with squamous cell lung carcinomas. Br J Cancer. 1998;77(4):663–669. | ||
Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol. 2009;6(6):352–366. | ||
Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 2003;21(12):2237–2246. | ||
Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290(16):2149–2158. | ||
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. | ||
Wakeling AE, Guy SP, Woodburn JR, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62(20):5749–5754. | ||
Moyer JD, Barbacci EG, Iwata KK, et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997;57(21):4838–4848. | ||
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | ||
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | ||
Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. | ||
Mitsudomi T, Suda K, Yatabe Y. Surgery for NSCLC in the era of personalized medicine. Nat Rev Clin Oncol. 2013;10(4):235–244. | ||
Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009;8(9):709–723. | ||
Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–774. | ||
Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. | ||
Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7(9):493–507. | ||
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. | ||
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. | ||
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. | ||
Doss GP, Rajith B, Chakraborty C, NagaSundaram N, Ali SK, Zhu H. Structural signature of the G719S-T790M double mutation in the EGFR kinase domain and its response to inhibitors. Sci Rep. 2014;4:5868. | ||
Yoshikawa S, Kukimoto-Niino M, Parker L, et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene. 2013;32(1):27–38. | ||
Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal. 2013;6(294):re6. | ||
Carey KD, Garton AJ, Romero MS, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66(16):8163–8171. | ||
Godin-Heymann N, Bryant I, Rivera MN, et al. Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation. Cancer Res. 2007;67(15):7319–7326. | ||
Mulloy R, Ferrand A, Kim Y, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67(5):2325–2330. | ||
Kong HJ, Mooney DJ. Microenvironmental regulation of biomacromolecular therapies. Nat Rev Drug Discov. 2007;6(6):455–463. | ||
McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov. 2013;12(3):217–228. | ||
Maeshima AM, Niki T, Maeshima A, Yamada T, Kondo H, Matsuno Y. Modified scar grade: a prognostic indicator in small peripheral lung adenocarcinoma. Cancer. 2002;95(12):2546–2554. | ||
Shimosato Y, Suzuki A, Hashimoto T, et al. Prognostic implications of fibrotic focus (scar) in small peripheral lung cancers. Am J Surg Pathol. 1980;4(4):365–373. | ||
Mueller MM, Fusenig NE. Friends or foes – bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–849. | ||
Xie M, Zhang L, He CS, et al. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. 2012;113(5):1501–1513. | ||
Sirica AE. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;9(1):44–54. | ||
Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’ – more than meets the eye. Trends Mol Med. 2013;19(8):447–453. | ||
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. | ||
Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2013;32(33):3846–3856. | ||
Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–290. | ||
Wang W, Li Q, Yamada T, et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15(21):6630–6638. | ||
Fuchs BC, Fujii T, Dorfman JD, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008;68(7):2391–2399. | ||
Yao Z, Fenoglio S, Gao DC, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A. 2010;107(35):15535–15540. | ||
Chung JH, Rho JK, Xu X, et al. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer. 2011;73(2):176–182. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Uramoto H, Shimokawa H, Hanagiri T, Kuwano M, Ono M. Expression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinoma. Lung Cancer. 2011;73(3):361–365. | ||
An J, Enomoto A, Weng L, et al. Significance of cancer-associated fibroblasts in the regulation of gene expression in the leading cells of invasive lung cancer. J Cancer Res Clin Oncol. 2013;139:379–388. | ||
Navab R, Strumpf D, Bandarchi B, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Nati Acad Sci U S A. 2011;108(17):7160–7165. | ||
Wald O, Izhar U, Amir G, et al. Interaction between neoplastic cells and cancer-associated fibroblasts through the CXCL12/CXCR4 axis: role in non-small cell lung cancer tumor proliferation. J Thorac Cardiovasc Surg. 2011;141(6):1503–1512. | ||
Vicent S, Sayles LC, Vaka D, et al. Cross-species functional analysis of cancer-associated fibroblasts identifies a critical role for CLCF1 and IL-6 in non-small cell lung cancer in vivo. Cancer Res. 2012;72(22):5744–5756. | ||
Choe C, Shin YS, Kim SH, et al. Tumor-stromal interactions with direct cell contacts enhance motility of non-small cell lung cancer cells through the hedgehog signaling pathway. Anticancer Res. 2013;33(9):3715–3723. | ||
Kim SH, Choe C, Shin YS, et al. Human lung cancer-associated fibroblasts enhance motility of non-small cell lung cancer cells in co-culture. Anticancer Res. 2013;33(5):2001–2009. | ||
Soh J, Okumura N, Lockwood WW, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4(10):e7464. | ||
Ayers KL, Therond PP. Evaluating smoothened as a G-protein-coupled receptor for hedgehog signalling. Trends Cell Biol. 2010;20(5):287–298. | ||
Ahmad A, Maitah MY, Ginnebaugh KR, et al. Inhibition of hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating miRNAs. J Hematol Oncol. 2013;6(1):77. | ||
Isohata N, Aoyagi K, Mabuchi T, et al. Hedgehog and epithelial-mesenchymal transition signaling in normal and malignant epithelial cells of the esophagus. Int J Cancer. 2009;125(5):1212–1221. | ||
Joost S, Almada LL, Rohnalter V, et al. GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 2012;72(1):88–99. | ||
Maitah MY, Ali S, Ahmad A, Gadgeel S, Sarkar FH. Up-regulation of sonic hedgehog contributes to TGF-beta1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS One. 2011;6(1):e16068. | ||
Ohta H, Aoyagi K, Fukaya M, et al. Cross talk between hedgehog and epithelial-mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br J Cancer. 2009;100(2):389–398. | ||
Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118(10):3331–3342. | ||
Yoo YA, Kang MH, Lee HJ, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71(22):7061–7070. | ||
Zheng X, Vittar NB, Gai X, et al. The transcription factor GLI1 mediates TGFbeta1 driven EMT in hepatocellular carcinoma via a SNAI1-dependent mechanism. PLoS One. 2012;7(11):e49581. | ||
Wong GS, Rustgi AK. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br J Cancer. 2013;108(4):755–761. | ||
Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 2002;16(21):2743–2748. | ||
Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in smoothened and patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–1009. | ||
Bijlsma MF, Borensztajn KS, Roelink H, Peppelenbosch MP, Spek CA. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007;19(12):2596–2604. | ||
Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi proteins link hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem. 2011;286(22):19589–19596. | ||
Sasaki N, Kurisu J, Kengaku M. Sonic hedgehog signaling regulates actin cytoskeleton via Tiam1-Rac1 cascade during spine formation. Mol Cell Neurosci. 2010;45(4):335–344. | ||
Zakowski MF, Ladanyi M, Kris MG; Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome Group: EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med. 2006;355(2):213–215. | ||
Robert G, Gaggioli C, Bailet O, et al. SPARC represses E-cadherin and induces mesenchymal transition during melanoma development. Cancer Res. 2006;66(15):7516–7523. | ||
Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–860. | ||
Huang S, Holzel M, Knijnenburg T, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell. 2012;151(5):937–950. | ||
Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat Med. 2013;19(11):1410–1422. | ||
Cufi S, Bonavia R, Vazquez-Martin A, et al. Silibinin suppresses EMT-driven erlotinib resistance by reversing the high miR-21/low miR-200c signature in vivo. Sci Rep. 2013;3:2459. | ||
Janji B, Vallar L, Al Tanoury Z, et al. The actin filament cross-linker L-plastin confers resistance to TNF-alpha in MCF-7 breast cancer cells in a phosphorylation-dependent manner. J Cell Mol Med. 2010;14(6A):1264–1275. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.