")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 4
Critical analysis of the use of β-site amyloid precursor protein-cleaving enzyme 1 inhibitors in the treatment of Alzheimer's disease
Received 13 October 2013
Accepted for publication 19 November 2013
Published 22 January 2014 Volume 2014:4 Pages 1—19
DOI https://doi.org/10.2147/DNND.S41056
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Genevieve Evin,1,2 Adel Barakat2
1Oxidation Biology Laboratory, Mental Health Research Institute, Florey Institute of Neuroscience and Mental Health, University of Melbourne, 2Department of Pathology, University of Melbourne, Parkville, VIC, Australia
Abstract: Alzheimer's disease (AD) is the major cause of dementia in the elderly and an unmet clinical challenge. A variety of therapies that are currently under development are directed to the amyloid cascade. Indeed, the accumulation and toxicity of amyloid-β (Aβ) is believed to play a central role in the etiology of the disease, and thus rational interventions are aimed at reducing the levels of Aβ in the brain. Targeting β-site amyloid precursor protein-cleaving enzyme (BACE)-1 represents an attractive strategy, as this enzyme catalyzes the initial and rate-limiting step in Aβ production. Observation of increased levels of BACE1 and enzymatic activity in the brain, cerebrospinal fluid, and platelets of patients with AD and mild cognitive impairment supports the potential benefits of BACE1 inhibition. Numerous potent inhibitors have been generated, and many of these have been proved to lower Aβ levels in the brain of animal models. Over 10 years of intensive research on BACE1 inhibitors has now culminated in advancing half a dozen of these drugs into human trials, yet translating the in vitro and cellular efficacy of BACE1 inhibitors into preclinical and clinical trials represents a challenge. This review addresses the promises and also the potential problems associated with BACE1 inhibitors for AD therapy, as the complex biological function of BACE1 in the brain is becoming unraveled.
Keywords: amyloid, dementia, secretase, aspartyl protease, neuregulin
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease of the central nervous system, which accounts for most cases of dementia in the elderly.1 AD has become an increasing socioeconomic burden, as it remains an unmet clinical challenge, with currently no treatment being available to halt or reverse its devastating effects. Various underlying factors, such as stroke, high blood pressure and hypercholesterolemia, diabetes, oxidative stress and inflammation, as well as sedentary lifestyle, which have been associated with AD, all converge to molecular mechanisms centered on the amyloid cascade theory.2,3 For over a century, AD has been traditionally diagnosed postmortem by the accumulation of amyloid plaques and τ-neurofibrillary tangles in the brain cortical regions and hippocampus.2 The major component of the amyloid plaques was identified nearly 30 years ago as a 4 kDa peptide fragment, termed amyloid-β (Aβ) peptide,4,5 and since then extensive research has aimed to reveal the pathological mechanisms that lead to neurodegeneration and AD pathogenesis.2 The most common hypothesis states that an imbalance between the production and clearance of Aβ peptides causes the formation of toxic Aβ species, and thereby triggers the dysfunction and death of neuronal cells.6,7 This is strongly supported by genetic studies that have identified disease-causative mutations in the genes encoding the Aβ precursor protein (APP) and the presenilins, which code for catalytic subunits of γ-secretase, the proteolytic enzyme that dictates the length of Aβ isoforms being generated, and thus their aggregating and toxic properties.8 An APP mutation, which impairs cleavage by β-secretase, and thereby Aβ production, was recently discovered in Finnish families.9,10 This mutation appears to protect its carriers against cognitive decline and AD, further supporting the amyloid theory.9,10 Other susceptibility genes are also implicated in the cellular pathways that control Aβ production and clearance, such as apolipoprotein E, clusterin, α-macroglobulin, or sortilin.11,12 Aβ peptides self-associate to form oligomers, which seed amyloid-plaque deposition, but also, more importantly, which operate as toxic species.13–15 Aβ toxicity induces a vicious circle of neuronal inflammation, oxidative stress, altered metal-ion homeostasis, and metal toxicity, which all in turn increase Aβ production.16,17 Aβ toxicity also alters kinase and phosphatase activities, leading to the hyperphosphorylation of microtubule-associated tau, resulting in microtubule destabilization and the formation of τ-neurofibrillary tangles.18 Therefore, present efforts from both academia and the pharmaceutical industry aim at implementing strategies to prevent Aβ accumulation in the brain. Currently, therapeutic approaches are targeted at either preventing Aβ production by inhibiting the secretase enzymes that are involved in its production, or at interfering with its aggregation by using metal modulators or competing molecules, or at facilitating its clearance through immunotherapy.3,19 The present review focuses on the first approach, more specifically on the inhibition of β-site APP-cleaving enzyme (BACE)-1.
BACE1 and Aβ amyloid production
Aβ is a proteolytic fragment derived from the APP. The APP membrane receptor is involved in the regulation of neuronal activity, synaptic function, neurogenesis, and metal homeostasis.20–24 APP undergoes proteolytic processing through two alternative cellular metabolic pathways, as illustrated in Figure 1. Aβ is produced through the so-called amyloidogenic pathway.25 This involves sequential cleavages by the β-secretase enzyme BACE1 and by γ-secretase. The initial cleavage by BACE1 secretes the APP extracellular domain (sAPPβ) and generates a membrane-tethered 99-amino acid carboxyl terminal fragment (C99).26,27 Subsequent γ-secretase processing of C99 releases Aβ in the intraluminal/extracellular side and the APP intracellular domain (AICD) in the cytosol. sAPPβ is further processed by an unknown protease to release a small N-terminal fragment that can activate death receptor 6 (DR6).28 In a competing cellular pathway, termed nonamyloidogenic, APP is processed by α-secretase that sheds the APP ectodomain sAPPα by cleaving at an alternate site within the Aβ sequence and sixteen residues downstream from the β-secretase cleavage site. Further processing of the corresponding C-terminal fragment (C83) by γ-secretase produces the 3 kDa p3 peptide and AICD.26 In the AD brain, the balance shifts towards the amyloidogenic pathway.
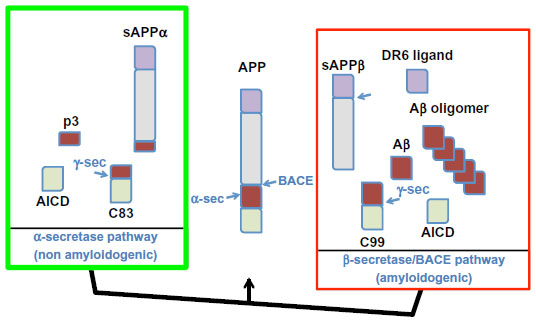 | Figure 1 Disruption of the processing balance of the amyloid precursor protein (APP) in Alzheimer’s disease. The APP can be processed through two alternative pathways. In the α-secretase (α-sec) pathway (or nonamyloidogenic pathway), APP is cleaved by α-sec, which releases the soluble sAPPα N-terminal fragment, creating a membrane-tethered, C-terminal fragment of 83 amino acids (C83). α-Secretase (α-sec) cleavage occurs within the amyloid-β (Aβ) domain (shown in dark red) and precludes Aβ formation. C83 is further processed by γ-secretase (γ-sec) to produce the 3 kDa, nonamyloidogenic peptide p3 and release the APP intracellular domain (AICD, shown in light green) in the cytosol. In the β-secretase pathway (or amyloidogenic pathway), cleavage by β-site APP-cleaving enzyme (BACE1) releases the soluble APP N-terminal fragment (sAPPβ), which can be further processed by an unknown protease to liberate a fragment (shown in lilac) that can activate DR6 receptors. BACE1 cleavage of APP also produces the membrane-tethered C-terminal fragment of 99 amino acids (C99), which is the direct precursor to Aβ. Further processing of C99 by γ-sec achieves the release of Aβ in the extracellular or intravesicular space and AICD in the cytosol. When Aβ production reaches a threshold, the peptide self-aggregates to form toxic oligomers that trigger degeneration of neuronal cells by a mechanism that involves free radical formation and shifts the balance of APP processing towards the amyloidogenic pathway. |
The cellular events that trigger APP processing through the amyloidogenic pathway remain unclear. Experimental evidence that derives from cellular and animal studies concurs on a role of BACE1 in the cellular stress response, as aging, oxidative stress, hypoxia, inflammation, energy deprivation, and other forms of stress, such as trauma and injury, stimulate BACE1 expression.29–33 Furthermore, our group has recently reported that low levels of oxidative stress, although not altering the levels of BACE1 expression, can induce the cellular redistribution of the enzyme, enhance its colocalization with APP, and increase the production of precursor fragments to Aβ, thereby initiating the amyloidogenic process.34
Postmortem studies, including data from our research group, have shown that AD sufferers have higher levels of BACE1 protein, BACE1 proteolytic products, and BACE1 enzymatic activity in cortical brain regions when compared to age-matched controls.35–41 This increase is observed in about half of AD cases, and is attributed to alterations of posttranscriptional and posttranslational mechanisms.36,38,42 BACE1 activity is also elevated in the cerebrospinal fluid (CSF) of patients with mild cognitive impairment (MCI) and with AD,43–45 but its levels may vary as the disease progresses, being actually lower at the more advanced stages of the illness, due to the loss of neurons that are the brain’s cell population with higher BACE1 expression.46 Increased CSF BACE1 activity in MCI patients correlates with changes in hippocampal volume and with the APOE ε4 genotype.47 BACE1 activity is also elevated in platelets of patients with AD and MCI.48,49 The other enzymatic activity that is directly implicated in Aβ formation is γ-secretase, a multimeric membrane-embedded proteolytic complex, where presenilin (either presenilin 1 or presenilin 2) acts as the catalytic subunit.50,51 Numerous presenilin mutations have been identified, particularly in the presenilin 1 gene, which segregate with early onset AD and alter cleavage of APP to produce longer and more aggregating Aβ isoforms.52 The discovery that γ-secretase was involved in a ubiquitous cellular mechanism of intracellular signaling by intramembrane proteolysis, and that it cleaved myriad cellular receptors, raised major concerns regarding the potential side effects of γ-secretase inhibitors in AD treatment.53–55 Modulating γ-secretase to modify its cleavage site on APP towards production of shorter Aβ peptides – without ablating its proteolytic activity – is currently the most sought-after therapeutic approach targeting γ-secretase for AD therapy.56,57 On the other hand, active site-directed γ-secretase inhibitors have found promise in cancer treatment.58 So far, BACE1 has been considered a much safer target than γ-secretase for reducing Aβ production and preventing its accumulation in the brain, principally from comparing phenotypes of presenilin 1 knockout (which is embryonically lethal) to BACE1 gene knockout (that has no major consequences on mouse development).
BACE1 biology
The enzyme responsible for β-secretase cleavage of APP, and which mediates the first step in Aβ production, was simultaneously identified by five research groups in 1999–2000 (reviewed in Cole and Vassar59). Its proposed denominations include BACE (or BACE1, to distinguish it from its homologue BACE2), memapsin 2, and aspartyl protease 2. BACE1 was demonstrated to have the expected tissue distribution and cleavage specificity of β-secretase, being highly expressed in the brain, particularly in neuronal cells, and capable of processing the APP sequence at the N-terminus of Aβ. This is in contrast to its homologue, BACE2, which does not have the correct cleavage specificity and is ubiquitously expressed, with higher expression in the peripheral tissues, such as the colon, kidney, and pancreas.60,61 BACE1 was further validated as the major β-secretase responsible for Aβ production by gene-knockout experiments, as the BACE1-/- mice had hardly detectable levels of Aβ in their brains and BACE1-/- cortical neuronal cultures did not produce Aβ.62–64
BACE1 is a membrane-anchored proteolytic enzyme of the aspartyl protease family, and is synthesized as a 501-amino acid protein with a signal peptide and a prosequence65–70 (Figure 2). BACE1 is inserted in cellular membranes in the same orientation as APP.65–70 The transmembrane domain, located near the C-terminus, helps regulate BACE1 subcellular distribution and access to its substrate.27,65,71–73 The maturation and trafficking of BACE1 have been thoroughly investigated for clues towards strategies to modulate BACE1 activity and/or restrict its access to APP.74 BACE1 undergoes extensive posttranslational modifications, including removal of the prosequence and extensive glycosylation.70,73,75–77 The prosequence helps to stabilize and correctly fold the newly synthesized protein. After maturation, BACE1 is sorted in the Golgi complex and packaged for export to the endosome, which represents its main cellular site of activity because of a low pH that is optimal for this protease.73,78 Sorting of BACE1 from the Golgi complex to the endosome is mediated by Golgi-associated γ-adaptin ear adapter proteins (GGAs), which recognize an aspartyl-X-X-leucyl-leucyl motif (DXXLL)– where X represents any amino acid – at residues 496–500 in its C-terminal region.79–81 BACE1 recycles between the endosome, plasma membrane, and trans-Golgi network. Phosphorylation at serine 498 facilitates BACE1 retrieval by GGA1 from the plasma membrane to the recycling endosomes and trans-Golgi network. Palmitoylation at cysteine residues 478, 482, and 485 contributes to segregating BACE1 into lipid-raft membrane domains, away from its APP substrate.77 BACE1 cellular trafficking, compartmentalization, and turnover are tightly regulated, thereby controlling its encounter with APP, as the two proteins follow independent trafficking routes.74 APP is endocytosed through a clathrin-dependent mechanism, whereas internalized BACE1 is sorted into vesicles containing the small guanosine triphosphatase (GTPase) adenosine diphosphate ribosylation factor 6 (ARF6), on its way to RAB GTPase 5 (RAB5)-positive early endosomes.82 Disruption of BACE1 trafficking increases its colocalization with APP and Aβ production, as exemplified when GGA expression is deficient and in AD.41,83,84 The cellular turnover of BACE1 is determined by ubiquitination at lysine 501, which tags its C-terminus for recognition by the GGA3 adaptor protein that directs its transport to lysosomal degradation, and also possibly facilitates its proteosomal degradation.85,86 Therefore, there are several stages where BACE1 maturation and trafficking could be altered to decrease its activity on APP and offer therapeutic avenues. However, the major approaches explored so far to decrease BACE1 activity on APP directly target its active site.
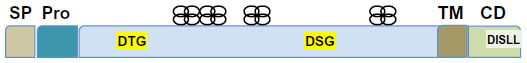 | Figure 2 Schematic diagram of β-site amyloid precursor protein-cleaving enzyme (BACE)-1. BACE1 is translated as a precursor protein, with a signal peptide (SP) and a prosequence (Pro). The mature protein consists of a large enzymatic domain (in blue, with the two motifs that constitute the active site in yellow boxes; DTG stands for aspartyl-threonyl-glycyl and DSG for aspartyl-seryl-glycyl), a transmembrane domain (TM), and a short cytoplasmic domain (CD) containing an endosomal sorting sequence (white box; DISLL stands for aspartyl-isoleucyl-seryl-leucyl-leucyl). The four-leaf designs represent the N-glycosylation sites. |
BACE1 inhibitors in preclinical and clinical trials
Publication of the crystal structure of BACE1 complexed with an inhibitor delineated its catalytic site and provided the bases for developing inhibitors.87 The active site of BACE1 contains two aspartates, which are essential for its proteolytic activity and represent the signature of the aspartyl protease class of enzymes, with these residues being included in canonical aspartyl-seryl/threonyl-glycyl motifs. The BACE1 active site is more elaborate and extended than that of other aspartyl proteases, such as renin or cathepsin D, making it challenging to design molecules capable of blocking its active site, yet small enough to penetrate cellular membranes and cross the blood–brain barrier.88–90 Most of the potent inhibitors reported so far have been selected for their inhibition of BACE1 in vitro in the cleavage reaction of a synthetic substrate based on the sequence surrounding the APP-cleavage site.91 A double mutation is introduced in the sequence, as identified in a Swedish family with early onset AD (KM670/671NL; lysyl670-methionyl671 mutated to asparaginyl-leucyl) to improve cleavage efficiency.92 The enzyme source most commonly used consists of a soluble form of BACE1 that is expressed in Escherichia coli or in baculovirus systems, and lacks the transmembrane and cytosolic domains.87 The fact that the native cellular BACE1 associates as a homodimer and that its enzymatic activity can be enhanced by dimerization93,94 may explain some discrepancies observed for some BACE1 inhibitors between potency in in vitro and cellular assays.
BACE1 inhibitors were initially designed as substrate analogs that mimicked the APP-cleavage sequence, with a nonhydrolyzable peptide bond, such as hydroxyethylene, statine analogue, and hydroxyethylamine isosteres.95,96 Sequence optimization revealed that wild-type APP was a poor substrate for BACE1, suggesting that other substrate proteins may exist.97 Inhibitor structures were fitted to maximize occupancy of the active site pockets, and were further refined to reduce their size and improve their pharmacological properties.90,98 In addition, a variety of classes of molecules capable of inhibiting BACE1 with a high potency were discovered through screening of drug libraries, particularly of aspartyl protease inhibitors prepared for drug-discovery programs targeting renin or human immunodeficiency virus protease, as well as through a fragment-based drug-discovery approach guided by X-ray and nuclear magnetic resonance structure analyses.88,99 Obtaining compounds with a selectivity of several orders of magnitude for BACE1 towards BACE2 and other aspartyl proteases, such as cathepsin D, remains a major challenge. Cathepsin D is a major lysosomal protease, and decreasing its activity would compromise the overall cellular degradation machinery.100 Whilst the function of BACE2 remains unclear, a recent publication of its substrate proteome suggests that it may play an important role in glucose metabolism in the pancreas,101 and this information should be taken into consideration when selecting a BACE1 inhibitor for clinical trials. Nonconventional inhibitors that work in an allosteric mechanism by displacing binding of the substrate have also been described.
Many published preclinical results have demonstrated the capability of proprietary compounds to block Aβ production and prevent or reverse amyloid deposition in animal models. A selection of compounds proven to be efficacious in vivo and which are undergoing or have undergone clinical trials is given in Table 1. GlaxoSmithKline was among the first companies to describe that a BACE1 inhibitor could lower Aβ levels in the brain of APP transgenic mice, using the hydroxyethylamine derivative GSK188909 (Figure 3).102 However, the application of this compound has been limited by its low brain penetration, as it is a substrate for the permeability glycoprotein, which prevents its brain delivery by active efflux.102 Ghosh et al developed the substrate analog hydroxyethylene derivative CTS-21166 (Figure 3), which can reduce Aβ levels in the brain and plasma in an AD animal model.103 Phase I clinical trials indicated that this drug was well tolerated and could decrease plasma Aβ in healthy volunteers.98 Further research led the same group to develop the hydroxyethylamine isostere GRL-8234 (Figure 3), which showed high potency against BACE1 in vitro and in cells, and also a good selectivity for BACE1 compared to BACE2 and cathepsin D. Furthermore, they demonstrated that this drug could penetrate the brain104 and sustain a reduction of plasma and brain Aβ40 for 12 hours in Tg2576 AD mice. Chronic administration over 6 months was well tolerated in the mice, and led to a large decrease of plasma Aβ40 and Aβ42. Behavioral tests following chronic administration showed improved cognitive performance that paralleled a decrease in brain amyloid burden. This study demonstrates that BACE1 inhibition can reduce amyloid deposition in the brain and rescue memory deficits in an animal model, and thus supports the feasibility of BACE1 inhibition for AD therapy. The major limitation of this drug is that it could not be administrated orally and was given by intravenous injection, or for prolonged treatment, through implanted osmotic pumps.
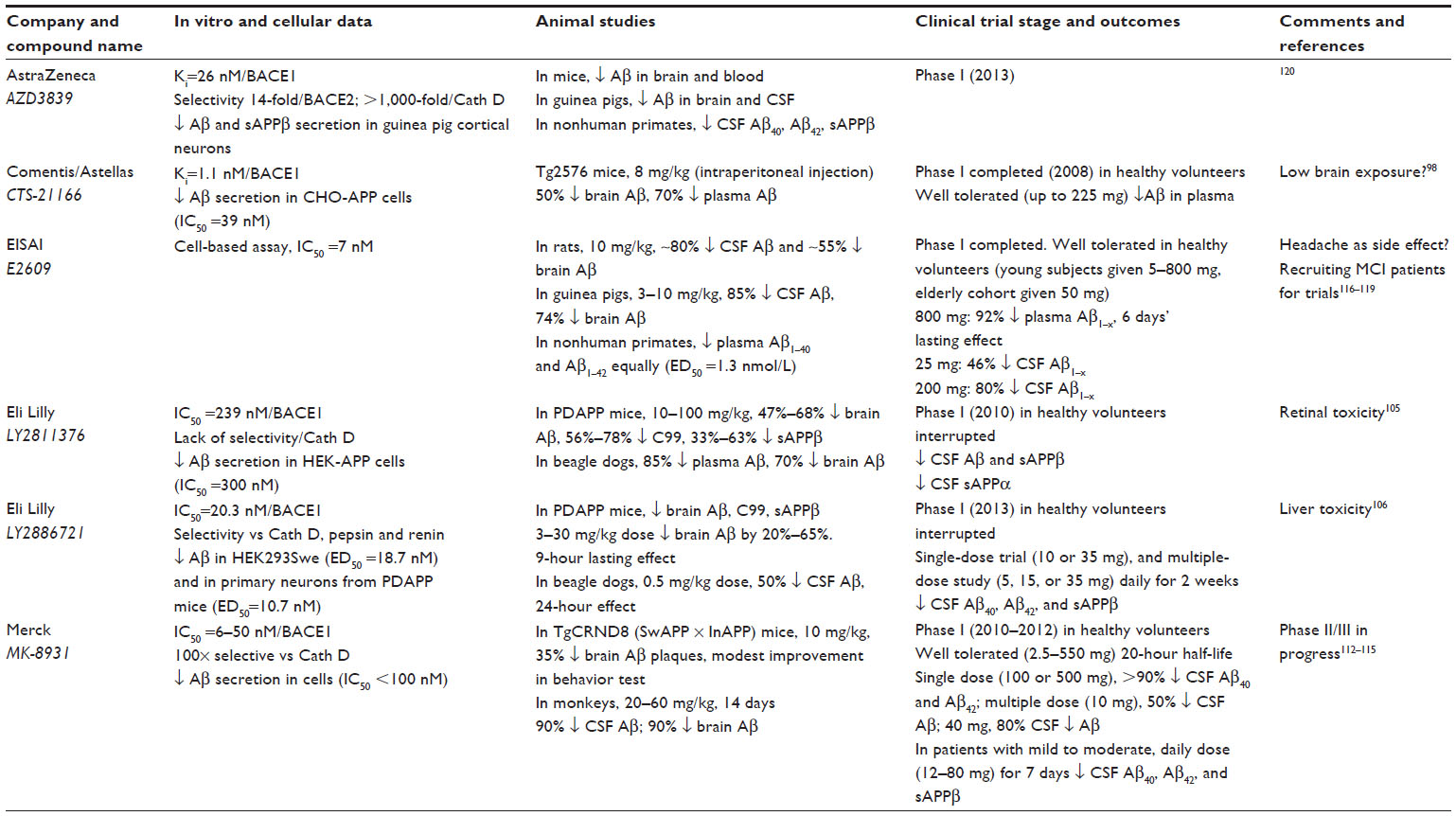 | Table 1 Data of BACE1 inhibitors in clinical trials |
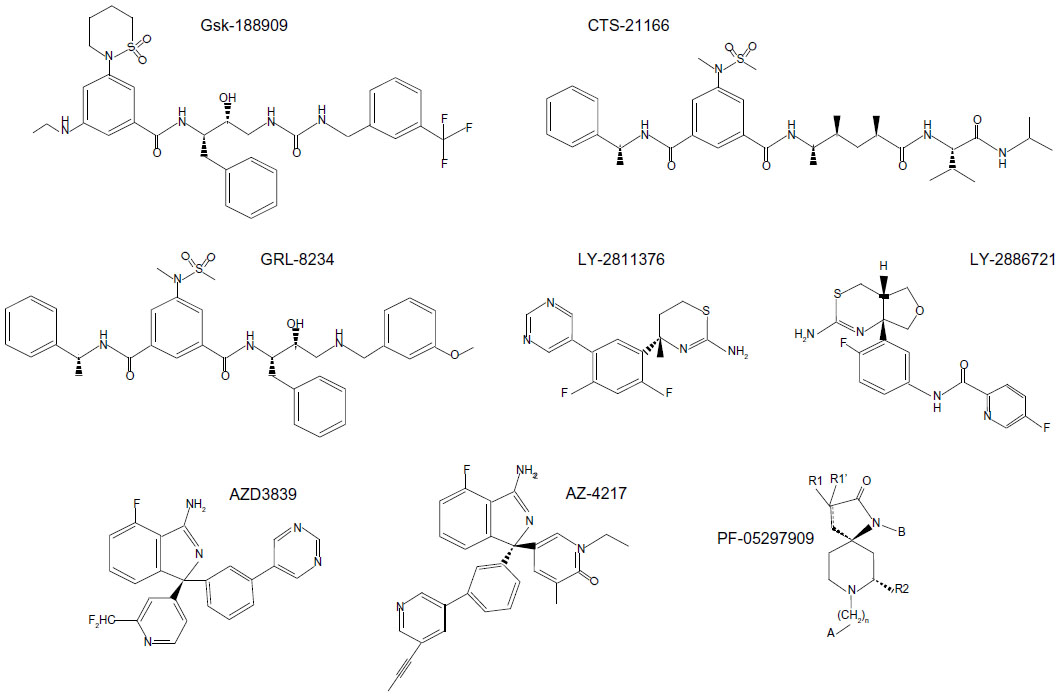 | Figure 3 Published chemical structure of β-site amyloid precursor protein-cleaving enzyme 1 inhibitors in preclinical and clinical trials. The precise structure of Pfizer compound PF-05297909 has not been disclosed, but it is presumably derived from the general formula given in Brodney’s United States Patent and Trademark Office application 20110224231, where R1 and R1′ are each independently hydrogen, alkyl, or alkenyl, R2 is alkyl, cycloalkyl, or alkenyl, B is alkyl, aryl, heteroaryl, cycloalkyl, or heterocycloalkyl, and A is independently aryl, cycloalkyl, heterocycloalkyl, or heteroaryl. |
Lilly Research Laboratories have developed several classes of potent BACE1 inhibitors. Preclinical trials of LY-2811376 (Figure 3), a nonpeptidic inhibitor designed from a fragment-based screening approach, were very promising, and showed that this drug had good bioavailability and high efficacy in vivo.105 A dose-dependent reduction of Aβ levels was demonstrated in the brain of an AD mouse model. The encouraging results of trials in beagle dogs prompted testing of this compound in humans. In Phase I clinical trials, the drug proved to be very efficacious at reducing Aβ and sAPPβ levels in the CSF of healthy volunteers, and seemed to be well tolerated.105 However, clinical trials were discontinued as a safety measure after parallel toxicity studies in rats pointed to retinal toxicity,105 which was attributed to a lack of specificity of the compound and its interference with cathepsin D. Soon afterwards, Lilly began Phase I trials of LY-2886721 (Figure 3), a compound with proven good selectivity for BACE1 against renin and cathepsin D and promising results in preclinical studies. In spite of a relatively short half-life of 3 hours, a dose-dependent inhibition of Aβ, C99, and sAPPβ was observed in the brain of AD mice that received the drug orally.106 Further preclinical trials in beagle dogs showed that a dose of 0.5 mg/kg could achieve a 9-hour-lasting 50% decrease in CSF Aβ and a 24-hour decrease in plasma Aβ.106 Phase I trials were then conducted, in which healthy volunteers received either a single dose or repeated doses of the drug over a period of 2 weeks. CSF analysis showed decreases of up to 74% in Aβ40, 71% in Aβ42, and 77% in sAPPβ concentrations, with a simultaneous increase in sAPPα (+~59%).106 These data show the proof of principle that BACE1 inhibition can achieve its expected outcomes in humans. However, reports of liver toxicity in some of the participants led to interruption of the human trials.
Merck researchers have developed several lead compounds that were identified through screening of a million compounds from drug libraries, and these have produced a large number of molecules that potently inhibit BACE1 in vitro.107–111 Trials of selected drugs in primate models have shown very promising potential, with near-complete inhibition of Aβ levels in the brain and CSF.112,113 Compound MK-8931 has now progressed to Phase II clinical trials, and presentations at conferences suggest this is currently the most advanced BACE1 inhibitor in human trials. Phase I was carried out in two separate centers on small cohorts of 18- to 45-year-old subjects, with healthy volunteers in Belgium receiving a single dose of the compound (2.5–550 mg), and another group of healthy volunteers in the US receiving multiple, rising doses (20, 100, 550 mg), and mild-to-moderate adverse effects being reported. Subjects who received a single dose had their CSF Aβ40 levels decrease by up to 79%, with a sustained 36-hour effect.114 Phase II studies are now being conducted, with no serious adverse effects reported to date. A recent trial in mild-to-moderate AD patients who were given 12–60 mg of MK-8931 for 7 days showed a dose-dependent and sustained reduction in CSF levels of Aβ40 (up to 84%), Aβ42 (up to 81%), and sAPPβ (up to 88%).115
Scientists at Eisai have reported detailed preclinical results of the small-molecule BACE1 inhibitor E2609. The compound showed a dose-dependent decrease of up to ~80% in CSF and up to ~75% in brain Aβ levels after a single-dose administration to rats and guinea pigs.116 An impressive and sustained reduction of CSF Aβ1–40 and Aβ1–42 levels was also observed in nonhuman primates after oral administration to cynomolgus monkeys.117 A Phase I trial of E2609 has also been completed successfully, with the compound being well tolerated. Single-dose administration of 5–800 mg of compound to healthy volunteers aged 30–55 years, and of 50 mg given to an elderly cohort aged 65–85 years, showed a dose-dependent prolonged reduction in plasma Aβ levels.118 Multiple ascending doses given for 2 weeks produced a sustained decrease of Aβ40 and Aβ42 levels in the CSF.119 This compound is now being trialed in Phase II in subjects with mild cognitive impairment.
AstraZeneca also has a development program on BACE1 inhibitors at an advanced stage, and has recently released preclinical data of drug candidates.120,121 Crystal structure-aided refinement of a lead compound that was identified through fragment-based screening achieved the design of AZD3839 (Figure 3), which is orally available and has a good selectivity for BACE1 compared to BACE2 (14-fold) and cathepsin D (>1,000-fold).120 Preclinical data showed low nanomolar in vitro potency of AZD3839 (Ki=26 nM) and its efficacy in cellular assays, including inhibition of Aβ and sAPPβ secretion from guinea pig primary cortical neurons. Studies in animal models indicated a sustained reduction in the levels of Aβ in the brain and plasma of wild-type mice and guinea pigs. CSF analysis in guinea pigs administered the drug showed a decline in Aβ40 that paralleled the decline of Aβ levels in the brain. Intravenous perfusion in cynomolgus monkeys reduced the levels of Aβ40, Aβ42, and sAPPβ in the CSF.120 Thus, AZD3839 appears to be a valid and promising drug candidate for lowering Aβ levels in the brain, and it is currently in Phase I human trials. Another compound, AZ-4217 (Figure 3), also proved to be highly potent in vitro and efficient at lowering Aβ levels and deposition in the brain of an AD mouse model, but it lacks selectivity for BACE1 compared to BACE2.121 Another AstraZeneca compound, AZD3293, was also recently reported to have the preclinical profile of a drug candidate.122
Pfizer researchers have also conducted Phase I safety evaluation of the spirocyclic lactam BACE1 inhibitor PF-05297909 (Figure 3). This compound was well tolerated at doses ranging from 25 to 325 mg, and was able to decrease Aβ levels in plasma; however, it did not induce any changes in CSF Aβ levels, suggesting that insufficient amounts were delivered to the brain.123
All the compounds described thus far are competitive inhibitors that directly target the BACE1-active site, and therefore block its enzymatic activity. In contrast, Takeda researchers have developed a noncompetitive inhibitor, TAK-070, which is a small lipophilic molecule that was uncovered through screening of drug libraries. TAK-070 is specific for BACE1, and disrupts the interaction of BACE1 with APP within the membrane.124 It also promotes the processing of APP through the α-secretase pathway.124 Chronic administration of TAK-070 to transgenic mice from 7 months of age reduced production of soluble Aβ40 and Aβ42 in the brain and decreased amyloid burden by up to 60%. The mice treated with 1 or 3 mg/kg doses of the drug performed better than their littermates in memory and novel object-recognition tests.124 Aged rats that were given the drug also showed improved performance in behavior cognitive tests.125 The compound was registered for Phase I clinical trials in 2005. Ferretti et al at McGill University have uncovered a novel approach to BACE1 inhibition by studying the effect of minocycline in an AD mouse model with abnormally high levels of BACE1.126 Minocycline, which is an anti-inflammatory and neuroprotective drug, was given to the mice at an early age, prior to amyloid deposition. Treatment between the ages of 1 and 3 months significantly decreased brain Aβ levels. Further analysis showed that minocycline not only decreased inflammation markers but also restored BACE1 expression and enzymatic activity to normal levels, through modulation of the nuclear factor κB pathway. These data are promising, although the toxicity of the drug was noticed.
Other approaches for interfering with BACE1 activity and cleavage of APP have been explored. These include silencing BACE1 gene expression by small interfering ribonucleic acid (siRNA),127–130 and immunotherapeutic strategies targeting BACE1 directly,131–134 or targeting the BACE1 cleavage site of APP.135,136
How safe is BACE1 inhibition? Lessons from gene-knockout models
Suppression of BACE1 expression in animal models has shed light on the potential consequences of the complete pharmacological inhibition of BACE1 and revealed that BACE1 cleaved substrates other than APP. The list of these substrates is given in Table 2, and the identified cleavage sites summarized in Table 3.
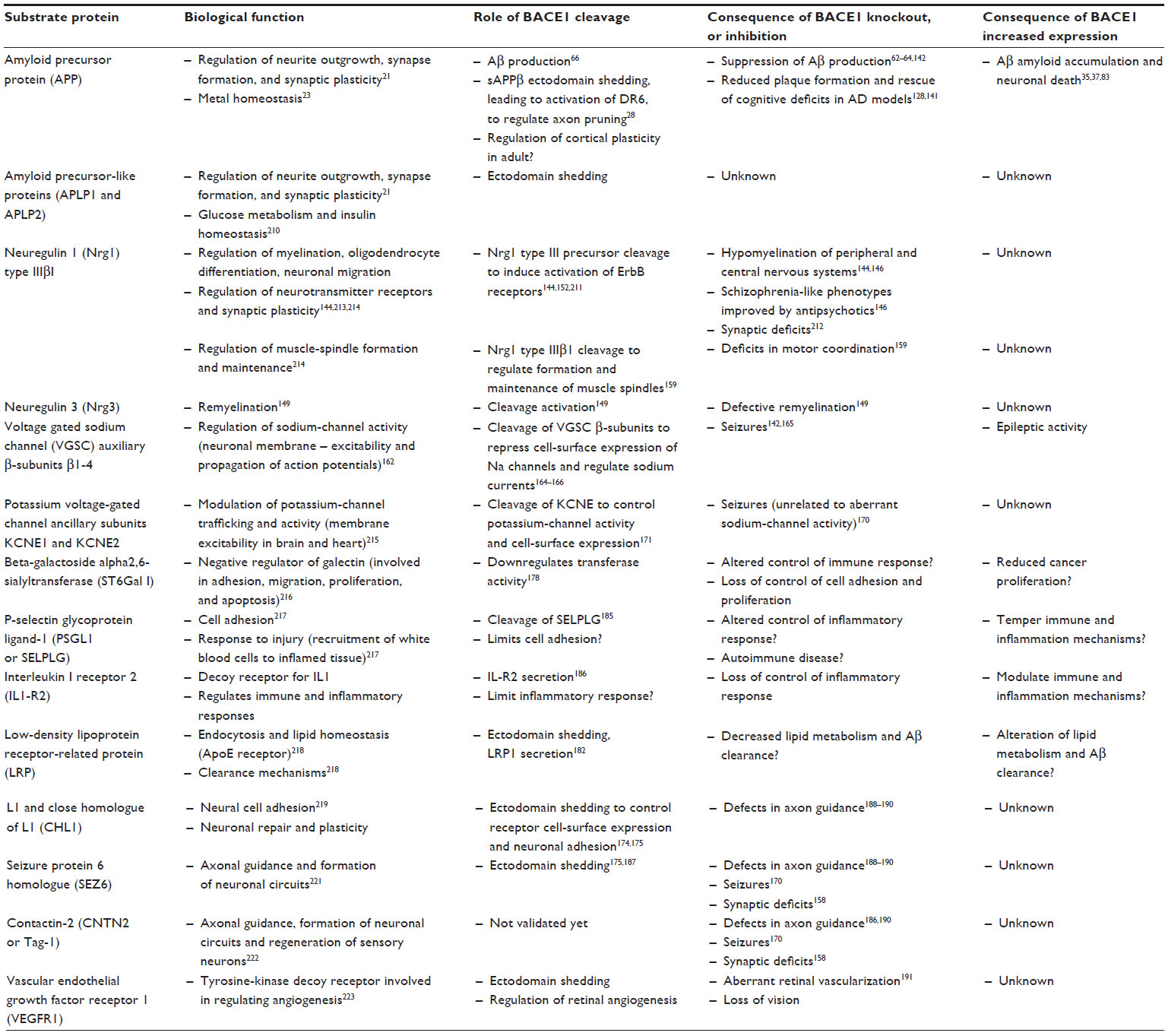 | Table 2 List of identified BACE1 substrates |
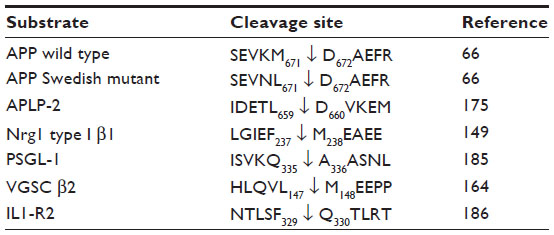 | Table 3 Identified BACE1 substrate-cleavage sites |
The first reports of BACE1 gene knockout in mice indicated no overt phenotype, and supported the safety of BACE1 inhibition in AD therapy. The mice were shown to develop normally, and by the age of 12 months showed no gross morphological defect or abnormality in tissue weights, hematology, or clinical chemistry.62,63 Importantly, BACE1 gene knockout abolished Aβ production in the brain and increased processing of APP by the α-secretase pathway.62 Neuronal cultures derived from the BACE1-/- mice did not produce any detectable amount of Aβ,64 thus establishing BACE1 as the major β-secretase activity in neurons, and supporting the view that inhibiting its activity would abolish Aβ production. Crossing the BACE1-/- mice with transgenic mouse models of AD, or partial genetic deletion of BACE1 in the AD mice, decreased brain amyloid load and rescued memory deficits and synaptic activity,137–141 thereby supporting the potential benefits of BACE1 inhibition in AD.
However, closer monitoring of the BACE1-null mice indicated an increased perinatal mortality, and the survivors were hyperactive and smaller than their wild-type counterparts.142,143 Thorough examination uncovered histological abnormalities as well as behavioral phenotypes, beginning to reveal that BACE1 cleaved substrates other than APP. BACE1-/- mice displayed hypomyelination in the peripheral nervous system and altered axonal sorting of unmyelinated neurons into Schwann cell bundles, akin to what is observed in the NRG1-knockout (NRG1−/+) mice or in mice that lack Schwann cells’ epidermal growth-factor receptor (ErbB) signaling.144 Willem et al further demonstrated that BACE1 was required for proper myelin-sheath formation through the processing of type III Nrg1, as full-length uncleaved type III Nrg1 accumulated in the BACE1-/- mice.144,145 Although BACE1 knockout did not prevent maturation of oligodendrocytes or construction of the myelin sheath, it reduced the expression of myelin proteins, such as myelin basic protein.145,146 Considering that the expression of BACE11 and type III Nrg1 is highest in the early postnatal stage, which is a critical period for neuron myelination, it has been proposed that BACE1 inhibitors would not affect neuron myelination in mature adults and elderly patients.147 However, recent research indicates that Nrg1 promotes remyelination in adults,148 and that the remyelination process after injury is decreased in the BACE1-knockout mice, with accumulation of unprocessed type III Nrg1 and its homologue Nrg3, which was thereby revealed to be another BACE1 substrate, and to also take part in the remyelination process.149 The BACE1 cleavage site of type III Nrg1 was identified at position 237–238, between a phenylalanine (F) and a methionine (M), on the C-terminal side near the juxtamembrane region (Table 3). The BACE1 cleavage site is located within a sequence that is only present in Nrg1 variants, type I, and type III, and in Nrg3, and it is totally conserved among vertebrates.149
The BACE1-null mice also showed defects in myelination in the central nervous system – principally in the hippocampus and cerebral cortex145 – and schizophrenia-like behavior phenotypes.146 These include novelty-induced hyperactivity and impaired social interactions, as well as a decrease in prepulse inhibition and cognitive deficits in the Morris water maze.146 These phenotypes can be attributed to disruptions of the Nrg1/ErbB4 signaling cascade, which has been associated with schizophrenia.150–152 Experimental studies have demonstrated that the Nrg1/ErbB4 system regulates synaptic activity and plasticity, and thus its perturbation causes cortical deficits that can result in glutamatergic hypofunction.153–155 Therefore, it was essential to investigate the consequences of BACE1 knockout on Nrg1 downstream signaling that involves the activation of ErbB4 receptors, which in turn activates a signaling kinase cascade that modulates synaptic plasticity, long-term potentiation, and Src kinase activation of N-methyl-d-aspartate (NMDA) receptors.155 Biochemical analysis of the brain of the BACE1-null mice revealed decreased phosphorylation of the Akt/phosphatidylinositide 3-kinase, which attenuates an interaction between ErbB4 and the postsynaptic density protein PSD95, and thus alters the postsynaptic membrane composition and the function of NMDA receptors.146 BACE1 knockout also decreased the expression of DISC1, a gene associated with psychotic disorders and involved in brain development and neuronal migration,156 and which has recently been shown to regulate NMDA receptors.157 A report of aberrant spine morphology and significantly reduced number of spine dendrites in the hippocampal CA3–CA1 pathway of BACE1-/- mice is consistent with the important role of Nrg1/ErbB4 signaling in the maintenance of synaptic function and plasticity.146 Severe deficits in mossy fiber synapses with an absence of long-term potentiation is another feature of BACE1-/- mice, which may be attributed to defective processing of both Nrg1 and the β-subunits of voltage-gated sodium channels (VGSCs), as developed below.158
A recent study has also reported deficits in muscle coordination in BACE1-/- mice, which are due to improper processing of Ig-Nrg1 type I β1, an Nrg1 isoform that plays an important role in the development, maturation, and maintenance of muscle spindles.159 Pharmacological or genetic ablation of BACE1 activity in adult mice demonstrated that BACE1 was required to sustain the muscle sensory system and regulate movement coordination.159
Other behavior phenotypes of the BACE1-null mice included hyperactivity and an increased susceptibility to seizures,142 which suggested improper sodium-channel activity. Electrophysiological experiments revealed subtle alterations in the inactivation of VGSC in BACE1-deficient neurons.143 Furthermore, when the AD mouse model PDAPP was crossed with BACE1-/-, it performed unexpectedly worse than the original PDAPP and the PDAPP/BACE1−/+ heterozygous, being slower at swimming, showing increased memory deficits, and being more frequently subject to seizures.160 Four-year monitoring of a BACE1-/- mouse colony showed that approximately 30% of these animals died soon after birth as a consequence of seizures. Among the survivors, 11% had seizures before the age of 1 month, and this frequency rose to 14.7% in mice aged 3–6 months and reached 21.9% in mice older than 10 months.161 Electrophysiological activity was altered in the brain of the overall BACE1-/- mouse population, as demonstrated by abnormal wave-spike discharges recorded by electroencephalography and an increased susceptibility to kainate-induced epileptic activity.161 These abnormalities reflect impaired sodium-channel activity. VGSCs are comprised of a heteromeric complex of one large α-subunit (Nav1) and one or two β-subunits (VGSCβ). The β-subunits, which show some homology to cell-adhesion molecules, are known to modulate gating of the channel and to regulate the surface expression of the α-subunit, thus leading to the formation of the ion-conducting pore.162,163 Several independent studies have identified each of the four VGSCβs as substrates for BACE1 cleavage.143,164–168 RNA-interference experiments in mouse embryonic fibroblasts overexpressing any of the four VGSCβ homologues have established their proteolytic processing by BACE1 and γ-secretase.168 Furthermore, biochemical analysis of the brain of BACE1-null mice showed altered processing of VGSCβ.168,169 Pharmacological manipulation of BACE1 activity as well as BACE1 gene knockout decreased the levels of Nav1.1 messenger RNA and protein in rat cortical and hippocampal neurons, supporting the view that BACE1 cleavage of the β subunits and subsequent release of their transcriptionally active intracellular domains controls the expression of the α-subunits and formation of the channel at the cell surface.169 A decrease of Nav1.1 in BACE1-null mice was observed at 1 and 3 months, indicating that endogenous BACE1 activity regulates the levels of Nav1.1 in developing and adult neurons. Furthermore, at 1 month, the BACE1-knockout mice showed increased expression of Nav1.2 and its accumulation at the neuronal surface, which would cause an imbalance in sodium-channel activity and may explain the seizure phenotypes observed in these animals.168 However, another study which corroborated seizure activity and hypersensitivity in the BACE1-/- mice, but to demonstrate a correlation between the expression and surface localization of Nav1.2 and seizure activity.170 It was also demonstrated that BACE1 regulates the excitability of cerebellar neurons by processing the Na+ channel β4-subunit.167 The recent identification of the auxiliary subunits of the potassium voltage-gated channels KCNE1 and KCNE2 and that of the seizure 6 protein as additional BACE1 substrates adds to the complexity of BACE1 physiological function in the brain.101,171
Functional diversity of BACE1 substrates
Besides the major substrates identified in the BACE1 knockout mice, additional substrates were also revealed by other approaches (a comprehensive list of identified BACE1 substrates and their function is given in Table 2 and the identified cleavage sites of BACE1 substrates are given in Table 3). The APP homologues, APLP1 and APLP2 were first investigated and experimentally demonstrated to undergo BACE1-dependent proteolysis by pharmacological and genetic manipulation of BACE1 activity,101,172–174 and their cleavage products were characterized.173,175 They were also harvested as BACE1 substrates in systematic proteomics studies.101,176 The function of the APP homologues remains unknown, but their expression in the central nervous system has been linked to synaptic plasticity and memory consolidation.21 Their role in glucose metabolism and insulin homeostasis has also been documented. And the function of APP itself remains to be taken into consideration. Although it is usually assumed that inhibiting BACE1 cleavage of APP would be beneficial by reducing Aβ production and preventing cell death, the apparently regulated and sophisticated processing of APP merits further investigation. It is important to consider that the cleavage of APP by BACE1 also releases sAPPβ, which is the precursor to a ligand for the death receptor 6 (DR6) that, not only regulates axon pruning during brain development,28 but has now been shown to contribute to regulating cortical plasticity in adults.177 The DR6 knockout mice show deficits in sensory experience caused by defects in axonal pruning that affect long-range horizontal excitatory connections and inhibitory neurons. Thus, the consequences of altering BACE1 activation of DR6 receptors will need further scrutiny.
BACE1 was also shown to process several proteins that contribute to cellular metabolism and trafficking. Among these is the Golgi-resident enzyme, β-galactoside alpha2,6-sialyltransferase (ST6Gal I),178,179 which modifies selected glycoproteins and acts as a negative regulator of galectin binding and function.180 This enzyme is abundantly expressed in the liver from where it becomes secreted into the plasma, particularly during hepatic acute inflammation. A marked decrease in ST6Gal I secretion was observed in the plasma of the BACE1 knockout mice. Furthermore, alterations of BACE1 expression in rodent models induced changes in plasma levels of ST6Gal I that paralleled changes in BACE1 levels.181 The multifunctional endocytic and signaling receptor, low-density lipoprotein receptor related protein (LRP) also undergoes processing by BACE1, which results in the secretion of its extracellular domain.182 Its homologue, LRP4 was also picked up by proteomics analysis.176 LRP1 was recently shown to be involved in neuronal mechanisms that clear apoptotic cells and myelin debris,183 and to mediate Schwann cell-axon interactions, particularly following peripheral nervous system (PNS) injury.184 Other identified BACE1 substrates participate in cell adhesion and in the inflammatory response. These include the P-selectin glycoprotein ligand 1 (PSGL-1), which promotes leukocyte adhesion in the inflammatory response,185 and the decoy receptor, interleukin-1 receptor 2 (IL1-R2), which modulates interleukin I in the brain.186 The neural cell adhesion molecules, L1, close homologue of L1 (CHL1) and contactin-1 were identified as BACE1 substrates through proteomics screens and further validated in vivo.186,187 The examination of BACE1 null mouse brain revealed axon guidance defects in the hippocampus and olfactory bulb,188–190 which closely resemble the CHLI-/- phenotype.188
Following report of the retinal toxicity of the Lilly BACE1 inhibitor LY-2811376, researchers have examined the retina of BACE1-/- mice for signs of pathology and discovered some abnormalities.191 The retina of the BACE1-/- mice is thinner compared to wild type mice, and displays defects in vascularization. These defects are associated with impaired shedding and signaling of the vascular endothelial growth factor receptor 1 (VEGFR1) and can be replicated by treatment with a BACE1 inhibitor of a different chemical class to LY-2811376. An accumulation of lipofuscin age pigment is also observed in retinal pigment epithelial cells that have been treated with BACE1 siRNA or with a BACE1 inhibitor, which reflects the perturbation of lysosomal function. These data suggest that the retinal toxicity of LY-2811376 was the direct consequence of BACE1 inhibition and highlight the importance of elucidating BACE1 biology and closely monitoring the side-effects of BACE1 inhibitors in clinical trials.
Proteomics screens of neuronal and non-neuronal cells, with either deficiency or overexpression of BACE1, have helped to define the BACE1 “sheddome” – the set of receptor proteins that are shed by BACE1 – and discover potential for a series of new substrates that, according to their function, can be subdivided into trafficking proteins (ie, sortilin 1 and sortilin receptor SorL1, semaphorins), proteases (ie, endothelin-converting enzyme 1, aminopeptidase A, carboxypeptidase D), cell adhesion molecules (ie, glypican-3, NCAM1, L1, contactin-1), receptors and co-receptors (mannose-6-phosphate receptor, syndecan-4), and importantly, proteins involved in cell–cell contact and intercellular communication (contactin-1, Jagged-1, ephrin receptors, protocadherins, reelin).101,176,192 These substrates consist mainly of type I integral proteins, and also of a few type II and glycosylphosphatidylinositol (GPI)-anchored proteins. Parallel characterization of the BACE2 “sheddome’ in pancreatic cells has revealed non-redundant roles for the two protease homologues, although some mutual compensatory mechanisms were also observed.101 Therefore the selectivity of BACE1 inhibitors remains an important issue as crossreactivity with BACE2 may lead to unwanted side-effects.
Overall, the findings from the BACE1 knockout mice warn of the potential dangers of eliminating BACE1 activity in humans. The risks of BACE1 ablation may even accrue when some other substrates that were recently uncovered by proteomics studies become further validated.
Detriments of BACE1 elevation and potential benefits of BACE1 inhibition
Studies of post-mortem brain tissue have pointed to a significant (up to two- or three-fold) increase in BACE1 protein levels in AD patients.35–41 The most obvious consequence of elevated BACE1 – which has been confirmed by experimental analysis – is the overproduction of Aβ peptides193 and amyloidogenic fragments.35 This has been replicated in several mouse models.194–198 However, insufficient effort has so far been dedicated towards investigating additional consequences of BACE1 overexpression. Indeed, considering the role of BACE1 substrates in fine-tuned signaling systems in the brain, it is equally important to examine adverse effects of a BACE1 gain-of-function, as those of a BACE1 loss-of-function, in order to fully evaluate the benefits of a BACE1-targeting therapy. BACE1 overexpression in transgenic mice causes excessive Navβ2 cleavage, which results in decreased surface expression of VGSC1, with consequent reduced propagation of action potentials and impaired neuronal activity.165 Excessive processing of Navβ2 due to elevated BACE1 may underlie the increased incidence of epileptic seizures, which has been observed in AD patients.199,200 Examination of an APP transgenic mouse model of AD has revealed high BACE1 expression, increased processing of Navβ2, and accumulation of Navβ2 fragments in the nucleus of GABAergic and non-GABAergic neurons, which were associated with defects in neuronal activity and impaired performance in cognitive behavior tests.201 In addition, the CD1 mouse, a pharmacological model of temporal epilepsy, showed increased BACE1 immunoreactivity and Navβ2 processing in the limbic system, and abnormal axonal sprouting.202 These data highlight potential harmful effects of increased BACE1 activity linked to aberrant sodium channel activity. Whether increased BACE1 activity and increased sAPPβ production may lead to over-activation of DR6 receptors and alter synaptic plasticity also remain to be investigated.
Can BACE1 inhibitors offer additional benefits to Aβ reduction? As described above, many studies have shown the benefits of BACE1 inhibitor treatment to reduce Aβ levels and prevent neuronal loss in animal models. Interestingly, the close observation of BACE1-/- mice showed a reduction in body weight, with increased energy expenditure and reduced metabolic efficiency, as well as increased glucose disposal and insulin sensitivity.203 These mice are also protected against high fat diet-induced obesity.203 Pharmacological reduction of BACE1 activity by Merck-3 inhibitor in a skeletal muscle cell line also increased insulin signaling and glucose uptake.203 These results suggest a role for BACE1 in glucose homoeostasis, which is further supported by proteomics studies.101 As type 2 diabetes is being proposed as a possible risk factor for AD,204 determining if BACE1 inhibition may help fight insulin resistance and decrease blood glucose levels would require future interest. Another positive effect of BACE1 reduction is the enhanced clearance of myelin debris and accelerated regeneration of peripheral axons and the reinnervation of neuromuscular junctions after injury, as evidenced in BACE1-/- mice.205 Treatment with a BACE1 inhibitor also improved axonal injury recovery in wild type mice.205
Conclusion
Cleavage of APP by BACE1 triggers Aβ production and initiates the amyloid cascade of AD. For this reason, BACE1 represents a rational and attractive therapeutic target. Extensive preclinical trials have proved the efficacy of BACE1 inhibitors at lowering Aβ levels in the brain and at rescuing cognitive deficits in AD animal models. However, BACE1 gene knockout experiments have offered some insight into the physiological function of BACE1 and warned of the risks associated with total eradication of its activity. BACE1 appears to play an important role in the developing central and peripheral nervous system, and to be also required for the maintenance and repair of these systems, particularly in response to injury and inflammation. Thus, we propose that safe therapies based on BACE1 inhibition should principally be addressed to patients with demonstrated high BACE1 activity, and be aimed to restore BACE1 to normal levels, but not suppress it. Recent findings from human genetic studies suggest that the position of the balance between the amyloidogenic and non-amyloidogenic processing of APP has direct implications to AD pathogenesis (refer to Figure 1). The Icelandic APP mutation, which shifts this balance towards the non-amyloidogenic pathway by lessening β-secretase cleavage was found to protect its carriers against age-related cognitive decline and dementia.9,10 Conversely, the newly identified missense mutations in the ADAM10, α-secretase gene increase the risk of developing late-onset AD by attenuating α-secretase activity, thus favoring APP processing through the β-secretase/BACE1 pathway.206,207 These results further validate the amyloid hypothesis and support a pharmacological intervention targeting BACE1 to restore the balance between α and β cleavages of APP. An early intervention will be recommended, as PET imaging studies have demonstrated that Aβ starts accumulating in the brain before the first signs of cognitive impairment are observed, and 20–30 years before a clinical AD diagnosis is established and irreparable neuronal loss has occurred.208 Thus, it will be essential to monitor early changes in biomarkers that indicate an increase in BACE1 levels, and to define the range of normal BACE1 activity in the brain or CSF. For this purpose, some of the newly identified BACE1 substrates may provide novel biomarkers to aid quantify BACE1 activity and control the effects, positive and negative, of BACE1 inhibitor therapy. Further development of strategies to modulate the interaction of BACE1 with APP should also be pursued.
Acknowledgments
The authors acknowledge the help of Duncan E Campbell with editing the revised version of the manuscript, and the funding support from the NHMRC (Dora Lusch postgraduate scholarship to AB).
Disclosure
The authors have no conflicts of interest in this work.
References
Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137–152. | |
Masters C, Beyreuther K. Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain. 2006;129(Pt 11):2823–2839. | |
Gandy S, DeKosky ST. Toward the treatment and prevention of Alzheimer’s disease: rational strategies and recent progress. Annu Rev Med. 2013;64(1):367–383. | |
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82(12):4245–4249. | |
Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–890. | |
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | |
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. | |
Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20(4):154–159. | |
Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–99. | |
Kero M, Paetau A, Polvikoski T, et al. Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol Aging. 2013;34(5):1518. e1–e3. | |
Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013;12(1):92–104. | |
Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(10). | |
Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30(4):552–557. | |
Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15(3):349–357. | |
McLean CA, Cherny RA, Fraser FW, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–866. | |
Cappai R, Barnham K. Delineating the mechanism of Alzheimer’s disease Abeta peptide neurotoxicity. Neurochem Res. 2008;33(3):526–532. | |
Yankner BA, Lu T. Amyloid-beta protein toxicity and the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284(8):4755–4759. | |
Gotz J, Lim YA, Ke YD, Eckert A, Ittner LM. Dissecting toxicity of tau and beta-amyloid. Neurodegener Dis. 2010;7(1–3):10–12. | |
Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712. | |
Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–736. | |
Korte M, Herrmann U, Zhang X, Draguhn A. The role of APP and APLP for synaptic transmission, plasticity, and network function: lessons from genetic mouse models. Exp Brain Res. 2012;217(3–4):435–440. | |
Kögel D, Deller T, Behl C. Roles of amyloid precursor protein family members in neuroprotection, stress signaling and aging. Exp Brain Res. 2012;217(3–4):471–479. | |
Duce JA, Tsatsanis A, Cater MA, et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 2010;142(6):857–867. | |
Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26(27):7212–7221. | |
Evin G, Weidemann A. Biogenesis and metabolism of Alzheimer’s disease Aβ amyloid peptides. Peptides. 2002;23(7):1285–1297. | |
De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6(2):99–107. | |
Creemers JW, Ines Dominguez D, Plets E, et al. Processing of beta-secretase by furin and other members of the proprotein convertase family. J Biol Chem. 2001;276(6):4211–4217. | |
Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457(7232):981–989. | |
Tamagno E, Guglielmotto M, Monteleone D, Tabaton M. Amyloid-beta production: major link between oxidative stress and BACE1. Neurotoxicity Res. 2012;22(3):208–219. | |
Chami L, Checler F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and beta-amyloid production in Alzheimer’s disease. Mol Neurodegener. 2012;7:52. | |
O’Connor T, Sadleir KR, Maus E, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60(6):988–1009. | |
Velliquette RA, O’Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J Neurosci. 2005;25(47):10874–10883. | |
Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol. 2004;164(2):719–725. | |
Tan J, Li QX, Ciccotosto G, et al. Mild oxidative stress induces redistribution of BACE1 in non-apoptotic conditions and promotes the amyloidogenic processing of Alzheimer’s disease amyloid precursor protein. PloS One. 2013;8(4):e61246. | |
Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol. 2002;51(6):783–786. | |
Hébert SS, Horré K, Nicolaï L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(17):6415–6420. | |
Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59(9):1381–1389. | |
Tesco G, Koh YH, Kang EL, et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54(5):721–737. | |
Yang LB, Lindholm K, Yan R, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9(1):3–4. | |
Li R, Lindholm K, Yang LB, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A. 2004;101(10):3632–3637. | |
Santosa C, Rasche S, Barakat A, et al. Decreased expression of GGA3 protein in Alzheimer’s disease frontal cortex and increased co-distribution of BACE with the amyloid precursor protein. Neurobiol Dis. 2011;43(1):176–183. | |
Faghihi MA, Modarresi F, Khalil AM, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14(7):723–730. | |
Holsinger R, McLean C, Collins S, Masters C, Evin G. Increased beta-secretase activity in cerebrospinal fluid of Alzheimer’s disease subjects. Ann Neurol. 2004;55(6):898–899. | |
Ewers M, Zhong Z, Burger K, et al. Increased CSF-BACE 1 activity is associated with ApoE-epsilon 4 genotype in subjects with mild cognitive impairment and Alzheimer’s disease. Brain. 2008;131(Pt 5):1252–1258. | |
Zhong Z, Ewers M, Teipel S, et al. Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch Gen Psychiatry. 2007;64(6):718–726. | |
Wu G, Sankaranarayanan S, Tugusheva K, et al. Decrease in age-adjusted cerebrospinal fluid beta-secretase activity in Alzheimer’s subjects. Clin Biochem. 2008;41(12):986–996. | |
Ewers M, Cheng X, Nural HF, et al. Increased CSF-BACE1 activity associated with decreased hippocampus volume in Alzheimer’s disease. J Alzheimers Dis. 2011;25(2):373–381. | |
Johnston J, Liu W, Coulson D, et al. Platelet beta-secretase activity is increased in Alzheimer’s disease. Neurobiol Aging. 2008;29(5):661–668. | |
Liu WW, Todd S, Craig D, et al. Elevated platelet beta-secretase activity in mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;24(6):464–468. | |
Wolfe MS, Haass C. The role of presenilins in gamma-secretase activity. J Biol Chem. 2001;276(8):5413–5416. | |
Iwatsubo T. The gamma-secretase complex: machinery for intramembrane proteolysis. Curr Opin Neurobiol. 2004;14(3):379–383. | |
Czech C, Tremp G, Pradier L. Presenilins and Alzheimer’s disease: biological functions and pathogenic mechanisms. Prog Neurobiol. 2000;60(4):363–384. | |
De Strooper B, Annaert W. Novel research horizons for presenilins and gamma-secretases in cell biology and disease. Ann Rev Cell Dev Biol. 2010;26(1):235–260. | |
Selkoe D, Kopan R. Notch and presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–597. | |
Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane? Nat Rev Mol Cell Biol. 2004;5(6):499–504. | |
Dimitrov M, Alattia JR, Lemmin T, et al. Alzheimer’s disease mutations in APP but not γ-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat Commun. 2013;4:2246. | |
Borgegard T, Juréus A, Olsson F, et al. First and second generation γ-secretase modulators (GSMs) modulate amyloid-beta (Abeta) peptide production through different mechanisms. J Biol Chem. 2012;287(15):11810–11819. | |
Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. γ-Secretase inhibitors and modulators. Biochim Biophys Acta. 2013;1828(12):2898–2907. | |
Cole SL, Vassar R. BACE1 structure and function in health and Alzheimer’s disease. Curr Alzheimer Res. 2008;5(2):100–120. | |
Bennett BD, Babu-Khan S, Loeloff R, et al. Expression analysis of BACE2 in brain and peripheral tissues. J Biol Chem. 2000;275(27):20647–20651. | |
Fluhrer R, Capell A, Westmeyer G, et al. A non-amyloidogenic function of BACE-2 in the secretory pathway. J Neurochem. 2002;81(5):1011–1020. | |
Luo Y, Bolon B, Kahn S, et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4(3):231–232. | |
Roberds SL, Anderson J, Basi G, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet. 2001;10(12):1317–1324. | |
Cai H, Wang Y, McCarthy D, et al. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4(3):233–234. | |
Sinha S, Anderson JP, Barbour R, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402(6761):537–540. | |
Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. | |
Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97(4):1456–1460. | |
Hussain I, Powell D, Howlett DR, et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14(6):419–427. | |
Yan R, Bienkowski MJ, Shuck ME, et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature. 1999;402(6761):533–537. | |
Haniu M, Denis P, Young Y, et al. Characterization of Alzheimer’s beta-secretase protein BACE. A pepsin family member with unusual properties. J Biol Chem. 2000;275(28):21099–21106. | |
Qahwash I, He W, Tomasselli A, Kletzien RF, Yan R. Processing amyloid precursor protein at the beta-site requires proper orientation to be accessed by BACE1. J Biol Chem. 2004;279(37):39010–39016. | |
Yan R, Han P, Miao H, Greengard P, Xu H. The transmembrane domain of the Alzheimer’s beta-secretase (BACE1) determines its late Golgi localization and access to beta-amyloid precursor protein (APP) substrate. J Biol Chem. 2001;276(39):36788–36796. | |
Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J Biol Chem. 2000;275(43):33729–33737. | |
Tan J, Evin G. β-Site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J Neurochem. 2012;120(6):869–880. | |
Capell A, Steiner H, Willem M, et al. Maturation and pro-peptide cleavage of beta-secretase. J Biol Chem. 2000;275(40):30849–30854. | |
Bennett BD, Denis P, Haniu M, et al. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer’s beta-secretase. J Biol Chem. 2000;275(48):37712–37717. | |
Benjannet S, Elagoz A, Wickham L, et al. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J Biol Chem. 2001;276(14):10879–10887. | |
Kandalepas PC, Vassar R. Identification and biology of beta-secretase. J Neurochem. 2012;120 Suppl 1:55–61. | |
He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J Biol Chem. 2004;280(12):11696–11703. | |
von Arnim CA, Tangredi MM, Peltan ID, et al. Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J Cell Sci. 2004;117(Pt 22):5437–5445. | |
Wahle T, Prager K, Raffler N, Haass C, Famulok M, Walter J. GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol Cell Neurosci. 2005;29(3):453–461. | |
Sannerud R, Declerck I, Peric A, et al. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A. 2011;108(34):E559–E568. | |
Tesco G, Koh YH, Kang EL, et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007;54(5):721–737. | |
Walker KR, Kang EL, Whalen MJ, Shen Y, Tesco G. Depletion of GGA1 and GGA3 mediates postinjury elevation of BACE1. J Neurosci. 2012.25;32(30):10423–10437. | |
Kang EL, Cameron AN, Piazza F, Walker KR, Tesco G. Ubiquitin regulates GGA3-mediated degradation of BACE1. J Biol Chem. 2010;285(31):24108–24119. | |
Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 2004;18(13):1571–1573. | |
Hong L, Koelsch G, Lin X, et al. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science. 2000;290(5489):150–153. | |
Evin G, Lessene G, Wilkins S. BACE inhibitors as potential drugs for the treatment of Alzheimer’s disease: focus on bioactivity. Recent Pat CNS Drug Discov. 2011;6(2):91–106. | |
Evin G, Kenche VB. BACE inhibitors as potential therapeutics for Alzheimer’s disease. Recent Pat CNS Drug Discov. 2007;2(3):188–199. | |
Cumming JN, Iserloh U, Kennedy ME. Design and development of BACE-1 inhibitors. Curr Opin Drug Discov Devel. 2004;7(4):536–556. | |
Ermolieff J, Loy JA, Koelsch G, Tang J. Proteolytic activation of recombinant pro-memapsin 2 (pro-beta-secretase) studied with new fluorogenic substrates. Biochemistry. 2000;39(40):12450–12456. | |
Mullan M, Crawford F, Axelman K, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1(5):345–347. | |
Schmechel A, Strauss M, Schlicksupp A, et al. Human BACE forms dimers and colocalizes with APP. J Biol Chem. 2004;279(38):39710–39717. | |
Westmeyer GG, Willem M, Lichtenthaler SF, et al. Dimerization of beta-site beta-amyloid precursor protein-cleaving enzyme. J Biol Chem. 2004;279(51):53205–53212. | |
Kimura T, Shuto D, Kasai S, et al. KMI-358 and KMI-370, highly potent and small-sized BACE1 inhibitors containing phenylnorstatine. Bioorg Med Chem Lett. 2004;14(6):1527–1531. | |
Ghosh A, Bilcer G, Harwood C, et al. Structure-based design: potent inhibitors of human brain memapsin 2 (beta-secretase). J Med Chem. 2001;44(18):2865–2868. | |
Turner RT 3rd, Koelsch G, Hong L, et al. Subsite specificity of memapsin 2 (beta-secretase): implications for inhibitor design. Biochemistry. 2001;40(34):10001–10006. | |
Ghosh AK, Brindisi M, Tang J. Developing β-secretase inhibitors for treatment of Alzheimer’s disease. J Neurochem. 2012;120 Suppl 1:71–83. | |
Wyss D, Wang YS, Eaton H, et al. Combining NMR and X-ray crystallography in fragment-based drug discovery: discovery of highly potent and selective BACE-1 inhibitors. Top Curr Chem. 2012;317:83–114. | |
Zaidi N, Maurer A, Nieke S, Kalbacher H. Cathepsin D: a cellular roadmap. Biochem Biophys Res Comm. 2008;376(1):5–9. | |
Stützer I, Selevsek N, Esterházy D, Schmidt A, Aebersold R, Stoffel M. Systematic proteomic analysis identifies beta-site amyloid precursor protein cleaving enzyme 2 and 1 (BACE2 and BACE1) substrates in pancreatic beta-cells. J Biol Chem. 2013;288(15):10536–10547. | |
Hussain I, Hawkins J, Harrison D, et al. Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases beta-cleavage of amyloid precursor protein and amyloid-beta production in vivo. J Neurochem. 2007;100(3):802–809. | |
Ghosh AK, Kumaragurubaran N, Hong L, et al. Design, synthesis, and X-ray structure of potent memapsin 2 (beta-secretase) inhibitors with isophthalamide derivatives as the P2-P3-ligands. J Med Chem. 2007;50(10):2399–2407. | |
Chang WP, Huang X, Downs D, et al. Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011;25(2):775–784. | |
May PC, Dean RA, Lowe SL, et al. Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci. 2011;31(46):16507–16516. | |
May P, Boggs L, Brier R, et al. Preclinical characterization of LY2886721: a BACE1 inhibitor in clinical development for early Alzheimer’s disease. Alzheimers Dement. 2012;8(Suppl 4):P95. | |
Stachel SJ, Steele TG, Petrocchi A, et al. Discovery of pyrrolidine-based beta-secretase inhibitors: lead advancement through conformational design for maintenance of ligand binding efficiency. Bioorg Med Chem Lett. 2012;22(1):240–244. | |
Treiber H, Hagemeyer N, Ehrenreich H, Simons M. BACE1 in central nervous system myelination revisited. Mol Psychiatry. 2012;17(3):237–239. | |
Coburn CA, Stachel SJ, Li YM, et al. Identification of a small molecule nonpeptide active site beta-secretase inhibitor that displays a nontraditional binding mode for aspartyl proteases. J Med Chem. 2004;47(25):6117–6119. | |
Steele TG, Hills ID, Nomland AA, et al. Identification of a small molecule beta-secretase inhibitor that binds without catalytic aspartate engagement. Bioorg Med Chem Lett. 2009;19(1):17–20. | |
Mandal M, Zhu Z, Cumming JN, et al. Design and validation of bicyclic iminopyrimidinones as beta amyloid cleaving enzyme-1 (BACE1) inhibitors: conformational constraint to favor a bioactive conformation. J Med Chem. 2012;55(21):9331–9345. | |
Sankaranarayanan S, Holahan MA, Colussi D, et al. First demonstration of cerebrospinal fluid and plasma Abeta lowering with oral administration of a beta-site amyloid precursor protein-cleaving enzyme 1 inhibitor in nonhuman primates. J Pharmacol Expl Ther. 2009;328(1):131–140. | |
Sankaranarayanan S, Price EA, Wu G, et al. In vivo beta-secretase 1 inhibition leads to brain Abeta lowering and increased beat-secretase processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther. 2008;324(3):957–969. | |
Forman M, Tseng J, Palcza J, et al. The novel BACE inhibitor MK-8931 dramatically lowers CSF Aβ peptides in healthy subjects: results from a rising single dose study (PL02.004). Neurology. 2012;78(Meeting Abstracts 1):PL02.004. | |
Forman M, Kleijn HJ, Dockendorf M, et al. The novel BACE inhibitor MK-8931 dramatically lowers CSF beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2013;9(Suppl 4):P139. | |
Fukushima T, Osada Y, Ishibashi A, Lucas F. Novel BACE1 inhibitor, E2609, lowers Aβ levels in the brain, cerebrospinal fluid and plasma in rats and guinea pigs. Alzheimers Dement. 2012;8(Suppl 4):P223–P224. | |
Lucas F, Fukushima T, Nozaki Y. Novel BACE1 inhibitor, E2609, lowers Aβ levels in the cerebrospinal fluid and plasma in nonhuman primates. Alzheimers Dement. 2012;8(Suppl 4):P224. | |
Lai R, Albala B, Kaplow JM, Aluri J, Yen M, Satlin A. First-in-human study of E2609, a novel BACE1 inhibitor, demonstrates prolonged reductions in plasma beta-amyloid levels after single dosing. Alzheimers Dement. 2012;8(Suppl 4):P96. | |
Lai R, Albala B, Kaplow J, et al. Novel BACE1 inhibitor E2609 reduces plasma and CSF amyloid in healthy subjects after 14 days oral administration. Poster presented at: 11th International Conference on Alzheimer’s and Parkinson’s Diseases; March 6–10, 2013; Florence, Italy. Abstract 51889. | |
Jeppsson F, Eketjall S, Janson J, et al. Discovery of AZD3839, a potent and selective BACE1 inhibitor clinical candidate for the treatment of Alzheimer disease. J Biol Chem. 2012;287(49):41245–41257. | |
Eketjäll S, Janson J, Jeppsson F, et al. AZ-4217: a high potency BACE inhibitor displaying acute central efficacy in different in vivo models and reduced amyloid deposition in Tg2576 mice. J Neurosci. 2013;33(24):10075–10084. | |
Poole M, Alexander R, Olsson T, et al. AZD3293, a potent and selective orally active, brain-permeable BACE1 Inhibitor. Alzheimers Dement. 2013;9(4):813. | |
Nicholas T, Ueckert S, Ito K, et al. A Novel BACE inhibitor (PF-05297909): a two-part adaptive design to evaluate safety, pharmacokinetics and pharmacodynamics for modifying beta-amyloid (Abeta) in a first-in-human study. Poster presented at: 11th International Conference on Alzheimer’s and Parkinson’s Diseases; March 6–10, 2013; Florence, Italy. Abstract PI-351. | |
Fukumoto H, Takahashi H, Tarui N, et al. A noncompetitive BACE1 inhibitor TAK-070 ameliorates Abeta pathology and behavioral deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2010;30(33):11157–11166. | |
Takahashi H, Fukumoto H, Maeda R, Terauchi J, Kato K, Miyamoto M. Ameliorative effects of a non-competitive BACE1 inhibitor TAK-070 on Abeta peptide levels and impaired learning behavior in aged rats. Brain Res. 2010;1361:146–156. | |
Ferretti M, Allard S, Partridge V, Ducatenzeiler A, Cuello AC. Minocycline corrects early, pre-plaque neuroinflammation and inhibits BACE-1 in a transgenic model of Alzheimer’s disease-like amyloid pathology. J Neuroinflammation. 2012;9(1):62. | |
Kao SC, Krichevsky AM, Kosik KS, Tsai LH. BACE1 suppression by RNA interference in primary cortical neurons. J Biol Chem. 2004;279(3):1942–1949. | |
Singer O, Marr RA, Rockenstein E, et al. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8(10):1343–1349. | |
Peng K, Masliah E. Lentivirus-expressed siRNA vectors against Alzheimer disease. In: Federico M, editor. Lentivirus Gene Engineering Protocols (Methods in Molecular Biology). 2nd ed. New York: Humana. 2010:215–224. | |
Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotech. 2011;29(4):341–345. | |
Chang WP, Downs D, Huang XP, Da H, Fung KM, Tang J. Amyloid-beta reduction by memapsin 2 (beta-secretase) immunization. FASEB J. 2007;21(12):3184–3196. | |
Yu Y, Zhang Y, Kenrick M, et al. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;3(84):84ra44. | |
Paul SM. Therapeutic antibodies for brain disorders. Sci Transl Med. 2011;3(84):84ps20. | |
Couch JA, Yu YJ, Zhang Y, et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci Transl Med. 2013;5(183):183ra157. | |
Arbel M, Yacoby I, Solomon B. Inhibition of amyloid precursor protein processing by beta-secretase through site-directed antibodies. Proc Nat Acad Sci U S A. 2005;102(21):7718–7723. | |
Rabinovich-Nikitin I, Rakover IS, Becker M, Solomon B. Beneficial effect of antibodies against β-secretase cleavage site of APP on Alzheimer’s-like pathology in triple-transgenic mice. PLoS One. 2012;7(10):e46650. | |
Ohno M, Sametsky E, Younkin L, et al. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron. 2004;41(1):27–33. | |
Giusti-Rodríguez P, Gao J, Graff J, Rei D, Soda T, Tsai LH. Synaptic deficits are rescued in the p25/Cdk5 model of neurodegeneration by the reduction of β-secretase (BACE1). J Neurosci. 2011;31(44):15751–15756. | |
Ohno M, Chang L, Tseng W, et al. Temporal memory deficits in Alzheimer’s mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23(1):251–260. | |
Ohno M, Cole S, Yasvoina M, et al. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26(1):134–145. | |
McConlogue L, Buttini M, Anderson JP, et al. Partial reduction of BACE1 has dramatic effects on alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem. 2007;282(36):26326–26334. | |
Dominguez D, Tournoy J, Hartmann D, et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280(35):30797–30806. | |
Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM. Reduced sodium channel Nav1.1 levels in BACE1-null mice. J Biol Chem. 2011;286(10):8106–8116. | |
Willem M, Garratt AN, Novak B, et al. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314(5799):664–666. | |
Hu X, Hicks C, He W, et al. BACE1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9(12):1520–1525. | |
Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Nat Acad Sci U S A. 2008;105(14):5585–5590. | |
Schubert C. Alzheimer disease: BACE1 branches out. Nat Med. 2006;12(10):1123. | |
Stassart RM, Fledrich R, Velanac V, et al. A role for Schwann cell-derived neuregulin-1 in remyelination. Nat Neurosci. 2013;16(1):48–54. | |
Hu X, He W, Diaconu C, et al. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J. 2008;22(8):2970–2980. | |
Harrison PJ, Law AJ. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiatry. 2006;60(2):132–140. | |
Buonanno A. The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull. 2010; 83(3–4):122–131. | |
Shamir A, Kwon OB, Karavanova I, et al. The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J Neurosci. 2012;32(9):2988–2997. | |
Hahn CG, Wang HY, Cho DS, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12(7):824–828. | |
Li B, Woo RS, Mei L, Malinow R. The neuregulin-1 receptor ErbB4 controls glutamatergic synapse maturation and plasticity. Neuron. 2007;54(4):583–597. | |
Hahn CG. A Src link in schizophrenia. Nat Med. 2011;17(4):425–427. | |
Seshadri S, Kamiya A, Yokota Y, et al. Disrupted-in-schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1 neuregulin cascade. Proc Nat Acad Sci U S A. 2010;107(12):5622–5627. | |
Wei J, Graziane NM, Wang H, et al. Regulation of N-methyl-D-aspartate receptors by disrupted-in-schizophrenia-1. Biol Psychiatry. Epub July 29, 2013. | |
Wang H, Song L, Laird F, Wong PC, Lee HK. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. J Neurosci. 2008;28(35):8677–8681. | |
Cheret C, Willem M, Fricker FR, et al. BACE1 and neuregulin-1 cooperate to control formation and maintenance of muscle spindles. EMBO J. 2013;32(14):2015–2028. | |
Kobayashi D, Zeller M, Cole T, et al. BACE1 gene deletion: impact on behavioral function in a model of Alzheimer’s disease. Neurobiol Aging. 2008;29(6):861–873. | |
Hu X, Zhou X, He W, et al. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci. 2010;30(26):8819–8829. | |
Yu EJ, Ko SH, Lenkowski PW, Pance A, Patel MK, Jackson AP. Distinct domains of the sodium channel beta3-subunit modulate channel-gating kinetics and subcellular location. Biochem J. 2005;392(Pt 3):519–526. | |
Isom LL. Beta subunits: players in neuronal hyperexcitability? Novartis Found Symp. 2002;241:124–138. | |
Gersbacher M, Kim D, Bhattacharyya R, Kovacs D. Identification of BACE1 cleavage sites in human voltage-gated sodium channel beta 2 subunit. Mol Neurodegener. 2010;5(1):61. | |
Kim D, Carey B, Wang H, et al. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9(7):755–764. | |
Huth T, Alzheimer C. Voltage-dependent Na+ channels as targets of BACE1 – implications for neuronal firing and beyond. Curr Alzheimer Res. 2012;9(2):184–188. | |
Huth T, Rittger A, Saftig P, Alzheimer C. β-Site APP-cleaving enzyme 1 (BACE1) cleaves cerebellar Na+ channel β4-subunit and promotes Purkinje cell firing by slowing the decay of resurgent Na+ current. Pflugers Arch. 2011;461(3):355–371. | |
Wong HK, Sakurai T, Oyama F, et al. β-Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280(24):23009–23017. | |
Kim DY, Gersbacher MT, Inquimbert P, Kovacs DM. Reduced sodium channel Na(v)1.1 levels in BACE1-null mice. J Biol Chem. 2011;286(10):8106–8116. | |
Hitt B, Jaramillo T, Chetkovich D, Vassar R. BACE1-/- mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol Neurodegener. 2010;5(1):31. | |
Sachse CC, Kim YH, Agsten M, et al. BACE1 and presenilin/γ-secretase regulate proteolytic processing of KCNE1 and 2, auxiliary subunits of voltage-gated potassium channels. FASEB J. 2013;27(6):2458–2467. | |
Eggert S, Paliga K, Soba P, et al. The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: modulation of APLP-1 processing by n-glycosylation. J Biol Chem. 2004;279(18):18146–18156. | |
Li Q, Südhof T. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem. 2004;279(11):10542–10550. | |
Pastorino L, Ikin A, Lamprianou S, et al. BACE (beta-secretase) modulates the processing of APLP2 in vivo. Mol Cell Neurosci. 2004;25(4):642–649. | |
Hogl S, Kuhn PH, Colombo A, Lichtenthaler SF. Determination of the proteolytic cleavage sites of the amyloid precursor-like protein 2 by the proteases ADAM10, BACE1 and gamma-secretase. PLoS One. 2011;6(6):e21337. | |
Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS One. 2009;4(12):e8477. | |
Marik SA, Olsen O, Tessier-Lavigne M, Gilbert CD. Death receptor 6 regulates adult experience-dependent cortical plasticity. J Neurosci. 2013;33(38):14998–15003. | |
Kitazume S, Tachida Y, Oka R, et al. Characterization of alpha 2,6-sialyltransferase cleavage by Alzheimer’s beta secretase (BACE1). J Biol Chem. 2003;278(17):14865–14871. | |
Kitazume S, Tachida Y, Oka R, Shirotani K, Saido T, Hashimoto Y. Alzheimer’s beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci U S A. 2001;98(24):13554–13559. | |
Zhuo Y, Bellis SL. Emerging role of beta-2,6-sialic acid as a negative regulator of galectin binding and function. J Biol Chem. 2011;286(8):5935–5941. | |
Kitazume S, Nakagawa K, Oka R, et al. In vivo cleavage of alpha2,6-sialyltransferase by Alzheimer beta-secretase. J Biol Chem. 2005;280(9):8589–8595. | |
von Arnim C, Kinoshita A, Peltan I, et al. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem. 2005;280(18):17777–17785. | |
Fernandez-Castaneda A, Arandjelovic S, Stiles TL, et al. Identification of the low density lipoprotein (LDL) receptor-related protein-1 interactome in central nervous system myelin suggests a role in the clearance of necrotic cell debris. J Biol Chem. 2013;288(7):4538–4548. | |
Orita S, Henry K, Mantuano E, et al. Schwann cell LRP1 regulates Remak bundle ultrastructure and axonal interactions to prevent neuropathic pain. J Neurosci. 2013;33(13):5590–5602. | |
Lichtenthaler S, Dominguez D, Westmeyer G, et al. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem. 2003;78(49):48713–48719. | |
Kuhn PH, Marjaux E, Imhof A, De Strooper B, Haass C, Lichtenthaler SF. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J Biol Chem. 2007;282(16):11982–11995. | |
Zhou L, Barao S, Laga M, et al. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem. 2012;287(31):25927–25940. | |
Hitt B, Riordan SM, Kukreja L, Eimer WA, Rajapaksha TW, Vassar R. β-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J Biol Chem. 2012;287(46):38408–38425. | |
Rajapaksha T, Eimer W, Bozza T, Vassar R. The Alzheimer’s β-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegen. 2011;6(1):88. | |
Cao L, Rickenbacher GT, Rodriguez S, Moulia TW, Albers MW. The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci Rep. 2012;2:231. | |
Cai J, Qi X, Kociok N, et al. β-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol Med. 2012;4(9):980–991. | |
Kuhn PH, Koroniak K, Hogl S, et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012;31(14):3157–3168. | |
Li R, Lindholm K, Yang LB, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A. 2004;101(10):3632–3637. | |
Chiocco M, Kulnane LS, Younkin L, Younkin S, Evin G, Lamb B. Altered amyloid-beta metabolism and deposition in genomic-based beta-secretase transgenic mice. J Biol Chem. 2004;279(50):52535–52542. | |
Ozmen L, Woolley M, Albientz A, et al. BACE/APPV717F double-transgenic mice develop cerebral amyloidosis and inflammation. Neurodegen Dis. 2005;2(6):284–298. | |
Bodendorf U, Danner S, Fischer F, et al. Expression of human β-secretase in the mouse brain increases the steady-state level of β-amyloid. J Neurochem. 2002;80(5):799–806. | |
Mohajeri MH, Saini KD, Nitsch RM. Transgenic BACE expression in mouse neurons accelerates amyloid plaque pathology. J Neural Transm. 2004;111(3):413–425. | |
Willem M, Dewachter I, Smyth N, et al. β-Site amyloid precursor protein cleaving enzyme 1 increases amyloid deposition in brain parenchyma but reduces cerebrovascular amyloid angiopathy in aging BACE × APP[V717I] double-transgenic mice. Am J Pathol. 2004;165(5):1621–1631. | |
Amatniek JC, Hauser WA, DelCastillo-Castaneda C, et al. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia. 2006;47(5):867–872. | |
Lozsadi DA, Larner AJ. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;22(2):121–124. | |
Corbett BF, Leiser SC, Ling HP, et al. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2013;33(16):7020–7026. | |
Yan XX, Cai Y, Zhang XM, et al. BACE1 elevation is associated with aberrant limbic axonal sprouting in epileptic CD1 mice. Exp Neurol. 2012;235(1):228–237. | |
Meakin PJ, Harper AJ, Hamilton DL, et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem J. 2012;441(1):285–296. | |
Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Brit J Clin Pharmacol. 2011;71(3):365–376. | |
Farah MH, Pan BH, Hoffman PN, et al. Reduced BACE1 activity enhances clearance of myelin debris and regeneration of axons in the injured peripheral nervous system. J Neurosci. 2011;31(15):5744–5754. | |
Kim M, Suh J, Romano D, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum Mol Genet. 2009;18(20):3987–3996. | |
Suh J, Choi SH, Romano DM, et al. ADAM10 missense mutations potentiate β-amyloid accumulation by impairing prodomain chaperone function. Neuron. 2013;80(2):385–401. | |
Doré V, Villemagne VL, Bourgeat P, et al. Cross-sectional and longitudinal analysis of the relationship between Aβ deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease. JAMA Neurol. 2013;70(7):903–911. | |
Ghosh A, Kumaragurubaran N, Hong L, et al. Design, synthesis, and X-ray structure of potent memapsin 2 (beta-secretase) inhibitors with isophthalamide derivatives as the P2-P3-ligands. J Med Chem. 2007;50(10):2399–2407. | |
Needham BE, Wlodek ME, Ciccotosto GD, et al. Identification of the Alzheimer’s disease amyloid precursor protein (APP) and its homologue APLP2 as essential modulators of glucose and insulin homeostasis and growth. J Pathol. 2008;215(2):155–163. | |
Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9(6):437–452. | |
Pitcher GM, Kalia LV, Ng D, et al. Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat Med. 2011;17(4):470–478. | |
Wen L, Lu YS, Zhu XH, et al. Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc Natl Acad Sci U S A. 2010;107(3):1211–1216. | |
Hippenmeyer S, Shneider NA, Birchmeier C, Burden SJ, Jessell TM, Arber S. A role for neuregulin1 signaling in muscle spindle differentiation. Neuron. 2002;36(6):1035–1049. | |
Kanda VA, Abbott GW. KCNE regulation of K+ channel trafficking – a Sisyphean task? Frontiers Physiol. 2012;3:231. | |
Lee M, Park JJ, Ko YG, Lee YS. Cleavage of ST6Gal I by radiation-induced BACE1 inhibits Golgi-anchored ST6Gal I-mediated sialylation of integrin beta1 and migration in colon cancer cells. Radiat Oncol. 2012;7(1):47. | |
Carlow DA, Gossens K, Naus S, Veerman KM, Seo W, Ziltener HJ. PSGL-1 function in immunity and steady state homeostasis. Immunol Rev. 2009;230(1):75–96. | |
May P, Woldt E, Matz RL, Boucher P. The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann Med. 2007;39(3):219–228. | |
Schmid RS, Maness PF. L1 and NCAM adhesion molecules as signaling coreceptors in neuronal migration and process outgrowth. Curr Opin Neurobiol. 2008;18(3):245–250. | |
Irintchev A, Schachner M. The injured and regenerating nervous system: immunoglobulin superfamily members as key players. Neuroscientist. 2012;18(5):452–466. | |
Gunnersen JM, Kim MH, Fuller SJ, et al. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56(4):621–639. | |
Stoeckli ET. Neural circuit formation in the cerebellum is controlled by cell adhesion molecules of the contactin family. Cell Adh Migr. 2010;4(4):523–526. | |
Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153(1):13–19. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.