")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Critical role of tedizolid in the treatment of acute bacterial skin and skin structure infections
Authors Ferrández O, Urbina O, Grau S
Received 22 May 2016
Accepted for publication 12 July 2016
Published 22 December 2016 Volume 2017:11 Pages 65—82
DOI https://doi.org/10.2147/DDDT.S84667
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Olivia Ferrández,1,2 Olatz Urbina,1 Santiago Grau1,3
1Hospital Pharmacy, Hospital Universitari del Mar, Barcelona, Spain; 2Nursing Department, Universitat Pompeu Fabra, Barcelona, Spain; 3Medicine Department, Universitat Autònoma de Barcelona, Barcelona, Spain
Abstract: Tedizolid phosphate has high activity against the Gram-positive microorganisms mainly involved in acute bacterial skin and skin structure infections, such as strains of Staphylococcus aureus (including methicillin-resistant S. aureus strains and methicillin-sensitive S. aureus strains), Streptococcus pyogenes, Streptococcus agalactiae, the Streptococcus anginosus group, and Enterococcus faecalis, including those with some mechanism of resistance limiting the use of linezolid. The area under the curve for time 0–24 hours/minimum inhibitory concentration (MIC) pharmacodynamic ratio has shown the best correlation with the efficacy of tedizolid, versus the time above MIC ratio and the maximum drug concentration/minimum inhibitory concentration ratio. Administration of this antibiotic for 6 days has shown its noninferiority versus administration of linezolid for 10 days in patients with skin and skin structure infections enrolled in two Phase III studies (ESTABLISH-1 and ESTABLISH-2). Tedizolid’s more favorable safety profile and dosage regimen, which allow once-daily administration, versus linezolid, position it as a good therapeutic alternative. However, whether or not the greater economic cost associated with this antibiotic is offset by its shorter treatment duration and possibility of oral administration in routine clinical practice has yet to be clarified.
Keywords: tedizolid, tedizolid phosphate, acute bacterial skin and skin structure infections, oxazolidinone, linezolid resistance
Introduction to the management of acute bacterial skin and skin structure infections
An acute bacterial skin and skin structure infection (ABSSSI) is defined as “a bacterial infection of the skin with a lesion size area of at least 75 cm2 (lesion size measured by the area of redness, edema, or induration).”1 This definition was included in a guidance document prepared by the US Food and Drug Administration (FDA) with the aim of guiding the development of new antimicrobial agents for ABSSSI. ABSSSIs include cellulitis and/or erysipelas, wound infection, and major cutaneous abscesses.
In recent years, there has been a considerable increase in the number of emergency department visits that may be attributed to bacterial skin and skin structure infections (SSSIs).2 This type of infection was diagnosed in 1.2 million visits in 1993 versus 3.4 million visits in 2005 (95% confidence interval [CI]: 2.8–4.1; P<0.001) in a study conducted in emergency departments in the USA.2 At the same time, SSSIs were diagnosed in 1.35% (95% CI: 1.07–1.64) of all emergency department visits in 1993 versus 2.98% (95% CI: 2.40–3.56) in 2005 (P<0.001).2
The microorganisms commonly involved in ABSSIs are Streptococcus pyogenes and Staphylococcus aureus strains, including methicillin-resistant strains. Streptococcus spp., Enterococcus faecalis, and Gram-negative bacteria must also be considered to a limited extent.1
The reported increase in the number of SSSIs paralleled the emergence of methicillin-resistant S. aureus (MRSA) strains in the community.2 In a study conducted in eleven emergency departments in the USA in August 2004, MRSA strains were isolated in 59% of patients with an SSSI (range: 15%–74%).3 MRSA was the main microorganism isolated in ten of the eleven emergency departments included in the study. A total of 216 (99%) of the 218 MRSA strains analyzed had characteristics of community-acquired MRSA. Among them, the USA 300 strain was isolated in the majority of cases (212 [98.1%]). These results were reproduced in a subsequent prevalence study conducted in 12 hospital emergency departments in the USA in August 2008.4 However, variability based on site was lower in this year (range: 38%–84%).
At the same time, the prevalence of MRSA in SSSIs was 46% in 96 patients with these infections enrolled in a study conducted in an emergency department in a Los Angeles hospital.5 The rate of MRSA SSSIs increased from 29% in 2001–2002 to 64% in 2003–2004. The prescription of antibiotics with activity against community-acquired MRSA strains has paralleled this microorganism’s epidemiology. From 1993 to 2001, this was limited, but in 2005, it increased to 38% (95% CI: 30–45; P<0.001).2
The data from studies conducted in Europe have shown a similar situation.6 In a study conducted at 106 sites in 19 European countries, S. aureus strains were isolated in 32.6% of 3,000 strains associated with SSSIs.7 More than 50% of these corresponded to MRSA strains.
The increase in the prevalence of community-acquired MRSA strains has greatly impacted the selection of empirical antibiotic treatment for ABSSSIs (Table 1). Thus, for treating an abscess, the Infectious Diseases Society of America guidelines recommend, in addition to incision and drainage, administering an antibiotic active against MRSA when the initial antibiotic treatment has failed or when the patient has immunosuppression or systemic inflammatory response syndrome and hypotension.8 In the case of cellulitis and erysipelas, including an antibiotic with activity against MRSA, as well as against streptococcus, is recommended when the infection is associated with penetrating trauma or when there is evidence of MRSA infection, MRSA colonization, parenteral drug use, or systemic inflammatory response syndrome.8 Finally, including an antimicrobial agent active against MRSA in the treatment of surgical wound infections is recommended in patients with risk factors for infection by this microorganism (nasal colonization or prior infection, hospitalization, or recent antibiotic administration).8
 | Table 1 Empirical treatment of acute bacterial skin and skin structure infections |
These guidelines were published before the FDA approved other antibiotics with activity against Gram-positive microorganisms, including MRSA, in ABSSSIs. These include tedizolid, belonging to the oxazolidinone family, and dalbavancin and oritavancin, both belonging to the lipoglycopeptide group. The therapeutic role of these compounds was evaluated in a previous review.9 Tedizolid’s different pharmacological and microbiological characteristics and efficacy in SSSIs will be discussed throughout this review. Regarding lipoglycopeptides, both oritavancin and dalbavancin may be suitable alternatives in patients with a history of prior catheter-related complications and patients who are candidates for receiving antibiotic treatment on an outpatient basis or with low adherence to oral antibiotic treatment.9 In addition, oritavancin is the only glycopeptide that shows activity against vanA-mediated vancomycin-resistant Enterococcus spp., which positions this antimicrobial agent as a good treatment option in infections caused by these microorganisms.9 However, some issues related to these compounds remain to be clarified.9 Notable among them is the absence of safety data related to the long-term administration of multiple doses of these antimicrobial agents, which is very important for being able to identify their positioning in ABSSSIs.9 However, studying this issue will not be free from complications in the case of some compounds such as oritavancin and dalbavancin, given that they are administered in one, or one or two doses, respectively.
Pharmacology
Tedizolid phosphate (TR-701) is an inactive synthetic compound that is rapidly hydrolyzed by phosphates in the body to the active compound tedizolid (TR-700), whose chemical structure is (R)-3-(4-(2-(2-methyltetrazole-5-yl)pyridine-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidinone-2-one.10,11
Similar to linezolid, tedizolid has a class pharmacophore group, the 3-(3-fluorophenyl)-oxazolidinone ring (ring A), which places these antimicrobial agents in the oxazolidinone family.
Both compounds have a lateral chain at C-5 of ring A that grants them greater potency against microorganisms susceptible to this family of antibiotics (Gram-positive microorganisms and mycobacteria). However, there are structural differences between the two antimicrobial agents. The C-5 methylcetamide chain of the linezolid oxazolidinone ring is replaced by a hydroxymethyl group in tedizolid. This structural modification was initially associated with a decrease in its potency. Subsequently, it was observed that the addition of the methyltetrazole ring to D not only offset this lower activity of tedizolid but also increased its antimicrobial potency versus linezolid.12
In addition, after evaluating several prodrugs with the aim of increasing the bioavailability of tedizolid, it was found that phosphorylation at C-5 (monophosphate ester) increased water solubility and stability in the pH range from 3 to 7 and facilitated rapid hydrolysis by nonspecific endogenous phosphatases.13
In addition, it will also represent an improvement in the interaction profile with monoamine oxidase (MAO) (Figure 1). As a result, it is administered in the form of tedizolid phosphate both orally and intravenously.
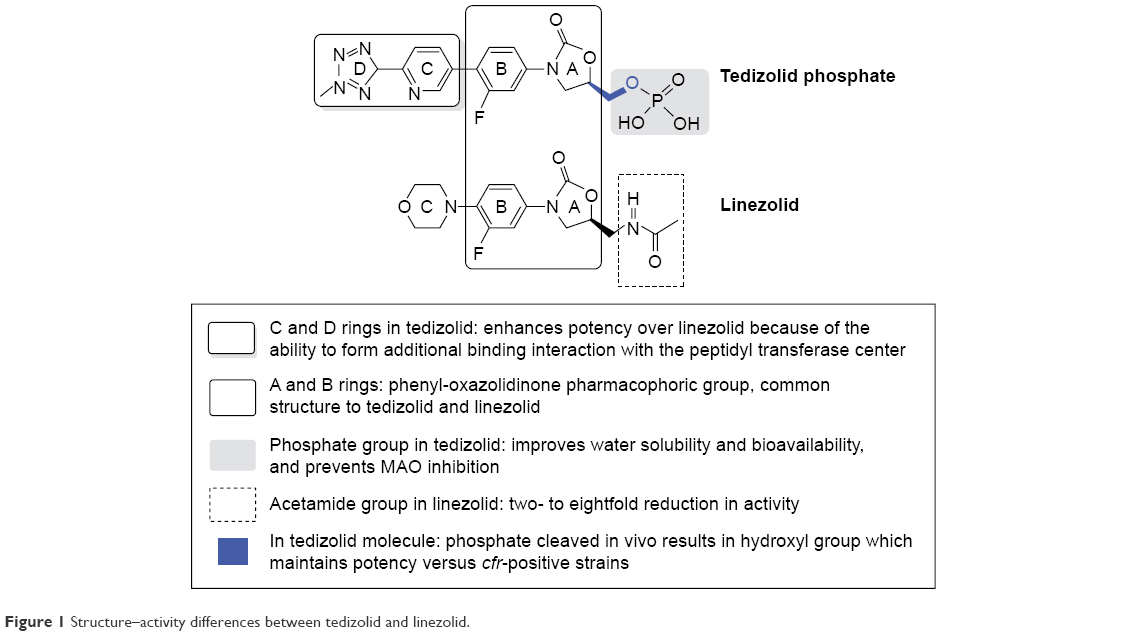 | Figure 1 Structure–activity differences between tedizolid and linezolid. |
Mode of action
Similar to linezolid, tedizolid exerts its bacteriostatic activity by inhibiting bacterial protein synthesis by binding to the 23S rRNA of the 50S ribosomal subunit, thus preventing the formation of the 70S ribosomal initiation complex formed by binding the 50S, 70S, and N-formylmethionine-tRNA subunits. It interrupts the translation process by blocking the alignment of aminoacyl-tRNA at the peptidyl transferase site, thereby interrupting peptide elongation and the start of protein synthesis.14–17
Pharmacokinetics
The use of tedizolid phosphate, the phosphorylated prodrug, increased the solubility of tedizolid, the active compound in water, which increased its bioavailability, given its lipophilic character.18 Following oral administration, tedizolid phosphate is rapidly converted to the active drug tedizolid by apical alkaline phosphatases, nonspecific enzymes located in the membranes of multiple cells in the body. Without these enzymes, intestinal absorption of the prodrug would be limited.19
The bioavailability of tedizolid following administration of a single oral dose of tedizolid phosphate 200 mg was 91.5%.20 The pharmacokinetics of tedizolid phosphate in healthy subjects was similar following oral and intravenous administration, so it is not necessary to perform a dose adjustment when switching from the intravenous route to the oral route.20
Lower bioavailability values have been reported in other studies that have included populations from far eastern countries such as the People’s Republic of China (85.5% [90% CI: 69.3–105])21 and Japan (82.6% [90% CI: 77.9–87.6]).22
Although one study observed a delay in absorption and lower maximum drug concentration (Cmax) values following administration of a single dose of tedizolid phosphate 600 mg together with food, the area under the curve (AUC) and t1/2 parameters remained unchanged, and so tedizolid phosphate may be administered with or without food.23
After analyzing the parameters from seven clinical trials, Flanagan et al24 established that the pharmacokinetics of tedizolid fits a two-compartment model with sigmoidal absorption, absolute bioavailability, and linear elimination.
An initial study by Flanagan et al20 sought to determine the pharmacokinetics of single escalating doses or multiple escalating doses of intravenous tedizolid phosphate as well as determine its oral bioavailability in healthy subjects.
First, the pharmacokinetics of single escalating doses of tedizolid phosphate administered intravenously (placebo or tedizolid phosphate 100, 200, or 400 mg) was studied. Second, placebo or tedizolid phosphate 200 or 300 mg was administered intravenously on a double-blind basis once daily for 7 days. Third, the absolute bioavailability of a 200 mg dose of oral tedizolid versus a 200 mg dose of intravenous tedizolid was evaluated. The two administrations were separated by a 7-day washout period.
Following intravenous administration of tedizolid phosphate 200 mg, plasma tedizolid phosphate concentrations reached a plateau at 15 minutes and subsequently its plasma levels decreased rapidly until arriving at plasma concentrations below the detection limit 2 hours after the start of the infusion. At the same time, plasma tedizolid phosphate levels were undetectable following oral administration of 200 mg of the antimicrobial agent. In addition, either tedizolid phosphate was not detected in urine or only 0.005% of the administered dose was detected.
The results of the aforementioned studies led to subsequent studies seeking to study the pharmacokinetics of tedizolid, the active compound.20
Unlike tedizolid phosphate, the plasma concentration of tedizolid increased in an escalating manner following administration of single ascending doses of tedizolid phosphate intravenous (100–400 mg).20 A dose-dependent increase of tedizolid was observed for tedizolid’s Cmax (1.2–5.1 μg/mL). Similar results were observed for mean AUC0–t and AUC0–∞, suggesting linear pharmacokinetics.
The mean steady-state apparent volume of distribution 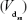 was 61.2–74.5 L, and the mean of the elimination kinetic parameters was 9.3–13.4 hours for t1/2 and 4.8–5.8 L/h for clearance (Cl). In these cases, the parameters were dose independent.
was 61.2–74.5 L, and the mean of the elimination kinetic parameters was 9.3–13.4 hours for t1/2 and 4.8–5.8 L/h for clearance (Cl). In these cases, the parameters were dose independent.
However, oral administration of single escalating doses of tedizolid phosphate (200, 400, 600, 800, and 1,200 mg) under fasting conditions (10 or more hours before and 4 hours after administration of the drug) yielded escalating Cmax values, but they were no longer proportional to the administered dose. By contrast, AUC values did retain this proportionality, mainly with the administration of lower doses. The t1/2 was higher at 10 hours for all administered doses.23
Tedizolid phosphate reached the steady state in 3 days,21 and once reached, it accumulated minimally and predictably. Drug accumulation ratio calculated in day 3 for Cmax was 1.16 (17.7%; range: 0.861–1.61) after intravenous administration. Similarly, the accumulation of tedizolid was 28% following intravenous administration of tedizolid phosphate 200 mg once daily for 7 days in a different study.20 Tedizolid exposure was slightly greater on day 7 than on day 1 (Cmax 3.0 [0.7] mg/mL and 2.3 [0.6] μg/mL, respectively; AUC0–24 29.2 (6.2) mg×h/mL and 22.3 [4.2] mg×h/mL, respectively). On day 1, tedizolid’s Cl was 6.4 (1.2) L/h and its Vd was 77.6 (15.9) L. On day 7, tedizolid’s Cl was 5.9 (1.4) L/h and its Vd was 80.1 (21.0) L. Again, these values were not proportional to the administered dose. The mean 24-hour urine recovery of tedizolid was approximately 1% on both day 1 and 7.20
After administering multiple doses of oral tedizolid phosphate (200, 300, or 400 mg for 21 days under fasting conditions), dose-proportional increases in tedizolid Cmax and AUC values at day 15 (200 mg) or at day 21 (300 and 400 mg) were observed.23 This contrasted with the kinetics of linezolid after administering doses of 600 mg every 12 hours for 21 days under fasting conditions, given that with linezolid a greater accumulation at steady state was observed (~72%).23 Linezolid steady-state Cmin were 3.9 and 8.02 mg/L, after multiple oral doses of 375 and 625 mg given every 12 h for 14 days, respectively.25 Cmax were 13.2 and 18.8 mg/L, respectively.
In the previous, tedizolid Cl was generally independent of both dose and the number of doses administered, while oral Cl of linezolid Cl decreased with the number of administrations.23 Clearance values at steady state were 10–30% lower with multiple oral administration and 11–19% lower with multiple intravenous administration compared with single dose.26 Values for t1/2 of linezolid 600 mg were 3.80 (1.67) hours on day 1 and 5.75 (1.15) hours on day 21.23 Mean tedizolid t1/2 was approximately 2-fold higher than that of linezolid.
The active drug tedizolid is a lipophilic compound with high penetration in plasma, interstitial fluid, muscle and adipose tissue, epithelial lining fluid, and pulmonary alveolar macrophages, and this is based on data from studies performed using microdialysis. Thus, plasma concentrations of tedizolid may be used as a direct surrogate for tissue concentrations.24,27,28 The ratio of penetration into the epithelial lining fluid and alveolar macrophages (AUC0–24/fAUC0–24) were approximately 40-fold and 20-fold, respectively, in 20 healthy adults.27
One study involved administering a single dose of tedizolid in both animals and humans.29 In vitro plasma protein binding of tedizolid was 97.7% in rats, 92.6% in mice, 78% in dogs, and 84.6% in humans.29
The Vd of tedizolid was more than double the value observed with linezolid (Vd: 101 L versus 40–50 L).29 After the dose, 87.6% of radioactivity was recovered at 96 hours, and 99.5% of radioactivity was recovered at 288 hours. Recovery in feces was 81.5%, and recovery in urine was 18%. In both feces and urine, tedizolid accounted for only 3% of recovery, whereas, in plasma, it was the main form.
Similar to another study in healthy adults, it showed plasma protein binding ranging from 86.1% to 91.9%.27 This plasma protein binding was considerably higher than that of linezolid (31%).25
Tedizolid is mainly metabolized by the liver. Tedizolid sulfate is the major isolated metabolite in both feces (69% [56.81–79.49%]) and urine (10% [7.17–14.2%]). This metabolite’s antimicrobial activity is very limited. Other minor isolated metabolites were carboxy tedizolid and desmethyltedizolid. Among these, only desmethyltedizolid seemed to retain antimicrobial activity.29
Regarding the formulation of oral tedizolid phosphate, Flanagan et al23 demonstrated similar pharmacokinetics after administration of tedizolid phosphate powder form from a capsule or tablet formulation. Kennedy et al30 evaluated the stability and recovery of the dose contained in the tedizolid phosphate capsule after it was crushed and dispersed in water. The recovery of tedizolid phosphate was greater than 93% of the initial dose after simulating its passage through two different types of nasogastric tubes. The reconstituted solution remained stable for 4 hours at room temperature, with minimal degradation of the initial administered dose. These findings ensure the possibility of crushing the tablet for patients with swallowing problems.
Special populations
Pharmacokinetic studies conducted by Flanagan et al24 in different populations (healthy adults, patients with ABSSSIs and patients with different degrees of organ damage enrolled in Phase I and III trials) indicated that none of the “clinical covariates” (age, sex, race, body mass index [BMI], renal failure, or hepatic failure) significantly affected tedizolid exposure.
The pharmacokinetic behavior of tedizolid showed no variation after it was evaluated in adolescent subjects (12–17 years).31 Mean tedizolid AUC values for adolescents were within 15% of previously reported adult values after oral or intravenous administration of a single dose of 200 mg tedizolid phosphate (geometric mean ratio (90% CI) of AUC adolescents/AUC adults after 200 mg of single dose oral administration was 0.847 mg×h/mL [0.736–0.975]).
Mean Cmax value was similar to adults after oral administration of a single dose of 200 mg. However, this value was ~43% higher in adolescents after intravenous administration than in adults (3.66 mg/mL versus 2.55 mg/mL, respectively).
There are no data in populations under 12 years of age. These results are in contrast with the pharmacokinetic data for linezolid in a population between 3 months and 16 years of age, in which availability of linezolid was age-dependent and Cl and Vd were higher compared to the adult population (mean values for Cl and Vd were 0.34±0.15 L/h/kg and 0.73±0.18 L/kg, respectively). This led to the need to apply a special dosage regimen in this age-group (10 mg/kg 2–3 times/d).32
At the same time, the pharmacokinetic data for administering a single dose of tedizolid 200 mg to 40 subjects over 65 years of age versus 14 subjects ranging from 18 to 45 years of age demonstrated an approximately 9% increase in the geometric mean ratio of the Cmax and an approximately 13% increase in the geometric mean ratio of the AUC.33 No discontinuation of treatment because of adverse events was required. For all these reasons, similar to linezolid,25 no dose adjustment seems necessary for oral administration of tedizolid phosphate in those over 65 years of age.
One study evaluated the pharmacokinetics of tedizolid in obese patients (BMI ≥30 kg/m2) and severely obese patients (BMI ≥35 kg/m2).34 According to the results, the pharmacokinetics was similar to those observed in nonobese patients, independently of the degree of obesity, and so a dose adjustment is not required in this population.
According to the results of this study, only ideal body weight (IBW) showed a statistically significant relationship to the pharmacokinetics of tedizolid.34 However, this finding was not considered to be clinically significant. Regarding linezolid, one study in 20 healthy obese volunteers (BMI: 30–54.9 kg/m2) demonstrated similar exposure to the antimicrobial agent after administering five doses of 600 mg IV every 12 hours (AUC for the 12 hour dosing interval was 130.3±60.1 mg×h/mL for moderately obese patients and 109.2±25.5 mg×h/mL for morbidly obese patients; P=0.032).35 This finding suggested that it is not necessary to adjust the dose of linezolid in people with a weight ≤150 kg.35 Another study compared the pharmacokinetics of linezolid in five obese adults (BMI >35 kg/m2) before and after Roux-en-Y gastric bypass surgery (RYGBS).36
According to the results, serum exposure for the antimicrobial agent was approximately 50% lower than that of the nonobese population, which suggested that obese patients could require a personalized dose adjustment. These differences were maintained when the pharmacokinetic parameters were compared prior to the surgical procedure with population data (mean [SD] AUC0–∞ with oral linezolid before RYGBS was 41.6 [20.9] mg×h/L compared with 98.9 [24.7] mg×h/L after RYGBS [P<0.001]).36 However, there is still no dosage regimen for oxazolidinones in obese people, given that pharmacokinetics in this population is commonly unpredictable and determined by other comorbidities such as renal failure.37
Regarding renal failure,38 the pharmacokinetics of administering a single dose of IV tedizolid was similar in patients with severe renal impairment without dialysis (estimated glomerular filtration rate <30 mL/min/1.73 m2)39 and in the control group.
In addition, it was observed that exposure to tedizolid was 25% lower in patients with end-stage renal failure than that observed in patients with renal failure who did not require hemodialysis and in the control group made up of healthy subjects. The authors observed no significant differences when they compared exposure to tedizolid prior to or subsequent to dialysis (Cmax geometric mean ratio 1.148 [90% CI: 1.053–1.252]; AUC0–∞ geometric mean ratio 0.913 [90% CI: 0.827–1.007]). The dosage loss of the antimicrobial agent was lower than 10% in subjects who received tedizolid prior to dialysis. This finding paralleled tedizolid’s high degree of plasma protein binding, and its elimination was less than the 30% observed with linezolid. Thus, its pharmacokinetics is independent of the time of administration with respect to dialysis (before or after). In this regard, it differs from linezolid, which should be administered after this technique.40
The authors concluded that no dose adjustment is required in patients with any degree of renal failure. In this regard, it is similar to linezolid, which does not seem to require an adjustment in the different stages of renal failure, even though approximately 35% of it is excreted by the kidneys (total apparent oral Cl of linezolid ranged from 92.5 to 109.6 mL/min in subjects with 24 hour urinary creatinine clearance of 10–39 mL/min, 40–80 mL/min, or >80 mL/min).25,41 However, having renal function deterioration has been associated with higher plasma concentrations of linezolid and the development of adverse events such as thrombocytopenia.42 Therefore, follow-up or even monitoring of the plasma concentration of linezolid when possible could be advisable in these patients.43 Linezolid has also been observed to be related to an insufficient plasma concentration (minimum drug concentration [Cmin] <2 mg/L) in patients with an estimated glomerular filtration rate higher than 80 mL/min/1.73 m2.44
This lower exposure to tedizolid in patients with end-stage chronic renal disease was examined again in a post hoc analysis of the data presented.45 To do this, AUC was determined for patients with different stages of renal failure and correlated with age, sex, and BMI. In addition, IBW was calculated for each participating subject. The mean (standard deviation) AUC0–∞ value was 23.27 (7.50) mg×h/mL in the end-stage renal disease patients, 29.99 (8.97) mg×h/mL in the severe renal impairment patients, and 32.43 (9.53) mg×h/mL in the control patients. Exposure to tedizolid was inversely proportional to IBW, which explained the lower AUC values in patients with end-stage renal disease who showed higher IBWs. The authors attributed the lower concentrations of tedizolid in these patients with end-stage chronic renal disease to the fact that they had a higher IBW. This covariate yielded greater variability in the pharmacokinetics of tedizolid, more than a different behavior on the part of the antibiotic in this population, and therefore they confirmed that it is not necessary to adjust the doses in renal disease.45
The same authors studied the pharmacokinetics of tedizolid after administering a single dose of oral tedizolid phosphate 200 mg in patients with moderate or severe hepatic failure according to the Child–Pugh classification.38 They observed minimal differences in the pharmacokinetic parameters calculated both between patients with moderate hepatic failure and severe hepatic failure and between these patients and their controls. The parameter with the most variation was the AUC0–∞ value, which was 34% higher in patients with severe hepatic failure (geometric mean ratio 1.341 [90% CI: 0.927–1.939]) and 22% higher in patients with moderate hepatic failure compared to their controls (geometric mean ratio 1.216 (90% CI: 0.862–1.716). These slight increases in exposure to tedizolid were not associated with clinical significance, given its good tolerance, and so the authors concluded that it is not necessary to adjust the dose of tedizolid in this group of patients. Similarly, exposure to linezolid increased by 1.3 times in subjects with mild-to-moderate hepatic failure compared to healthy subjects. However, this increase was not associated with the need for dose titration in this population.46
Pharmacodynamics
The pharmacodynamic behavior of neither tedizolid phosphate nor tedizolid has been fully established. One of the main studies that sought to evaluate this behavior involved tedizolid phosphate/tedizolid dose fractionation studies in a neutropenic mouse model with a thigh infection caused by methicillin-sensitive S. aureus (MSSA) and MRSA strains.47 It also compared their activity in vivo versus linezolid. The total dose of tedizolid phosphate was provided as equivalent doses of tedizolid. The AUC0–24/minimum inhibitory concentration (MIC), T>MIC, and Cmax/MIC free drug ratios were calculated for daily doses of tedizolid phosphate of 10, 20, 36, and 72 mg/kg/24 h, fractionated into one, two, or four daily doses. According to the results, the AUC0–24/MIC pharmacodynamic ratio achieved the best correlation with the efficacy of tedizolid (r2: 0.984), versus the T>MIC ratio (r2: 0.624) and the Cmax/MIC ratio (r2: 0.757). Thus, a value for the AUC0–24/MIC ratio of 49.3 for tedizolid and 105.9 for tedizolid phosphate resulted in stasis and in a 1-log CFU/g reduction of the bacterial concentration in the thigh at 24 hours against the ATCC 33591 MRSA strain. At the same time, a value for the AUC0–24/MIC ratio of 43.1 for tedizolid and 56.9 for tedizolid phosphate resulted in stasis and in a 1-log CFU/g reduction of the bacterial concentration in the thigh at 48 hours against the ATCC 33591 MRSA strain. In addition, it was estimated that a value for the AUC0–24/MIC ratio of 307.6 for TR-700/TR-701 would result in a 4.53-log CFU/g reduction and a 5.92-log CFU/g reduction in the bacterial concentration in the thigh at 24 and 48 hours, respectively. By contrast, a value for the AUC0–24/MIC ratio of 35.7 obtained after administering 120 mg/kg of linezolid did not result in stasis. In addition, no greater microbiological effect of linezolid was observed at 48 hours versus at 24 hours.
Furthermore, a dose fractionation study was conducted for TR-701/TR-700 and for linezolid against an MSSA strain and against a community-acquired MRSA strain. The linezolid doses evaluated resulted in a lower reduction of the bacterial load versus the same doses of TR-701/700 for both S. aureus strains. Regarding the MRSA strain, the 150-mg/kg linezolid dose yielded a value for the AUC0–24/MIC ratio of 44.6, while stasis was achieved with a TR-701/TR-700 dose of 33.8 mg/kg, corresponding to a value for the AUC0–24/MIC ratio of 44.0. The difference between the two dosage regimens roughly corresponded to a 1.1-log CFU/g higher reduction in the bacterial load for TR-701/700. Regarding the MSSA strain, the 40.6-mg/kg TR-701/700 dose, corresponding to a value for the AUC0–24/MIC ratio of 52.8, resulted in stasis, while the 150-mg/kg linezolid dose (AUC0–24/MIC of 44.6) did not achieve this state.
Subsequently, another study was conducted in a nonneutropenic mouse model with an infection caused by the ATCC 33591 MRSA strain (used in the aforementioned study) with the aim of evaluating the impact of granulocytes on the antimicrobial activity of TR-700.48 According to the results, the presence of granulocytes considerably increased the activity of TR-700. Thus, administration of TR-701 at an equivalent dose of 1,600 mg daily in humans did not result in stasis at 72 hours when it was administered to the neutropenic model. However, the equivalent dose in humans of 3,200 mg daily resulted in a reduction of 2.75 log CFU/g in the bacterial load (killing rate) at 72 hours, of 1.73 log CFU/g at 48 hours, and of 1.0 log CFU/g at 24 hours.
Moreover, administering TR-701 at an equivalent dose of 1,200 mg daily in humans resulted in the maximum effect at 24 hours in the nonneutropenic model, while this was observed with the 800 mg daily dose at 48 hours. Finally, the 200 mg daily dose in the nonneutropenic mouse resulted in an effect near the maximum at 72 hours. Practically, no differences were observed with the 3,200 mg daily dose.
From these studies, it appears that the value for the AUC0–24/MIC ratio should be lower in immunocompetent animal models.49 Thus, a value for the AUC0–24/MIC ratio of 3 was extrapolated in immunocompetent animals based on a value of 50 in a neutropenic model.
Given this, a Monte Carlo simulation was performed with the aim of estimating the probability of obtaining a value for the AUC0–24/MIC ratio of 3 based on pharmacokinetic parameters in humans.50 The estimated probability approached zero when the microorganism’s MIC was 2 μg/mL or higher. Moreover, the probability was above 98% when the microorganism’s MIC was 0.5 μg/mL or lower.
Review of microbiology
In general, tedizolid shows high activity against Gram-positive microorganisms, including those with limited sensitivity to linezolid and other commonly used antibiotics in the treatment of infections caused by these bacteria. Tedizolid-susceptible Staphylococcus spp. and group A, B, C, and G Streptococcus strains were considered to be those against which the antibiotic’s MIC90 was 0.5 mg/L or lower. Resistant strains were considered to be those whose MIC90 was above this value.51 Tedizolid-susceptible Streptococcus anginosus group strains were considered to be those against which the antibiotic’s MIC90 was 0.25 mg/L or lower. Resistant strains were considered to be those whose MIC90 was above this value.51 No MIC90 breakpoints were established for Gram-positive anaerobic microorganisms.51 As with linezolid, tedizolid activity against Gram-negative microorganisms is very limited.52,53
Gram-positive microorganisms
Tedizolid shows high activity in vitro against strains of S. aureus (including MRSA and MSSA strains), S. pyogenes, Streptococcus agalactiae, the Streptococcus anginosus group (including S. anginosus, Streptococcus intermedius, and Streptococcus constellatus), and E. faecalis (FDA fact sheet). Tedizolid’s MIC is 0.5 mg/L or lower against these microorganisms and achieves a value of 0.25 mg/L or lower in the case of S. anginosus group strains (FDA fact sheet).
These values demonstrate activity that is generally two to four times higher than that of linezolid. 54–59 In one study, tedizolid’s MIC range was 0.125–0.5 mg/L against MRSA strains from SSSIs, while linezolid’s MIC range was 0.25–4 mg/L.54 The most effective drugs against MRSA were tedizolid (MIC90 of 0.5 mg/L), linezolid (MIC90 of 2 mg/L), and vancomycin (MIC90 of 2 mg/L). Similar results were observed in other studies, where vancomycin’s MIC90 and tedizolid’s MIC90 for both MRSA and MSSA were 2 mg/L,55 and 0.5 mg/L,55,56 respectively. Lower values have been reported, with tedizolid’s MIC90 for both MRSA and MSSA of 0.25 mg/L, compared with a MIC90 of 2 mg/L for linezolid.57,58
The MIC90 for both methicillin-resistant (MRSE) and methicillin-susceptible S. epidermidis (MSSE) was 0.5 mg/L against 74 clinical isolates of this microorganism (MIC range 0.06–0.5 mg/L).55 The MIC90 values were 0.25 and 0.5 mg/L for S. epidermidis and other coagulase-negative staphylococci included in other study, respectively.56 These values were lower than those for vancomycin (MIC90 was 4 and 2 mg/L for MRSE and MSSE, respectively)55 or linezolid (MIC90 was 1 mg/L for S. epidermidis and 2 mg/L for other coagulase-negative staphylococci).56
At the same time, tedizolid’s MIC range was 0.25–0.5 mg/L for Enterococcus spp. strains, while linezolid’s MIC range was 0.5–2 mg/L.54 Tedizolid MIC90 value was 0.5 mg/L against 873 enterococci included in other study while linezolid’s MIC90 was 2 mg/L.56
Finally, tedizolid’s MIC range was 0.25–0.5 mg/L for S. agalactiae strains and 0.125–0.5 mg/L for S. pyogenes strains, while linezolid’s MIC range was 2–4 mg/L for S. agalactiae strains and 1–2 mg/L for S. pyogenes strains.54 Tedizolid MIC90 values for S. agalactiae, S. pyogenes, and other β-hemolytic streptococci were each 0.25 mg/L, whereas linezolid’s MIC90 was 1 mg/L.56,58
Although tedizolid has high activity in vitro against Gram-positive microorganisms, this activity is very limited against biofilm-forming bacteria.55 Similar to linezolid, tedizolid could show synergistic activity with rifampin.
Mechanisms of resistance to tedizolid
Various mechanisms of resistance against oxazolidinones have been reported. Notable among them are mutations in the genes that encode 23S rRNA60,61 or the ribosomal proteins L3 (rplC) and L4 (rplD)62–64 and horizontal transmission of the chloramphenicol–florfenicol resistance (cfr) gene.65,66
In general, the microorganisms that show resistance against oxazolidinones because of mutations in the chromosomal genes that encode 23S rRNA or the L3 and L4 ribosomal proteins show cross-resistance to tedizolid (FDA fact sheet). However, in the absence of these chromosomal mutations, the presence of the cfr gene did not result in resistance to tedizolid in the limited number of S. aureus strains evaluated (FDA fact sheet). It has been observed that the frequency of spontaneous mutation that grants resistance to tedizolid is approximately 10−10 (FDA fact sheet).
Similar values were observed in another study that sought to evaluate the potential of one strain of MSSA and another strain of MRSA to develop resistance against tedizolid and linezolid by analyzing the spontaneous mutation rates and serial passage of these strains in media with an escalating concentration gradient of these oxazolidinones.62 The median spontaneous mutation rate that resulted in a reduction in sensitivity to tedizolid was 1.1×10−10 for the MSSA strain and 1.9×10−10 for the MRSA strain. These values were approximately 16 times lower than those reported for linezolid because a median spontaneous mutation rate of 2.0×10−9 was found for the MSSA strain and 3.0×10−9 was found for the MRSA strain. The mutant strains selected through exposure to tedizolid had the T2500A mutation and the coupled T2571C/G2576T mutation at the 23S rRNA, while the mutant strains selected through exposure to linezolid had the G2447T, G2576T, and T2500A mutations. In addition, mutations were identified in the rplC gene, which encodes the L3 ribosomal protein, and the rplD gene, which encodes the L4 ribosomal protein. The Gly152Asp, Gly155Arg, Gly155Arg/Met169Leu, and ΔPhe127-His146 mutations were identified in L3, and the Lys68Gln mutation was identified in L4.
In addition, serial passage of the MSSA and MRSA strains was performed in a concentration gradient of tedizolid and linezolid. Following 30 passages in these media, tedizolid’s MIC against the MSSA strain remained at 0.5 μg/mL, while linezolid’s MIC increased from 2 to 128 μg/mL. A reduction in sensitivity to linezolid was observed following five passages in a medium with linezolid, associated with the Gly155Arg mutation in L3. Subsequent passages in this medium resulted in mutant MSSA strains with the G2447T mutation in the rRNA or the G2447T mutation coupled with the Gly152Asp mutation in L3. Similarly, against the MRSA strain, tedizolid’s MIC increased by eight times (from 0.25 to 2 μg/mL), while linezolid’s MIC increased 32 times (from 1 to 32 μg/mL). The reduction in sensitivity to these antibiotics in the MRSA strain was associated with the Lys68Gln mutation in L4 and the G2576T mutation in the rRNA in the case of linezolid and the double T25761C/G2576T mutation at the 23S rRNA in the case of tedizolid.
Recently, another study sought to characterize the mechanism of resistance against oxazolidinones of 27 strains of Gram-positive linezolid-resistant microorganisms, as well as to evaluate the microbiological activity of tedizolid against these bacteria.67 Altogether, five S. aureus strains, 21 E. faecium strains, and one S. epidermidis strain were included.
The IDRL-10060, NRS271, and NRS119 S. aureus strains and all E. faecium strains had the G2576T mutation at the 23S rRNA. This mutation was identified in at least five of the six alleles. In both S. aureus and E. faecium and E. faecalis strains, a correlation has been reported between successive accumulation of the G2576T mutation in the different copies of the gene that encodes the 23S rRNA and a gradual increase in the level of resistance to linezolid.68,69 In this study, tedizolid’s MIC against the aforementioned strains ranged from 8 to 64 mg/L. Moreover, the S. epidermidis strain had the L101V, H146Q, V154L, and A157R mutations in the gene that codes for the L3 protein, and the 71G72 insertion and the N158S mutation in the gene that codes for the L4 protein. Among them, the L101V mutation and the N158S mutation seem to be associated with a lower impact on the development of resistance to oxazolidinones.
Finally, the NRS127 S. aureus strain had a deletion of the serine in position 145 of the gene that codes for L3, the IDRL-0025 S. aureus strain had the G152D mutation, and the NRS271 S. aureus strain had the Q3K mutation in the gene that codes for this protein.
In addition to the aforementioned mechanisms of resistance, a new mechanism of resistance to oxazolidinones was recently identified in E. faecalis and E. faecium strains.70 This mechanism consists of the presence of the optrA gene, located in plasmids, which codes for an ABC transporter that grants resistance to phenicols as well as to oxazolidinones. With the aim of finding out the prevalence of this gene, 595 Enterococcus spp. strains from humans and 290 strains from animals were included. The optrA gene was identified in 12 (2%) of the strains from humans and in 46 (15.9%) of the strains from animals. These data highlight the significance of monitoring the use of florfenicol in the production of animal feed. In addition, they emphasize the importance of monitoring the presence of this gene in Enterococcus spp. strains with a high MIC for linezolid and tedizolid, in addition to cfr gene carrier status and mutations at the 23S rRNA.
Gram-positive microorganisms with limited susceptibility versus commonly used antibiotics
Tedizolid has shown high activity in vitro against strains of Gram-positive microorganisms with limited sensitivity to commonly used antibiotics in infections caused by these microorganisms.56,57,71 Tedizolid activity was four to eight times higher than that observed with linezolid in one study that included 302 MRSA strains and 220 vancomycin-resistant (VR) Enterococcus spp. strains.71 Tedizolid MIC90 values for heterogenous vancomycin-intermediate MRSA (n=120), vancomycin-intermediate MRSA (n=100), and daptomycin-non-susceptible MRSA (n=75) were 0.5 mg/L versus 4, 4, and 2 mg/L for linezolid, respectively.71 When only the linezolid-resistant MRSA strains were evaluated (n=7), tedizolid MIC ranged from <0.063 to 1 mg/L. Three of these strains had a MIC of 1 mg/L versus tedizolid and all lacked the cfr gene, so the reduction in sensitivity to tedizolid could be attributed to an alternative mechanism, such as mutations in the 23S ribosomal RNA or in L3 and L4 ribosomal proteins.
The 98.3% of all staphylococci strains were inhibited by tedizolid MIC value of 0.5 mg/L. Rates of susceptibility to other common antimicrobials were: clindamycin (32.3%), levofloxacin (19.9%), oxacillin (0%), tigecycline (98.9%), and trimethoprim/sulfamethoxazole (79.1%).
Tedizolid MIC90 values for VR E. faecium (n=120) and E. faecalis (n=100) were 1 mg/L and 0.25 mg/L, respectively. These values were two dilutions lower for E. faecium and three dilutions lower for E. faecalis compared with linezolid. When linezolid-resistant enterococcal strains were evaluated (10 E. faecium strains), tedizolid MIC values ranged from 1 to 4 mg/L (linezolid MIC values ranged from 8 to 32 mg/L). None of these 10 strains was cfr positive. Similarly, among daptomycin-non-susceptible VR enterococcal strains (25 E. faecium strains), tedizolid MIC values ranged from 0.25 to 4 mg/L (56% with MIC values <1 mg/L) (linezolid MIC range was 1 to 32 mg/L).
Tedizolid’s MIC was 0.5 mg/L against three of four S. aureus strains that were linezolid resistant owing to being cfr gene carriers, and 1 mg/L for the fourth strain carrying this gene.56 These values were 16–32 times lower than those observed with linezolid. However, tedizolid showed no activity against three strains that were linezolid-resistant owing to a mutation at the 23S rRNA (one strain carrying a mutant G2405A allele and two mutant G2576T alleles, one strain carrying three mutated G2576T alleles, and one strain carrying four mutated G2576T alleles). Tedizolid’s MIC against these strains was 2 mg/L, and linezolid’s MIC against these strains was 16 mg/L.
Tedizolid MIC values were 4 mg/L for four of five linezolid-resistant S. epidermidis strains and 8 mg/L for the remaining strain (linezolid MIC values ranged from 32 to 128 mg/L).56
In another study, tedizolid maintained its activity against linezolid-resistant coagulase-negative Staphylococcus strains when the mechanism involved was cfr gene carrier status.57 However, higher MICs for tedizolid were observed when the mechanism of resistance identified was the G2576T mutation. Finally, linezolid’s MIC was 2–5 doubling dilutions higher than that of tedizolid against Staphylococcus spp. and linezolid-resistant Enterococcus spp. strains included in another study.67 Tedizolid’s MIC ranged from 1 to 8 mg/L against five linezolid-resistant S. aureus strains, while linezolid’s MIC ranged from 8 to 64 mg/L. At the same time, tedizolid’s MIC was 1 to 4 mg/L against 21 linezolid-resistant E. faecium strains, while linezolid’s MIC was 8 to 64 mg/L. Finally, tedizolid’s MIC was 2 mg/L against the linezolid-resistant S. epidermidis strain included in the study, while linezolid’s MIC was 32 mg/L.
In general, although activity of tedizolid is higher against linezolid-resistant Gram-positive microorganisms, limited sensitivity to tedizolid must be considered when the mechanisms of resistance involved are mutations in the genes that code for the 23S rRNA, the rplC gene, and/or the rplD gene.56,57,67,71
Anaerobic microorganisms
Tedizolid shows excellent activity against anaerobic Gram-positive microorganisms.54 Tedizolid’s MIC range against these microorganisms was four to eight times lower than that observed with linezolid.
Clinical efficacy
A limited number of studies have evaluated the efficacy of tedizolid in ABSSSIs. To date, one Phase II study and two Phase III studies have been completed. All were conducted according to the FDA guidance document for the development of new compounds in ABSSSIs.
Phase II study
A Phase II, randomized, double-blind, clinical trial sought to evaluate the efficacy of tedizolid phosphate administered at doses of 200, 300, and 400 mg once daily for 5–7 days in patients with SSSIs.72 Subsequently, the microbiological activity of tedizolid and linezolid and the microbiological efficacy of tedizolid against isolated Gram-positive microorganisms in cultures from patients enrolled in the aforementioned study were evaluated.58
Between September 2008 and January 2009, a total of 192 patients were randomized; 188 of them received at least one dose of tedizolid phosphate and had a diagnosis of complicated SSSI (modified intention-to-treat [IIT] and clinical modified IIT, respectively).72 In addition, another three study populations were identified: the microbiological modified intention-to-treat (mMITT) population, which comprised patients enrolled in the clinical modified IIT population who also had isolation of a Gram-positive microorganism at baseline, and the clinically evaluable population, which comprised those patients who received the minimum requirement of study drug and were clinically evaluated at the test-of-cure (TOC) visit. Finally, the microbiologically evaluable (ME) population was made up of those patients included in both the clinically evaluable population and the mMITT population. Thus, 164 patients were included in the clinically evaluable population, 154 were included in the mMITT population, and 133 were included in the ME population.
The clinical cure rate at the (TOC) visit (7–14 days after the end of treatment [EOT]) in clinically evaluable patients was 98.2% for the group that received tedizolid phosphate 200 mg and 94.4% for the group that received 300 or 400 mg of the antibiotic. No differences were observed based on the type and size of the lesion or the seriousness of the infection.
S. aureus strains were isolated in 139 (90.3%) of the 154 patients included in the mMITT population; 112 (80.6%) of these were MRSA strains.58
Similarly, S. aureus strains were isolated in 119 (89.5%) of the 133 patients included in the ME population; 94 (79.0%) of these were MRSA strains.58 In this group, a similar rate of microbiological eradication was observed for all tedizolid dose groups at the TOC visit. A value of 97.7% was achieved when all isolated microorganisms were taken into account, a value of 97.9% was achieved when only MRSA strains were taken into account, and a value of 95.7% was achieved for MSSA strains.58 When only S. aureus strains were considered, the clinical cure rate in the ME population was 96.6% at the TOC visit, and reached a value of 96.9% when only MRSA strains were considered and a value of 95.7% for MSSA strains.58
Phase III studies
ESTABLISH-1
A randomized, double-blind, double-dummy, multicenter, multinational study designed with the aim of establishing the noninferiority of once-daily oral administration of tedizolid phosphate 200 mg for 6 days versus oral administration of linezolid 600 mg every 12 hours for 10 days in the treatment of ABSSSIs.73 The main objective of the study was to evaluate early clinical response to treatment at 48–72 hours after the first dose of study antibiotic in the ITT analysis set. Secondary objectives included evaluating clinical response in the ITT analysis set and in the clinically evaluable analysis set both at the EOT (day 11) and at 7–14 days from the EOT. The clinically evaluable analysis set included all patients included in the ITT analysis set who complied with the protocol without major violations and who completed specific evaluations for a particular objective. A total of 667 patients aged 18 years or older were randomized to receive tedizolid phosphate or linezolid between August 12, 2010 and September 30, 2011 and formed the ITT analysis set. At the same time, the clinically evaluable analysis set both at the EOT and at 7–14 days from the EOT was formed by 559 patients. In the ITT analysis set, the clinical response rates at 48–72 hours were 79.5% (95% CI: 74.8%–83.7%) of 332 patients in the tedizolid phosphate group and 79.4% (95% CI: 74.7%–83.6%) of 335 patients in the linezolid group (a treatment difference of 0.1% [95% CI: –6.1%–6.2%]). Sustained clinical treatment response rates at the EOT were similar in the tezidolid phosphate and linezolid groups in the ITT analysis set (69.3% and 71.9%, respectively) and in the clinically evaluable analysis set (80.2% and 81.1%, respectively). Results of investigator-assessed clinical treatment response 1–2 weeks after the EOT were also similar in the tedizolid phosphate and linezolid groups in the ITT analysis set (85.5% and 86.0%, respectively) and in the clinically evaluable analysis set (94.6% and 95.4%, respectively). In addition, no differences in clinical response rate were observed at 7–14 days after the EOT assessed by the investigator based on the isolated S. aureus strain (MRSA strains: 75/88 [85.2%] for tedizolid phosphate versus 77/90 [85.6%] for linezolid; MSSA strains: 73/83 [88.0%] for tedizolid phosphate versus 82/87 [94.3%] for linezolid; strains positive for the Panton–Valentine leukocidin gene: 83/97 [85.6%] for tedizolid phosphate versus 86/102 [84.3%] for linezolid).
ESTABLISH-2
A randomized, double-blind, multinational, noninferiority, parallel-group design study with the aim of evaluating the efficacy of once-daily intravenous administration of tedizolid phosphate 200 mg for 6 days versus intravenous administration of linezolid 600 mg every 12 hours for 10 days in the treatment of ABSSSIs, with the possibility of sequential oral therapy.74 The main objective of the study was to evaluate early clinical response at 48–72 hours after the first dose of study antibiotic in the IIT analysis set. Secondary objectives included evaluating clinical response on day 7 (assessed by the investigator), at the EOT (determined by the schedule and assessed by the investigator) and at 7–14 days after the EOT (assessed by the investigator). A total of 666 patients were randomized to receive tedizolid (n=332) or linezolid (n=334) between September 28, 2011 and January 10, 2013. Sequential therapy from intravenous tedizolid to oral tedizolid was not inferior to linezolid (283 [85%] participants in the tedizolid group and 276 [83%] of those in the linezolid group achieved early clinical response). Results were consistent with investigator assessment (Table 2).
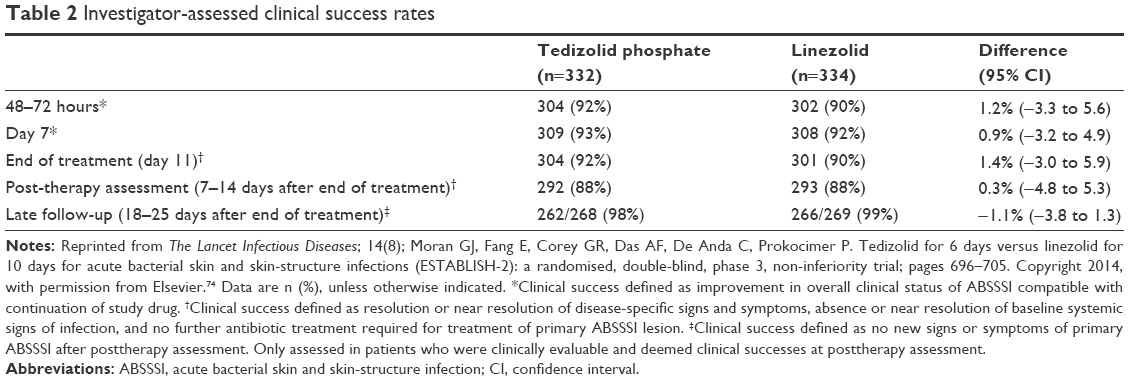 | Table 2 Investigator-assessed clinical success rates |
These results were maintained when microbiological response was evaluated at 7–14 days after the EOT in patients enrolled in the microbiological IIT population, which comprised those patients who received at least one dose of antimicrobial agent who also had isolation of a Gram-positive microorganism at baseline. In total, 168 (88%) of 192 patients enrolled in the tedizolid phosphate group versus 177 (89%) of 199 patients enrolled in the linezolid group had a favorable microbiological response at 7–14 days following the EOT (−1.4 [95% CI: −8.0 to 5.1]). The noninferiority of tedizolid phosphate was maintained when the isolated Gram-positive microorganism corresponded to MRSA strains (43/53 [81.0%] versus 43/56 [77.0%]; 95% CI: 4.3 [−11.4 to 19.8]).
With the aim of optimizing evaluation of the efficacy of tedizolid and linezolid, one study conducted an aggregate data analysis for both Phase III trials.59 A total of 1,333 patients were randomized to receive treatment with tedizolid 200 mg once daily for 6 days (n=664) or linezolid 600 mg every 12 hours for 10 days (n=669). A total of 1,225 patients (91.9%) completed the study; 612 of them belonged to the tedizolid arm and 613 of them belonged to the linezolid arm. In the aggregate data analysis, tedizolid was not inferior to linezolid in the clinical response at 48–72 hours evaluated in the IIT population (81.6% versus 79.4%; difference: 2.2% [95% CI: −2.0 to 6.5]). Regarding the secondary objectives, tedizolid was not inferior to linezolid in the clinical response determined by the schedule at the EOT (87.0% versus 87.9%; difference: −0.8% [95% CI: −4.4 to 2.7]) or in the clinical response assessed by the investigator at 7–14 days following the EOT (86.7% versus 86.8%; difference: −0.1 [95% CI: −3.8 to 3.6]). The clinical response at 48–72 hours and at 7–14 days following the EOT was similarly independent of the Gram-positive microorganism isolated. Regarding patients with MRSA isolation, 114 (80.9%) of 141 patients who received treatment with tedizolid had a clinical response at 48–72 hours, versus 111 (76.0%) of 146 patients who received treatment with linezolid. At the same time, in this group, clinical response was observed at 7–14 days from the EOT in 151 (84.8%) of 178 patients who received tedizolid versus 119 (81.5%) of 146 patients who received linezolid.
The noninferiority of tedizolid versus linezolid was also demonstrated in a post hoc analysis of Latino patients enrolled in the ESTABLISH-1 and ESTABLISH-2 trials.75
Currently, further clinical trials are being conducted with the aim of evaluating the efficacy of tedizolid in ABSSSIs.76
Tolerability
In a study by Leach et al,17 oxazolidinones were observed to be cross-linked to RNA in the peptidyltransferase center of mitochondrial ribosomes instead of cytoplasmic ribosomes. This led to the inhibition of mitochondrial protein synthesis, which has been associated with mitochondrial toxicity, and to a wide variety of adverse events, such as myelosuppression, lactic acidosis, optic neuropathy, and peripheral neuropathy.77–79
McKee et al80 evaluated the capacity of different oxazolidinones to inhibit mitochondrial protein synthesis in the heart and liver in a mouse model, and in the heart and bone marrow in rabbits. According to the results, mitochondrial inhibition was similar in all tissues.80 In addition, their data indicated that oxazolidinones with higher antimicrobial activity (lower MICs) also demonstrated a greater capacity for protein synthesis inhibition. However, some oxazolidinones that demonstrated a high capacity for inhibiting mitochondrial protein synthesis did not have powerful antibacterial activity.
Flanagan et al81 found a relationship between trough-free concentrations (Cmin) of linezolid and a greater risk of adverse effects. This is because a Cmin higher than the IC50 without recovery time has been associated with mitochondrial toxicity.81 Similarly, an association between an higher Cmin of linezolid and lower mitochondrial function has been observed in other studies,82 and this fact has been associated with a greater proportion of adverse events related to mitochondrial toxicity.
In addition to Cmin, treatment duration83 is among the variables related to the potential onset of adverse events associated with mitochondrial toxicity. Therefore, the usefulness of monitoring plasma levels of linezolid, mainly in patients with risk factors for toxicity, such as variation in its regular dosage regimen in certain clinical situations (use of high doses or continuous infusion), and hepatic and/or renal failure, has been established.37,84
Given these data, Flanagan et al85 evaluated the capacity of the new oxazolidinone tedizolid to cause mitochondrial toxicity in a mouse model through pharmacokinetic studies. Tedizolid in vitro demonstrated more powerful dose- and time-dependent mitochondrial protein synthesis inhibition than linezolid.86 However, in the results, which compared the effect of several supratherapeutic doses of tedizolid, with plasma exposure up to eight times greater than human therapeutic plasma exposure versus placebo, there was no evidence of abnormalities in behavior caused by neuropathic damage or of histopathological changes in the central or peripheral nervous system caused by oxazolidinone following a detailed analysis of brain, eye, optic nerve, spinal cord, and peripheral nerve tissue.86
This contrasted with the development of optic and peripheral neuropathy observed in rats following 3 months of administration of linezolid at doses that provided plasma levels comparable to those provided by therapeutic doses in humans, which suggested a lower neuropathological profile for tedizolid versus linezolid.
Further analyses performed by the same group suggested more rapid dissociation and partial migration of mitochondrial impairment over the course of a dosing interval for tedizolid versus linezolid. The pharmacokinetic profile of tedizolid, which allows for once-daily administration, and an effective therapeutic dose of 200 mg, six times lower than linezolid (1,200 mg), would explain this behavior.85 However, caution is required in prolonged treatments, given the absence of data for tedizolid administered for periods longer than 21 days.
Recently, Milosevic et al87 evaluated tedizolid’s capacity to inhibit the expression of proteins encoded by mitochondrial genes in cell cultures, with the aim of simulating administration in human beings to evaluate the impact of oxazolidinone on mitochondrial metabolism. The authors of this study concluded that tedizolid caused mitochondrial metabolic abnormalities in vitro that were related to the dysfunction observed in protein expression encoded by the mitochondrial genome. However, the clinical impact was limited by fast recovery upon washout of the drug, especially when tedizolid was used for short periods of time and owing to its pharmacokinetic characteristics that allowed its administration in a single daily dose.
One of the main consequences of mitochondrial toxicity is potential myelosuppression. The hematological toxicity of tedizolid was evaluated in detail in a study by Shorr et al73 that comprised 1,333 patients previously enrolled in two Phase III studies73,74 that compared this antimicrobial agent (n=664) versus linezolid (n=669). This study also evaluated treatment-emergent adverse events and clinical laboratory parameters.
Regarding hematological parameters, changes over time in platelet counts, neutrophil counts, and hemoglobin were analyzed. According to the results, a platelet count lower than 150,000/mm3 was observed on days 7–9 of treatment in 3.7% of patients who received tedizolid versus 5.8% of those who received linezolid. This difference increased to 4.9% of patients who received tedizolid and 10.8% of patients who received linezolid at the EOT. The authors believed that this difference between tedizolid and linezolid was substantial and paralleled the results of Phase I studies, in which hematological toxicity was not developed in any cell line following administration of tedizolid 200 mg for 21 days.88 However, these results should be confirmed in prolonged treatments with tedizolid.
Regarding treatment-emergent adverse events, similar rates were reported in the two groups (42.8% for tedizolid versus 43.2% for linezolid). Only 2% of all these adverse events were considered to be serious, requiring discontinuation of treatment in 0.5% of patients treated with tedizolid and 0.9% of patients treated with linezolid. The most commonly reported adverse events were gastrointestinal abnormalities (16% for tedizolid versus 23% for linezolid), notably nausea (8.2% for tedizolid versus 12.2% for linezolid). Also reported were headache (6.2% for tedizolid versus 5.9% for linezolid) and abscess (5.3% for tedizolid versus 3.9% for linezolid). No differences were observed in terms of physical examination or electrocardiogram data.89
Drug interactions
One characteristic shared by oxazolidinones is their capacity to inhibit MAO, an enzyme responsible for the metabolism of the neurotransmitters epinephrine, norepinephrine, and serotonin (MAO A) and dopamine (MAO B). MAO inhibitors may give rise to interactions with foods and other drugs, resulting in the so-called serotonin syndrome.90 This syndrome results from the accumulation of serotonin, which causes an increase in serotonin neurotransmission in both the central nervous system central and peripheral serotonin receptors. It is very serious, given that it may be fatal in some cases.
Linezolid is a competitive and reversible nonselective MAO inhibitor, and the onset of serotonin syndrome has been reported following its administration together with certain serotonergic drugs.91,92 Given this, the FDA indicated that “linezolid should generally not be given to patients taking serotonergic drugs unless the benefit is deemed to outweigh the risk.”93
Although tedizolid has shown a capacity to nonselectively inhibit MAO in in vitro studies, similar to linezolid, studies in a mouse model have not managed to demonstrate serotoninergic activity with clinical repercussions.94 However, owing to the short duration of the clinical trials, the limited number of participants, and the exclusion of other concomitant serotonergic drugs, some authors have not ruled out the potential onset of serotonin toxicity in the future.95
MAO inhibitors may increase blood pressure when they are administered together with vasoconstrictors such as tyramine or pseudoephedrine, and they may trigger hypertensive crises. The potential interaction between tedizolid and oral tyramine and pseudoephedrine was evaluated in two randomized, double-blind, placebo-controlled crossover studies.94 In the first day, subjects were randomized to administration of tedizolid 200 mg or placebo. On the third day, they received tyramine at the dose required to cause an increase in systolic blood pressure ≥30 mmHg. According to the results, administration of tedizolid did not result in a potentiation of the hypertensive effect of tyramine, given that the sensitivity ratio reached was 1.33, <2, a ratio with clinical significance. Only one subject reached a sensitivity ratio >2 (2.1), and palpitations were reported at some point in the study in 25 (83.3%) for 30 subjects enrolled. This effect may be attributed to tyramine. By contrast, eight (80%) of ten subjects who received linezolid had a ratio >2 (range: 1.50–5.0).96
The authors concluded that it is not necessary to take special precautions when tedizolid is administered with tyramine-rich foods, given that foods do not tend to contain more than 40 mg of the amino acid.
When coadministration of tedizolid with pseudoephedrine was studied, no significant changes in blood pressure or heart rate were observed between the control group and the group that received placebo.94 The profile found was somewhat more beneficial than that observed with linezolid, which was associated with increases in blood pressure when it was administered together with pseudoephedrine.97
Benefits and disadvantages compared to other oxazolidinone antibiotics
Tedizolid shows higher activity than linezolid against Gram-positive microorganisms, including those with limited sensitivity against commonly used antibiotics in infections caused by these bacteria.
One of the characteristics of tedizolid that makes it more attractive is its activity against staphylococci strains that contain the cfr gene, given the high capacity for transmission of this mechanism of resistance between strains of this species and other Gram-positive microorganisms.98 However, the frequency of the appearance of strains with this mechanism of resistance is limited, and for this reason, linezolid’s MIC against Gram-positive microorganisms has remained stable in recent years.99 The bioavailability of tedizolid is high, both orally and intravenously, similar to linezolid. However, tedizolid has more favorable pharmacokinetics, especially because of its prolonged biological half-life, which allows it to be administered in a single daily dose. In addition, administration of tedizolid seems to be associated with a lower risk of hematological adverse effects as well as developing serotonin syndrome when it is administered with other drugs related to this syndrome.
According to the results from the clinical trials, administration of tedizolid phosphate for 6 days was not inferior to administration of linezolid for 10 days. Currently, it is being evaluated whether this difference in treatment duration maintains its efficacy in other infectious diseases such as pneumonia, with the aim of positioning tedizolid in the available antimicrobial arsenal.100 Should these findings be confirmed, the use of tedizolid could be associated with reduced economic resources mainly deriving from a shorter hospital stay.
Acknowledgment
The authors thank Enrique Gómez-Bengoa (Departamento de Química Orgánica I, Facultad de Química, Universidad del País Vasco, San Sebastián, Spain) for his assistance in editing of the figures.
Disclosure
The authors report no conflicts of interest in this work.
References
U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for Industry Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment. Silver Spring, MD: US Food and Drug Administration; 2013. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071185.pdf. Accessed May 1, 2016. | ||
Pallin DJ, Egan DJ, Pelletier AJ, Espinola JA, Hooper DC, Camargo CA Jr. Increased US emergency department visits for skin and soft tissue infections, and changes in antibiotic choices, during the emergence of community-associated methicillin-resistant Staphylococcus aureus. Ann Emerg Med. 2008;51(3):291–298. | ||
Moran GJ, Krishnadasan A, Gorwitz RJ, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666–674. | ||
Talan DA, Krishnadasan A, Gorwitz RJ, et al. Comparison of Staphylococcus aureus from skin and soft-tissue infections in US emergency department patients, 2004 and 2008. Clin Infect Dis. 2011;53(2):144–149. | ||
Moran GJ, Amii RN, Abrahamian FM, Talan DA. Methicillin-resistant Staphylococcus aureus in community-acquired skin infections. Emerg Infect Dis. 2005;11(6):928–930. | ||
Bassetti M, Baguneid M, Bouza E, Dryden M, Nathwani D, Wilcox M. European perspective and update on the management of complicated skin and soft tissue infections due to methicillin-resistant Staphylococcus aureus after more than 10 years of experience with linezolid. Clin Microbiol Infect. 2014;20(Suppl 4):3–18. | ||
Morrissey I, Leakey A, Northwood JB. In vitro activity of ceftaroline and comparator antimicrobials against European and Middle East isolates from complicated skin and skin-structure infections collected in 2008–2009. Int J Antimicrob Agents. 2012;40(3):227–234. | ||
Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):147–159. | ||
Crotty MP, Krekel T, Burnham CA, Ritchie DJ. New gram-positive agents: the next generation of oxazolidinones and lipoglycopeptides. J Clin Microbiol. 2016. | ||
Shaw KJ, Barbachyn MR. The oxazolidinones: past, present, and future. Ann N Y Acad Sci. 2011;1241:48–70. | ||
Bae SK, Yang SH, Shin KN, Rhee JK, Yoo M, Lee MG. Pharmacokinetics of DA-7218, a new oxazolidinone, and its active metabolite, DA-7157, after intravenous and oral administration of DA-7218 and DA-7157 to rats. J Pharm Pharmacol. 2007;59(7):955–963. | ||
Rybak JM, Roberts K. Tedizolid phosphate: a next-generation oxazolidinone. Infect Dis Ther. Epub 2015 Feb 24. | ||
Locke JB, Zurenko GE, Shaw KJ, Bartizal K. Tedizolid for the management of human infections: in vitro characteristics. Clin Infect Dis. 2014;58(Suppl 1):S35–S42. | ||
Colca JR, McDonald WG, Waldon DJ, et al. Cross-linking in the living cell locates the site of action of oxazolidinone antibiotics. J Biol Chem. 2003;278(24):21972–21979. | ||
Kanafani ZA, Corey GR. Tedizolid (TR-701): a new oxazolidinone with enhanced potency. Expert Opin Investig Drugs. 2012;21(4):515–522. | ||
Wilson DN, Schluenzen F, Harms JM, Starosta AL, Connell SR, Fucini P. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc Natl Acad Sci U S A. 2008;105(36):13339–13344. | ||
Leach KL, Swaney SM, Colca JR, et al. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol Cell. 2007;26(3):393–402. | ||
Huttunen KM, Raunio H, Rautio J. Prodrugs – from serendipity to rational design. Pharmacol Rev. 2011;63(3):750–771. | ||
Heimbach T, Oh DM, Li LY, et al. Absorption rate limit considerations for oral phosphate prodrugs. Pharm Res. 2003;20(6):848–856. | ||
Flanagan S, Fang E, Munoz KA, Minassian SL, Prokocimer PG. Single- and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacotherapy. 2014;34(9):891–900. | ||
Chen R, Shen K, Chang X, Tanaka T, Hu P. Pharmacokinetics of 7-day multiple dose tedizolid phosphate in healthy chinese subjects [Abstract A-452]. Poster presented at: 55th Interscience Conference on Antimicrobial Agents and Chemotheraphy; September 17–21; 2015; San Diego, CA. | ||
Tanaka T, Hayashi Y, Okumura K, et al. Oral bioavailability of tedizolid in healthy Japenese subjects in a phase I study [Abstract P1718]. Poster presented at: European Congress of Clinical Microbiology and Infectious Diseases; May 10–13; 2014; Barcelona, Spain. | ||
Flanagan SD, Bien PA, Munoz KA, Minassian SL, Prokocimer PG. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy. 2014;34(3):240–250. | ||
Flanagan S, Passarell J, Lu Q, Fiedler-Kelly J, Ludwig E, Prokocimer P. Tedizolid population pharmacokinetics, exposure response, and target attainment. Antimicrob Agents Chemother. 2014;58(11):6462–6470. | ||
Stalker DJ, Jungbluth GL. Clinical pharmacokinetics of linezolid, a novel oxazolidinone antibacterial. Clin Pharmacokinet. 2003;42(13):1129–1140. | ||
Stalker DJ, Jungbluth GL, Hopkins NK, Batts DH. Pharmacokinetics and tolerance of single- and multiple-dose oral or intravenous linezolid, an oxazolidinone antibiotic, in healthy volunteers. J Antimicrob Chemother. 2003;51(5):1239–1246. | ||
Housman ST, Pope JS, Russomanno J, et al. Pulmonary disposition of tedizolid following administration of once-daily oral 200-milligram tedizolid phosphate in healthy adult volunteers. Antimicrob Agents Chemother. 2012;56(5):2627–2634. | ||
Sahre M, Sabarinath S, Grant M, et al. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int J Antimicrob Agents. 2012;40(1):51–54. | ||
Ong V, Flanagan S, Fang E, et al. Absorption, distribution, metabolism, and excretion of the novel antibacterial prodrug tedizolid phosphate. Drug Metab Dispos. 2014;42(8):1275–1284. | ||
Kennedy G, Osborn J, Flanagan S, Alsayed N, Bertolami S. Stability of crushed tedizolid phosphate tablets for nasogastric tube administration. Drugs R D. 2015;15(4):329–333. | ||
Bradley JS, Flanagan SD, Arrieta AC, Jacobs R, Capparelli E, Prokocimer P. Pharmacokinetics, safety and tolerability of single oral or intravenous administration of 200 mg tedizolid phosphate in adolescents. Pediatr Infect Dis J. 2016;35(6):628–633. | ||
Kearns GL, Abdel-Rahman SM, Blumer JL, et al. Single dose pharmacokinetics of linezolid in infants and children. Pediatr Infect Dis J. 2000;19(12):1178–1184. | ||
Dreskin H, Muñoz KA, Fang E, et al. Safety and pharmacokinetics of single oral administration of tedizolid phosphate in healthy elderly subjects and adult control subjects [Abstract A-1293]. Poster presented at: 52nd Interscience Conference on Antimicrobial Agents and Chemotheraphy; September 9–12; 2012; San Francisco, CA. | ||
Flanagan S, Minassian SL, Passarell J, Fiedler-Kelly J, Prokocimer P. Tedizolid plasma pharmacokinetics are comparable in obese and nonobese patients and healty patients [Abstract P1703]. Poster presented at: European Congress of Clinical Microbiology and Infectious Diseases; May 10–13; 2014; Barcelona, Spain. | ||
Bhalodi AA, Papasavas PK, Tishler DS, Nicolau DP, Kuti JL. Pharmacokinetics of intravenous linezolid in moderately to morbidly obese adults. Antimicrob Agents Chemother. 2013;57(3):1144–1149. | ||
Hamilton R, Thai XC, Ameri D, Pai MP. Oral bioavailability of linezolid before and after Roux-en-Y gastric bypass surgery: is dose modification necessary in obese subjects? J Antimicrob Chemother. 2013;68(3):666–673. | ||
Lopez-Garcia B, Luque S, Roberts JA, Grau S. Pharmacokinetics and preliminary safety of high dose linezolid for the treatment of gram-positive bacterial infections. J Infect. 2015;71(5):604–607. | ||
Flanagan S, Minassian SL, Morris D, et al. Pharmacokinetics of tedizolid in subjects with renal or hepatic impairment. Antimicrob Agents Chemother. 2014;58(11):6471–6476. | ||
Levey AS, Coresh J, Greene T, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. 2006; 145(4):247–254. | ||
MacGowan AP. Pharmacokinetic and pharmacodynamic profile of linezolid in healthy volunteers and patients with Gram-positive infections. J Antimicrob Chemother. 2003;51(Suppl 2):ii17–ii25. | ||
Brier ME, Stalker DJ, Aronoff GR, et al. Pharmacokinetics of linezolid in subjects with renal dysfunction. Antimicrob Agents Chemother. 2003;47(9):2775–2780. | ||
Nukui Y, Hatakeyama S, Okamoto K, et al. High plasma linezolid concentration and impaired renal function affect development of linezolid-induced thrombocytopenia. J Antimicrob Chemother. 2013;68(9):2128–2133. | ||
Lopez-Garcia B, Luque S, Maseda E, Grau S. Comment on: pharmacokinetics of high dosage of linezolid in two morbidly obese patients. J Antimicrob Chemother. 2015;70(11):3168. | ||
Morata L, Cuesta M, Rojas JF, et al. Risk factors for a low linezolid trough plasma concentration in acute infections. Antimicrob Agents Chemother. 2013;57(4):1913–1917. | ||
Flanagan S, Prokocimer P. Reduction in tedizolid plasma exposure among end-stage renal disease patients undergoing dialysis is explained by variations in ideal body weight. Antimicrob Agents Chemother. 2016;60(5):3246–3247. | ||
Hendershot PE, Jungbluth GL, Cammarata SK, Hopkins NJ. Pharmacokinetics of linezolid in patients with liver disease. J Antimicrob Chemother. 1999;44(Suppl A):55. | ||
Louie A, Liu W, Kulawy R, Drusano GL. In vivo pharmacodynamics of torezolid phosphate (TR-701), a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(7):3453–3460. | ||
Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300–5305. | ||
Lodise TP, Drusano GL. Use of pharmacokinetic/pharmacodynamic systems analyses to inform dose selection of tedizolid phosphate. Clin Infect Dis. 2014;58(Suppl 1):S28–S34. | ||
Anti-infective drugs advisory committee. Tedizolid Phosphate for the Treatment of Acute Bacterial Skin and Skin Structure Infections. Silver Spring, MD: US Food and Drug Administration; 2014. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM390790.pdf. Accessed May 1, 2016. | ||
The European Committee on Antimicrobial Susceptibility Testing (Eucast). Breakpoint tables for interpretation of MICs and zone diameters. Version 6.0; 2016. Available from: http://www.eucast.org. Accessed May 21, 2016. | ||
Brown SD, Traczewski MM. Comparative in vitro antimicrobial activities of torezolid (TR-700), the active moiety of a new oxazolidinone, torezolid phosphate (TR-701), determination of tentative disk diffusion interpretive criteria, and quality control ranges. Antimicrob Agents Chemother. 2010;54(5):2063–2069. | ||
Schaadt R, Sweeney D, Shinabarger D, Zurenko G. In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent. Antimicrob Agents Chemother. 2009;53(8):3236–3239. | ||
Lee Y, Hong SK, Choi S, Im W, Yong D, Lee K. In vitro activity of tedizolid against gram-positive bacteria in patients with skin and skin structure infections and hospital-acquired pneumonia: a Korean multicenter study. Ann Lab Med. 2015;35(5):523–530. | ||
Schmidt-Malan SM, Greenwood Quaintance KE, Karau MJ, Patel R. In vitro activity of tedizolid against staphylococci isolated from prosthetic joint infections. Diagn Microbiol Infect Dis. 2016;85(1):77–79. | ||
Sahm DF, Deane J, Bien PA, et al. Results of the surveillance of Tedizolid activity and resistance program: in vitro susceptibility of gram-positive pathogens collected in 2011 and 2012 from the United States and Europe. Diagn Microbiol Infect Dis. 2015;81(2):112–118. | ||
Chen H, Yang Q, Zhang R, et al. In vitro antimicrobial activity of the novel oxazolidinone tedizolid and comparator agents against Staphylococcus aureus and linezolid-resistant Gram-positive pathogens: a multicentre study in China. Int J Antimicrob Agents. 2014;44(3): 276–277. | ||
Prokocimer P, Bien P, Deanda C, Pillar CM, Bartizal K. In vitro activity and microbiological efficacy of tedizolid (TR-700) against Gram-positive clinical isolates from a phase 2 study of oral tedizolid phosphate (TR-701) in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2012;56(9):4608–4613. | ||
Shorr AF, Lodise TP, Corey GR, et al. Analysis of the phase 3 ESTABLISH trials of tedizolid versus linezolid in acute bacterial skin and skin structure infections. Antimicrob Agents Chemother. 2015;59(2):864–871. | ||
Gonzales RD, Schreckenberger PC, Graham MB, Kelkar S, DenBesten K, Quinn JP. Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet. 2001;357(9263): 1179. | ||
Tsiodras S, Gold HS, Sakoulas G, et al. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet. 2001;358(9277):207–208. | ||
Locke JB, Hilgers M, Shaw KJ. Novel ribosomal mutations in Staphylococcus aureus strains identified through selection with the oxazolidinones linezolid and torezolid (TR-700). Antimicrob Agents Chemother. 2009;53(12):5265–5274. | ||
Kosowska-Shick K, Julian KG, McGhee PL, Appelbaum PC, Whitener CJ. Molecular and epidemiologic characteristics of linezolid-resistant coagulase-negative staphylococci at a tertiary care hospital. Diagn Microbiol Infect Dis. 2010;68(1):34–39. | ||
Pournaras S, Ntokou E, Zarkotou O, et al. Linezolid dependence in Staphylococcus epidermidis bloodstream isolates. Emerg Infect Dis. 2013;19(1):129–132. | ||
Kehrenberg C, Schwarz S, Jacobsen L, Hansen LH, Vester B. A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Mol Microbiol. 2005;57(4):1064–1073. | ||
Toh SM, Xiong L, Arias CA, et al. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol Microbiol. 2007;64(6):1506–1514. | ||
Silva-Del Toro SL, Greenwood-Quaintance KE, Patel R. In vitro activity of tedizolid against linezolid-resistant staphylococci and enterococci. Diagn Microbiol Infect Dis. 2016;85(1):102–104. | ||
Besier S, Ludwig A, Zander J, Brade V, Wichelhaus TA. Linezolid resistance in Staphylococcus aureus: gene dosage effect, stability, fitness costs, and cross-resistances. Antimicrob Agents Chemother. 2008;52(4):1570–1572. | ||
Marshall SH, Donskey CJ, Hutton-Thomas R, Salata RA, Rice LB. Gene dosage and linezolid resistance in Enterococcus faecium and Enterococcus faecalis. Antimicrob Agents Chemother. 2002;46(10):3334–3336. | ||
Wang Y, Lv Y, Cai J, et al. A novel gene, optrA, that confers transferable resistance to oxazolidinones and phenicols and its presence in Enterococcus faecalis and Enterococcus faecium of human and animal origin. J Antimicrob Chemother. 2015;70(8):2182–2190. | ||
Barber KE, Smith JR, Raut A, Rybak MJ. Evaluation of tedizolid against Staphylococcus aureus and enterococci with reduced susceptibility to vancomycin, daptomycin or linezolid. J Antimicrob Chemother. 2016;71(1):152–155. | ||
Prokocimer P, Bien P, Surber J, et al. Phase 2, randomized, double-blind, dose-ranging study evaluating the safety, tolerability, population pharmacokinetics, and efficacy of oral torezolid phosphate in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2011;55(2):583–592. | ||
Prokocimer P, De Anda C, Fang E, Mehra P, Das A. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA. 2013;309(6):559–569. | ||
Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8): 696–705. | ||
Ortiz-Covarrubias A, Fang E, Prokocimer PG, et al. Efficacy, safety, tolerability and population pharmacokinetics of tedizolid, a novel antibiotic, in Latino patients with acute bacterial skin and skin structure infections. Braz J Infect Dis. 2016;20(2):184–192. | ||
Bayer. Efficacy and safety of intravenous to oral 6-day tedizolid phosphate vs. Intravenous to oral 10-day linezolid in patients with acute bacterial skin and skin structure infection (ABSSSI). Available from: https://clinicaltrials.gov/ct2/show/NCT02066402?term=tr701&rank=26. NLM identifier: NCT02066402. Accessed May 1, 2016. | ||
Soriano A, Miro O, Mensa J. Mitochondrial toxicity associated with linezolid. N Engl J Med. 2005;353(21):2305–2306. | ||
Narita M, Tsuji BT, Yu VL. Linezolid-associated peripheral and optic neuropathy, lactic acidosis, and serotonin syndrome. Pharmacotherapy. 2007;27(8):1189–1197. | ||
Beekmann SE, Gilbert DN, Polgreen PM. Toxicity of extended courses of linezolid: results of an Infectious Diseases Society of America Emerging Infections Network survey. Diagn Microbiol Infect Dis. 2008;62(4):407–410. | ||
McKee EE, Ferguson M, Bentley AT, Marks TA. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob Agents Chemother. 2006;50(6):2042–2049. | ||
Flanagan S, Dreskin H, Prokocimer P. Comparison of tedizolid and linezolid pharmacokinetics in Phase 1 studies [Abstract A-692]. Poster presented at: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; September 5–19; 2014; Washington, DC. | ||
Song T, Lee M, Jeon HS, et al. Linezolid trough concentrations correlate with mitochondrial toxicity-related adverse events in the treatment of chronic extensively drug-resistant tuberculosis. EBioMedicine. 2015;2(11):1627–1633. | ||
Gould FK. Linezolid: safety and efficacy in special populations. J Antimicrob Chemother. 2011;66(Suppl 4):iv3–iv6. | ||
Pea F, Viale P, Cojutti P, Del Pin B, Zamparini E, Furlanut M. Therapeutic drug monitoring may improve safety outcomes of long-term treatment with linezolid in adult patients. J Antimicrob Chemother. 2012;67(8):2034–2042. | ||
Flanagan S, McKee EE, Das D, et al. Nonclinical and pharmacokinetic assessments to evaluate the potential of tedizolid and linezolid to affect mitochondrial function. Antimicrob Agents Chemother. 2015;59(1):178–185. | ||
Schlosser MJ, Hosako H, Radovsky A, et al. Lack of neuropathological changes in rats administered tedizolid phosphate for nine months. Antimicrob Agents Chemother. 2015;59(1):475–481. | ||
Milosevic T, Payen VL, Brisson L, Sonveaux P, Van Vambeke F, Tulkens PM. Tedizolid inhibits mitochondrial metabolism and reversibly impairs Cytochrome c-oxydase expression in cultured human cells: impact of a discontinous exposure [Abstract A-974]. Poster presented at: 55 th Interscience Conference on Antimicrobial Agents and Chemotheraphy; September 17–21; 2015; San Diego, CA. | ||
Prokocimer P, Bien P, Muñoz KA, Aster R. Haematological effects of TR-701, linezolid and placebo administered for 21 days in healthy subjects [Abstract A-2069]. Poster presented at: 48th Interscience Conference on Antimicrobial Agents and Chemotheraphy; October 25–28; 2008; Washington, DC. | ||
Fang E, De Anda C, Das A, Prokocimer P. Safety and tolerability of tedizolid phosphate, a novel oxazolidinone versus linezolid in two phase III studies in skin/skin structure infections [Abstract PS-295]. Poster presented at: 18th Congress of the Asian Pacific Society of Respirology; November 11–14; 2013; Yokohama, Japan. | ||
Rybak JM, Marx K, Martin CA. Early experience with tedizolid: clinical efficacy, pharmacodynamics, and resistance. Pharmacotherapy. 2014;34(11):1198–1208. | ||
Morales-Molina JA, Mateu-de Antonio J, Marin-Casino M, Grau S. Linezolid-associated serotonin syndrome: what we can learn from cases reported so far. J Antimicrob Chemother. 2005;56(6):1176–1178. | ||
Clark DB, Andrus MR, Byrd DC. Drug interactions between linezolid and selective serotonin reuptake inhibitors: case report involving sertraline and review of the literature. Pharmacotherapy. 2006;26(2): 269–276. | ||
FDA. FDA drug safety communication: serious CNS reactions possible when linezolid (Zyvox) is given to patients taking certain psychiatric medications. Silver Spring, MD: FDA; October 21, 2011. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm265305.htm. Accessed March 27, 2016. | ||
Flanagan S, Bartizal K, Minassian SL, Fang E, Prokocimer P. In vitro, in vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob Agents Chemother. 2013;57(7):3060–3066. | ||
Douros A, Grabowski K, Stahlmann R. Drug-drug interactions and safety of linezolid, tedizolid, and other oxazolidinones. Expert Opin Drug Metab Toxicol. 2015;11(12):1849–1859. | ||
Antal EJ, Hendershot PE, Batts DH, Sheu WP, Hopkins NK, Donaldson KM. Linezolid, a novel oxazolidinone antibiotic: assessment of monoamine oxidase inhibition using pressor response to oral tyramine. J Clin Pharmacol. 2001;41(5):552–562. | ||
Hendershot PE, Antal EJ, Welshman IR, Batts DH, Hopkins NK. Linezolid: pharmacokinetic and pharmacodynamic evaluation of coadministration with pseudoephedrine HCl, phenylpropanolamine HCl, and dextromethorpan HBr. J Clin Pharmacol. 2001; 41(5):563–572. | ||
Moellering RC Jr. Tedizolid: a novel oxazolidinone for gram-positive infections. Clin Infect Dis. 2014;58(Suppl 1):S1–S3. | ||
Flamm RK, Mendes RE, Hogan PA, Streit JM, Ross JE, Jones RN. Linezolid surveillance results for the United States (LEADER Surveillance Program 2014). Antimicrob Agents Chemother. 2016;60(4): 2273–2280. | ||
Burdette SD, Trotman R. Tedizolid: The first once-daily oxazolidinone class antibiotic. Clin Infect Dis. 2015;61(8):1315–1321. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.