")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
COVID’s Razor: RAS Imbalance, the Common Denominator Across Disparate, Unexpected Aspects of COVID-19
Authors Czick M , Shapter C , Shapter R
Received 1 June 2020
Accepted for publication 4 August 2020
Published 11 September 2020 Volume 2020:13 Pages 3169—3192
DOI https://doi.org/10.2147/DMSO.S265518
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ming-Hui Zou
Maureen Czick,1 Christine Shapter,2 Robert Shapter3
1University of Connecticut, Department of Anesthesia, Farmington, CT, USA; 2 University of Saint Joseph, West Hartford, CT, USA; 3Independent Consultant ( Medical Research, Medical Communications, and Medical Education), Hartford, CT, USA
Correspondence: Maureen Czick Email [email protected]
Abstract: A modern iteration of Occam’s Razor posits that “the simplest explanation is usually correct.” Coronavirus Disease 2019 involves widespread organ damage and uneven mortality demographics, deemed unexpected from what was originally thought to be “a straightforward respiratory virus.” The simplest explanation is that both the expected and unexpected aspects of COVID-19 share a common mechanism. Silent hypoxia, atypical acute respiratory distress syndrome (ARDS), stroke, olfactory loss, myocarditis, and increased mortality rates in the elderly, in men, in African-Americans, and in patients with obesity, diabetes, and cancer—all bear the fingerprints of the renin-angiotensin system (RAS) imbalance, suggesting that RAS is the common culprit. This article examines what RAS is and how it works, then from that baseline, the article presents the evidence suggesting RAS involvement in the disparate manifestations of COVID-19. Understanding the deeper workings of RAS helps one make sense of severe COVID-19. In addition, recognizing the role of RAS imbalance suggests potential routes to mitigate COVID-19 severity.
Keywords: ACE, ACE2, silent hypoxia, atypical ARDS, stroke, myocarditis
Introduction: Full of Bad Surprises
Medieval English theologian William of Occam is widely credited with the eponymous principle known as Occam’s Razor: “Entities should not be multiplied without necessity.” Or, in modern terms, “the simplest explanation is usually correct.”
In late winter/early spring of 2020, as novel coronavirus case numbers exploded in Europe and parts of the United States, prominent newspapers and online news-media outlets reported with alarm a broad array of Coronavirus Disease 2019 (COVID-19) demographic and clinical outcomes deemed to be surprising and unexpected, even by some of the physicians interviewed. As one Washington Post headline exclaimed: “Once thought a relatively straightforward respiratory virus, COVID-19 is proving to be much more frightening.”1
Outcomes that initially jolted both the press and the medical community include cases of severe COVID-19 in young, apparently healthy adults,2 mortality rates among African-Americans and Latinos doubled compared to Caucasians,3,4 similarly doubled mortality among men compared to women,5 and a tripled death rate among certain cancer patients.6 In the UK, government epidemiologic data suggested that Blacks there are four times more likely to die of COVID-19 compared to Whites, with UK residents of Indian/Pakistani/Bangladeshi ancestry also showing increased mortality rates.7
Journalists warned as COVID-19 patients developed renal failure requiring dialysis,8 and obesity was linked to higher mortality.9 Subsequent reports revealed occurrences of fatal COVID-19 myocarditis,10 strokes in young patients,1,11,12 “silent hypoxia,”13 a mortality rate among ventilated COVID-19 patients approaching 90%,14 and a Kawasaki-like syndrome in children.15,16
In the early days of the pandemic, the clear association of pre-existing hypertension with more severe COVID-19 outcomes triggered consternation among physicians,17 as did cases of the virus presenting with seemingly incongruent hyperglycemia,18 and loss of taste and smell sensation.8
The widespread organ involvement and demographic perplexities do at first seem to be a bewildering pathophysiologic maelstrom. But Occam’s Razor posits that the most likely explanation is that a common underlying mechanism unites them all. When viewed through the lens of the renin-angiotensin system (RAS), the COVID-19 disarray slips into crystal clear focus. In addition to its better-known circulatory volume/pressure regulatory role, RAS is also a cellular-level system, present in the tissues of practically every organ of the body, and mediating a broad array of inflammatory, metabolic, osmotic, growth, and repair functions.19
The hallmarks of RAS imbalance, either pre-existing or COVID-19-mediated, are a common thread interwoven through wide-ranging COVID-19 symptomatology and disparate sub-population outcomes. The commonality of RAS dysregulation does not prove that RAS is the sole mediator of COVID-19 pathophysiology; there may be multiple factors that synergize to worsen COVID-19 severity (Table 1). Yet the underlying presence of RAS imbalance in so many aspects of COVID-19 does suggest that RAS may be the key to understanding and potentially mitigating COVID-19. Only after taking a deeper look at what RAS is and how it works, does severe COVID-19 itself begin to make sense.
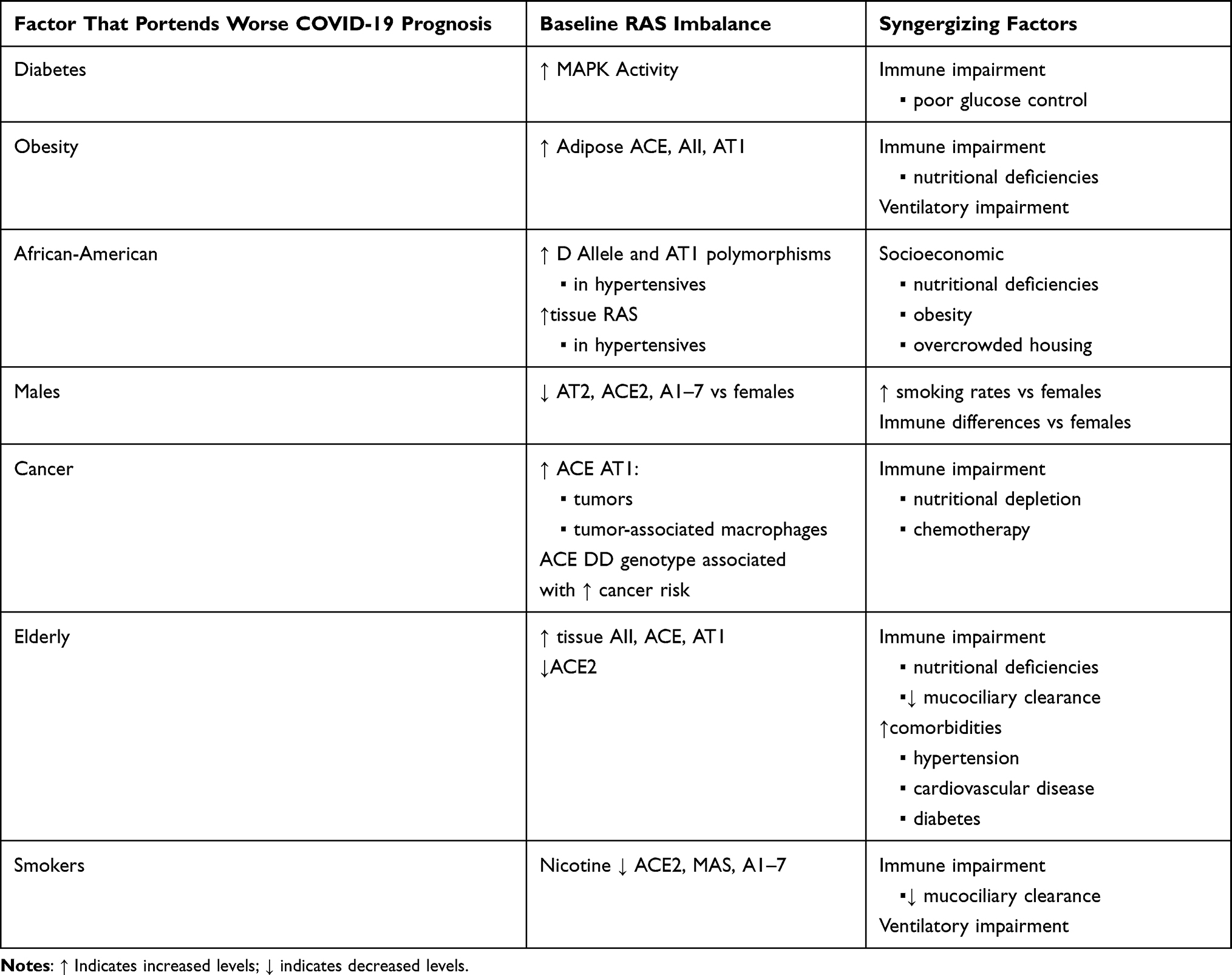 |
Table 1 This Table Lists Patient Subpopulations Reported to Have Worse COVID-19 Prognosis and Relates the Known Aspects of Renin-Angiotensin System (RAS) Imbalance in Those Populations, as Well as Potential Non-RAS Factors That May Also Be Contributing to Worse Prognosis |
The Original Concept of RAS
RAS was first conceptualized as an exclusively endocrine effector system, activated in response to decreased arterial blood pressure, increased sympathetic nervous system tone, or decreased filtered solute delivery within the kidney tubules as, for instance, during hemorrhage or dehydration.20–23 RAS was depicted as a linear sequence of enzymatic activations, with substrates circulating in the bloodstream to reach both their enzymatic activators and their target receptors. Renin, released into circulation by renal juxtaglomerular (JG) cells, cleaves hepatically produced angiotensinogen to generate angiotensin I (AI), an intermediate and inactive compound, which circulates to encounter membrane-bound angiotensin converting enzyme (ACE) on vascular endothelial cells,24 primarily within the pulmonary circulation.25 ACE converts AI to angiotensin II (AII), which transits to the adrenal cortex, triggering the release of aldosterone. Aldosterone subsequently stimulates increasing renal reabsorption of sodium and water, thereby augmenting blood volume.26 This sequence of RAS activations has been dubbed the Renin/ACE/Angiotensin/AT1 axis or, more succinctly, the ACE axis.27
AII also binds to angiotensin type-1 receptors (AT1) on vascular smooth muscle cells to mediate vasoconstriction and increase blood pressure.26 AT1 dispatches a G-protein to activate phospholipase C, which splits the membrane lipid phosphatidyl inositol bisphosphate (PIP2) into two products, inositol tri-phosphate (ITP) and diacylglycerol (DAG), both of which increase intracellular calcium levels.28 Calcium-bound calmodulin next activates myosin light-chain kinase (MLCK), which phosphorylates smooth muscle myosin, enabling actin binding, smooth muscle contraction, and vasoconstriction28 (Figure 1).
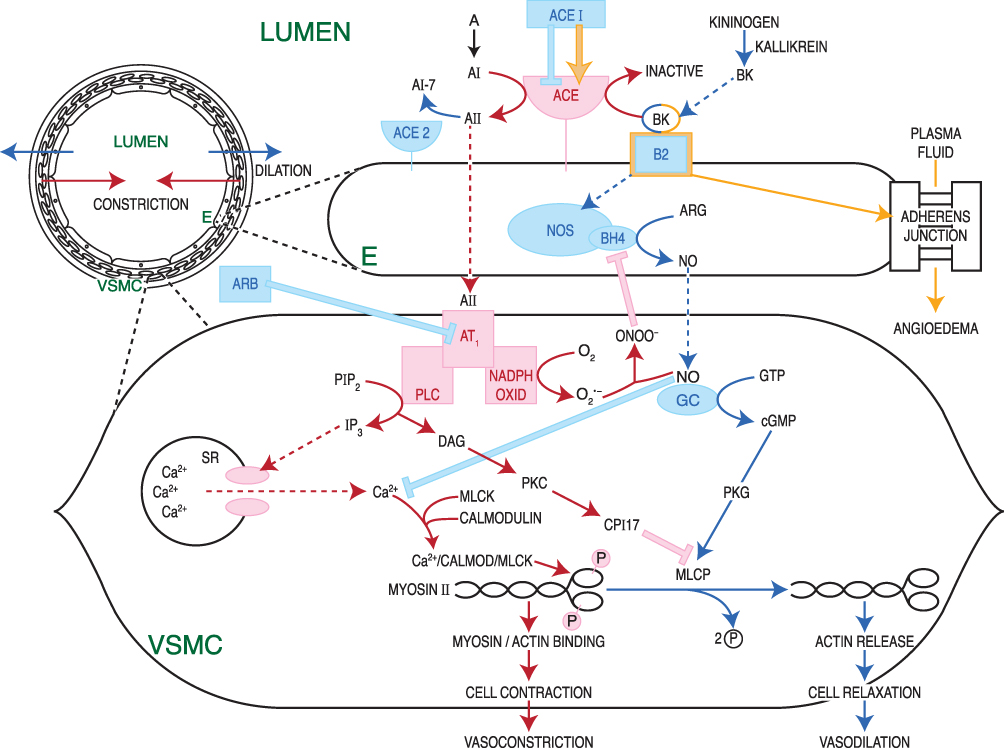 |
Figure 1 Second messenger pathways of the ACE axis and bradykinin in vascular wall. To mediate vasoconstriction and increase blood pressure, AII binds to AT1 receptors on vascular smooth muscle cells.26 AT1 activates PLC, which splits the membrane lipid PIP2 into two products, ITP and DAG, resulting in SR calcium release and increased intracellular calcium levels.28 Calcium binds to calmodulin; together they activate MLCK, which phosphorylates smooth muscle myosin. Phosphorylated myosin then binds actin, triggering smooth muscle contraction, and vasoconstriction.28 BK initiates a pathway opposing the vasoconstrictive action of AII. BK binding to the B2 receptor218 activates NOS to produce NO.43 NO diffuses from its site of production in vascular endothelial cells, to neighboring vascular smooth muscle, where it activates a vasodilatory second messenger cascade, culminating in activation of MLCP, which strips phosphate groups from smooth muscle myosin, such that it can no longer bind to actin.43 NO also inhibits calcium release from the SR, blocking this critical step in the vasoconstriction cascade. Smooth muscle relaxation and vasodilation ensue. AT1 also activates NADPH oxidase, generating reactive oxygen species such as O2−.44 O2− binds to and quenches existing NO, in the process creating ONOO−, which disables NOS so that it cannot manufacture new NO.43 Hence, crosstalk between the vasodilatory BK pathway and the vasoconstrictive AII pathway enables each pathway to inhibit the other. ACE metabolizes BK to inactive fragments, thus ACEIs may prolong the action of BK.221–225 BK dysregulation with use of ACEIs can cause angioedema,226–228 as B2 receptors inhibit the function of adherens junctions between vascular endothelial cells, leading to plasma fluid extravasation into the interstitium.229 Figure property of the authors.Abbreviations: A, Angiotensinogen; AI, Angiotensin I, AII, Angiotensin II; A1-7, Angiotensin 1–7; ACE, Angiotensin Converting Enzyme; ACE2, Angiotensin Converting Enzyme 2; ACEI, ACE Inhibitor; ARB, Angiotensin Type 1 Receptor Blocker; ARG, arginine; AT1, Angiotensin Type 1 Receptor; BH4, tetrahydrobiopterin, cofactor for NOS; BK, Bradykinin; B2, Bradykinin Receptor 2; Ca2+, calcium ion; cGMP, cyclic guanosine monophosphate; CPI17, PKC-dependent inhibitor (−17) of MLCP, DAG, diacylglycerol; E, vascular endothelial cell; GC, guanylate cyclase; GTP, guanosine triphosphate; IP3, inositol triphosphate; MLCK, myosin light-chain kinase; MLCP, myosin light-chain phosphatase; NADPH OXID, NADPH oxidase; NO, nitric oxide; NOS, nitric oxide synthase; O2-, superoxide anion; ONOO-, peroxynitrite anion; PIP2, phosphatidyl inositol 4,5 bisphosphate; PLC, phospholipase C; PKC, protein kinase C; PKG, protein kinase G; SR, sarcoplasmic reticulum; VSMC, vascular smooth muscle cell. |
The “Evolving” Concept of RAS
Circulating pressure/volume regulation within the vascular system is not the only function of RAS.29 After much debate,30,31 there is now widespread agreement that local tissue-level RAS systems administer paracrine and autocrine effects in various structures including adipose tissue, the pancreas, vascular walls, and the heart, kidneys, and brain.19,32
Evidence suggests that locally produced RAS components affect physiology independently of blood pressure.33 Although this premise remains controversial, the original purpose of some RAS components may not have involved blood pressure regulation and might even pre-date the evolution of blood vessels;34 for example, the modern invertebrate fruit fly, which has an open circulatory system and therefore lacks discrete blood vessels, has an ACE-like enzyme,35 and a renin-like enzyme is found in the leech.26
Invertebrates demonstrate the value of a synergistic proinflammatory and procoagulant injury response system. They possess a sole cell-type having both platelet-like and macrophage-like functions which speeds injury response.36 In open circulatory systems, blood directly contacts cells, so pathogens entering via a breach in the exoskeleton would rapidly begin cellular invasion. In the absence of blood vessels, there is no mechanism to restrict blood flow to the injury site, and this would be fatal without prompt coagulation.36 With an open circulatory system, there is no time to lose.
The evolution of vertebrate closed-circulatory systems interposed the vascular wall as a barrier to shield cells from blood-borne invaders.36 Coagulation and inflammation evolved along separate, specialized cell lines,36 but the advantages of integration between coagulation and inflammation during injury response were preserved through crosstalk and mutual co-activation.37–41 Vasoconstriction to limit blood flow to injured areas is intertwined with inflammation and coagulation to establish a fully integrated injury-response mechanism. The vascular endothelium became the pivotal regulatory interface, with the ACE axis mediating all three processes.
The Darker Side of RAS: Endothelial Dysfunction, Hypertrophy, Fibrosis, Cell Death
RAS is a critical survival mechanism, yet when dysregulated, it poses a significant danger. Under normal conditions, the healthy vascular endothelium promotes vasodilation and inhibits coagulation and inflammation.42,43 However, when injury occurs, AII, together with proinflammatory cytokines from innate immune cells, transforms endothelium into an “activated” state that promotes vasoconstriction, inflammation, and coagulation.37,42,43 Endothelial activation is intended to be temporary; failure to recover a vasodilatory, anti-inflammatory, and anticoagulant baseline is termed endothelial dysfunction.44 It results from excessive generation of reactive oxygen species (ROS) by overactive AII, with consequent loss of nitric oxide (NO) production capacity.44 (Figure 1) Dysfunctional endothelium, due to RAS dysregulation, promotes inflammatory and thrombotic damage in disease states such as diabetes, hypertension, and atherosclerosis.45–47
The ACE axis has direct growth-promoting effects on cells and cell matrix for post-injury repair. AT1 activation of mitogen-activated protein kinase (MAP Kinase) upregulates growth factors such as vascular endothelial growth factor (VEGF), and inflammatory mediators including tumor necrosis factor (TNF) alpha, interleukin-1 (IL1) beta, and nuclear factor kappa B (NF-κB). This upregulated signaling contributes not only to repair but also to inflammation, cancer, and nervous system function.48 Acting again via MAP Kinase, AT1 upregulates cytokines, such as transforming growth factor (TGF) beta, that drive fibrotic collagen deposition by myofibroblasts.48,49 Fibrosis and hypertrophy can at first be reversible repair responses, but when dysregulated they progressively damage vascular smooth muscle, myocardium, and kidneys.48,50
In addition, some cellular damage may be too extensive for recovery, in which case the ACE axis contributes to activating cell death pathways.48,51,52 ROS derived from AT1 activation can inhibit anti-apoptotic BCL-2 and activate pro-apoptotic Bax pathways, triggering programmed cell death.52 AII can also impair mitochondrial function and increase protein misfolding, causing pathological protein aggregation.48 And AII enhances autophagic catabolism of damaged organelles and protein aggregates, necessary for homeostasis, but capable of inciting cell death when dysregulated.51
Reining in RAS: AT2 and ACE2
It is evident that a system as powerful and far-reaching as RAS must be tightly regulated, to prevent potentially lethal misapplication of vasoconstriction, inflammation, and coagulation.
The AT2 receptor is generally described as a protective arm of RAS, opposing the actions of the AT1 receptor.53,54 The functional roles of AT2 have been challenging to tease out, in part because of low adult AT2 expression levels compared to AT1, whose effects may overwhelm the more subtle alterations prompted by AT2.54 AT2 has extensive expression during fetal development, but comparatively low-level expression in adults in select sites: heart, kidney, adrenal, brain, uterus, pancreas, retina, endothelium, and vascular smooth muscle.55
AII also binds to AT2, and in doing so, it counteracts AT1 effects, promoting instead natriuresis, which can slightly lower blood pressure, as well as stimulating anti-inflammatory, antithrombotic, and antifibrotic effects.54 AT2 activates superoxide dismutase (SOD), which neutralizes ROS and defends against oxidative stress created by AT1.52 AT2 knockout mice have increased vascular hypertrophy, indicating that AT2 has protective anti-hypertrophy functions,55 congruent with AT2 upregulation in the heart during disease states including heart failure, cardiac ischemia, and dilated cardiomyopathy.55 Some of the benefits of angiotensin receptor blockers (ARBs), such as reducing vascular and cardiac hypertrophy, appear to be AT2-mediated, by shunting AII away from AT1.55
The principal AT2 second messenger pathway involves production of nitric oxide (NO), cyclic GMP (cGMP), and bradykinin, all of which promote vasodilation and oppose inflammation.55 Endothelial NO upregulates AT2 which then decreases ACE activity.54 cGMP also deactivates RhoA, an AT1 second messenger, thereby antagonizing AT1-mediated vasoconstriction.55
ACE has a counterbalancing “younger brother” enzyme known as ACE2.56,57 ACE2 converts AII into angiotensin 1–7 (A1–7), so named because it possesses only seven of AII’s original eight amino acids. A1–7 binds to Mas receptors, yielding vasodilatory, anti-inflammatory, and antifibrotic effects.27,32,33 In sum, ACE2 depletes AII levels by converting it to A1–7, which binds to Mas receptors, thereby counteracting AT1 actions.56–58
Through a side-pathway, ACE2 can also convert AI into angiotensin 1–9 (A1–9),55 which possesses nine of AI’s original ten amino acids. Interestingly, ACE then converts A1–9 into A1–7; therefore, ACE plays a role in producing AII, but also in decreasing AII levels by shunting substrates toward A1–7 instead of AII. Rather than being a linear cascade of enzymatic reactions, RAS is now understood as a web of counterbalancing activations and deactivations26,55 (Figure 2).
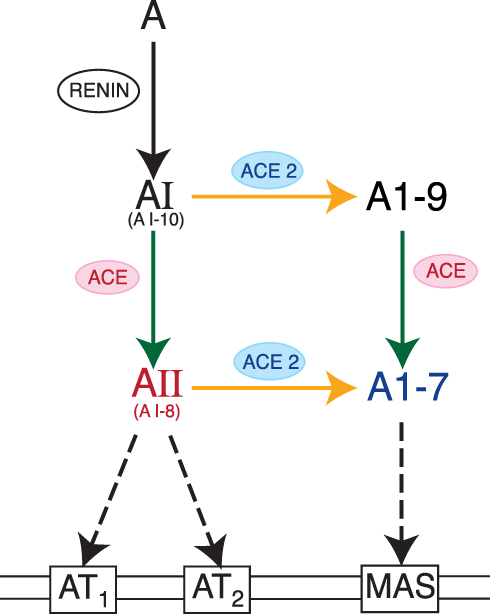 |
Figure 2 The RAS web. The RAS system is now more appropriately conceptualized as a web of activations and counterbalancing deactivations, rather than its original depiction as a linear cascade. Figure property of the authors. Abbreviations: ACE, Angiotensin Converting Enzyme; ACE2, Angiotensin Converting Enzyme 2; A, Angiotensinogen; AI, Angiotensin I; AII, Angiotensin II; A1-9, Angiotensin 1–9; A1–7, Angiotensin 1–7; AT1, Angiotensin Receptor Type 1; AT2, Angiotensin Receptor Type 2; MAS, MAS Receptor. |
SARS, SARS2: Runaway RAS
The original SARS coronavirus (SARS-CoV or SARS), which caused severe acute respiratory syndrome, (also called SARS) hitchhiked inside cells using cell-surface ACE2.56 The new, closely related coronavirus SARS-CoV-2 or SARS2 (which is responsible for COVID-19) uses the same method.59 Once inside the host cell, original SARS virus dramatically reduces cellular expression of ACE2.60 SARS2 virus evidently functions the same way.61 Because ACE2 is critical for regulating the balance between the effects of AII and A1–7,62 reduced ACE2 expression leaves the “big brother” enzyme ACE, unchecked, enabling unopposed oxidative, inflammatory and procoagulant effects61 (Figure 3).
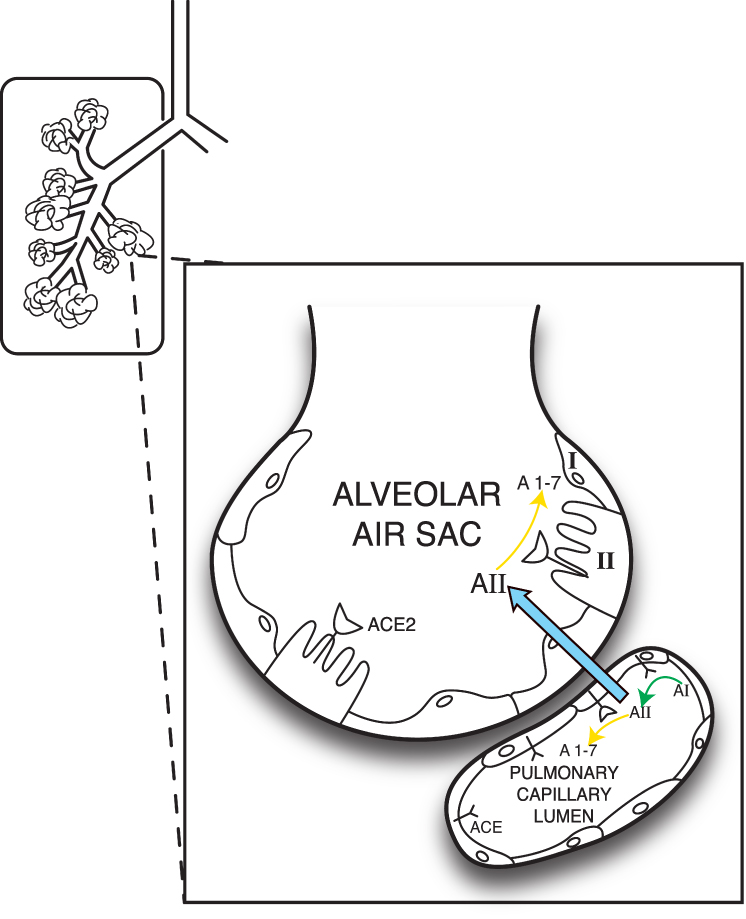 |
Figure 3 ACE, ACE2 expression, action in alveolar units. Pulmonary vasculature has the highest levels of ACE expression in the body. In contrast, ACE2 expression is more restricted in the lungs, largely to type II pneumocytes,61 although there is evidence of some expression in pulmonary capillary endothelium.240 ACE2 deactivates AII, by converting it to A1–7, thereby blunting the pro-inflammatory, pro-fibrotic effects of the ACE axis. In COVID-19, novel coronavirus SARS-CoV-2 can enter type II pneumocytes utilizing ACE2 and then downregulate ACE2 expression,61 leaving the lungs vulnerable to damaging, unbalanced proinflammatory effects of ACE/AII.61 Type II pneumocytes have multiple protective functions, including producing surfactant, and serving as stem cells to replenish damaged Type I pneumocytes (which function in gas exchange).61 Viral impairment of Type II pneumocytes, coupled with AII inflammatory damage to Type I pneumocytes, would thus severely damage multiple aspects of alveolar function in COVID-19, including oxygen exchange and capacity for repair. Figure property of the authors. Abbreviations: ACE, Angiotensin Converting Enzyme; ACE2, Angiotensin Converting Enzyme 2; AI, Angiotensin I; AII, Angiotensin II; A1–7, Angiotensin 1–7; I, Type I pneumocytes; II, Type II pneumocytes. |
In animal models and human cases of acute respiratory distress syndrome (ARDS), ACE2 levels are severely decreased,63 and ACE/ACE2 balance is lost.57 Overactive AII/AT1 contributes to severe oxidative and inflammatory lung injury,63 which in mouse models is blunted by ACE2.64 ACE inhibitors (ACEIs) and ARBs lessen pulmonary damage in mice injected with the original SARS surface spike-protein,65,66 suggesting that unbalanced ACE axis activity, after ACE2 is downregulated, is the root of original SARS lung pathophysiology.
Close assessment of COVID-19 pathophysiology suggests the same mechanism.61 As the following sections show, fingerprints of RAS imbalance with ACE/AII overactivation and diminished ACE2/A1–7 are spread across the array of unexpected COVID-19 organ system involvement. Subpopulations manifesting higher rates of COVID-19 mortality—including hypertensives, the elderly, the obese, diabetics, men, and African-Americans—correlate with preexisting RAS imbalance, with ACE overactivity and/or ACE2 underactivity priming these patients for more severe COVID-19 outcomes.
Silent Hypoxia: RAS in the Right Places
Many hospitalized COVID-19 patients reportedly display silent hypoxia: severe arterial hypoxemia without evident dyspnea, air hunger, or breathlessness. The neural reflex pathways mediating dyspnea have not been fully delineated, but likely involve integration among lung mechanosensory receptors, central and peripheral chemoreceptors, and emotion-regulation centers of the limbic system.67,68
The sensation of dyspnea has been reported to require intact chemosensory function.67 The carotid bodies (neural crest-derived structures located at the carotid bifurcations), are the peripheral chemoreceptors, regulating the ventilatory response to hypoxia.69 Carotid body glomus cells scan their extensive blood supply for adequacy of oxygenation:69,70 if arterial partial pressure (PaO2) falls below approximately 60 mmHg, potassium background leak channels close, halting K+ exit and depolarizing the glomus cells.69 Next, calcium influx through voltage-gated channels triggers the release of glomus cell neurotransmitters which initiate firing of action potentials by adjacent afferent terminals of the carotid sinus branch of cranial nerve IX.69,70 The action potentials travel first to the nucleus tractus solitarius (NTS), and then to the Prebotzinger complex in the medulla, augmenting respiratory drive as an attempt to correct the oxygen shortfall,68,69 (Figure 4) and simultaneously invoking the reflex pathways that mediate air hunger.
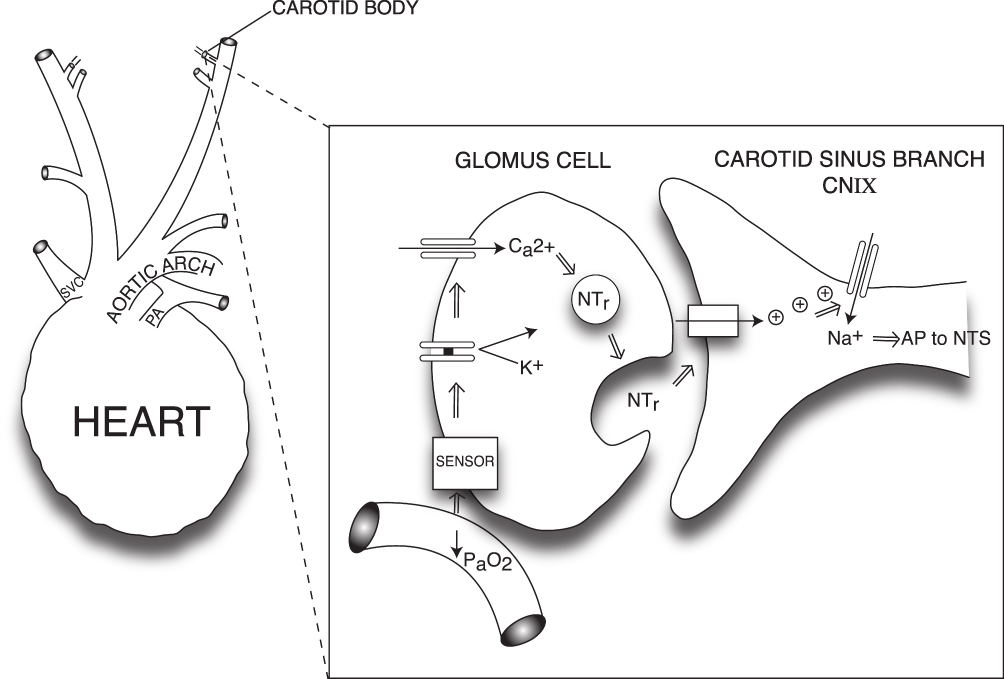 |
Figure 4 Cellular mechanism of glomus cell hypoxic signaling. Glomus cells, located within the carotid body, are neural crest-derived, and they exhibit voltage-dependent neurotransmitter release upon detection of hypoxemia.69,70 Glomus cells detect hypoxemia by an as-yet undefined sensor, which is thought to close K+ channels, halting K+ exit and thereby depolarizing the Glomus cell.69,70,241,242 Depolarization leads to opening of voltage-gated Ca2+ channels and neurotransmitter release, which triggers action potential firing in nearby cranial nerve IX.69,70,241,242 Figure property of the authors. Abbreviations: AP, action potential; CNIX, cranial nerve IX; NTr, neurotransmitter; NTS, nucleus tractus solitarius; SVC, superior vena cava; PA pulmonary artery; PaO2, arterial partial pressure of oxygen. |
The oxygen sensor which initiates glomus depolarization is still debated, but it may be ROS-producing NADPH oxidase,69 which also happens to participate in AT1 receptor signaling. Whether or not AT1 and NADPH oxidase are involved in hypoxia sensing, RAS is definitively activated during the hypoxic response. The carotid body is not only a driver of increased ventilatory effort; it is also a nexus for homeostasis regulation.70 Oxygen perturbations signify severe systemic stress with profound metabolic implications, requiring anti-stress responses including renal compensation to maintain pH, adjustment of cardiovascular functions, and autonomic and endocrine effectors, all of which RAS integrates into the ventilatory response.70
To mediate this integration, components of RAS are expressed constitutively in glomus cells, including ACE, ACE2, AT1, Mas, AT2.71,72 Hypoxia increases expression of ACE, angiotensinogen, AT1 and AT2.72 Hypoxia also increases sympathetic tone, which triggers renin release and elevates plasma AII levels. In turn, plasma AII has a stimulating effect on carotid body glomus cells, augmenting intracellular influx of calcium ions, and neurotransmitter release.72 Blood pressure and ventilatory responses crosstalk and reinforce each other.72
SARS2 coronavirus would be able to enter glomus cells utilizing membrane-bound ACE2. The subsequent ACE2 downregulation, amid hypoxemic pulmonary infection, would result in significant ACE imbalance,61 leading to carotid body hypoxic-signaling failure via two potential routes. First, AII overactivation could drive infected glomus cells into programmed cell death. This would be consistent with experimental AII/AT1 apoptosis induction in rat and human alveolar epithelial cells.73–75 Alternatively, glomus failure could be akin to cellular dysfunction in heart failure, in which excessive AII triggers cell volume overload and impairment of cardiac electrical function.76 Although AII increases cell volume, A1–7 decreases it.33 Thus, with loss of both ACE2 and A1–7, AII overactivity would be expected to volume overload glomus cells, impairing depolarization, and blocking neurotransmitter release. Either scenario, or both acting synergistically, would result in failure of hypoxic signaling, with an absent dyspnea response despite severe hypoxemia in COVID-19 patients.
Incidentally, those same two scenarios could also be at work in loss of olfaction and taste, both of these neural structures requiring depolarization and neurotransmitter release to mediate their sensory functions. Both express tissue RAS components.77,78
“Atypical” ARDS: RAS “Mismatched”
But why are COVID-19 patients so severely hypoxemic in the first place? Critical care anesthesiologists in the thick of the COVID-19 crisis in Italy suggested that severe COVID-19 patients may not have classical ARDS.79 Unlike the situation in ARDS, lung compliance is preserved in COVID-19. Instead, COVID-19 patients seemingly displayed catastrophic loss of hypoxic pulmonary vasoconstriction (HPV), with resulting ventilation-perfusion (V/Q) mismatch hypoxemia.79
In ARDS, lung compliance is poor because of decreased alveolar elasticity from noncardiogenic pulmonary edema.80,81 This decreased alveolar elasticity impairs gas entry into alveoli, lowering the alveolar partial pressure (PAO2), the top end of the gradient which drives the transfer of oxygen into pulmonary blood. Oxygenation in ARDS patients often improves with ventilation using low tidal volumes and high positive end-expiratory pressure (PEEP) which increases PAO2.80,81 But severe COVID-19 patients have not shown a similar benefit with this ventilation strategy.82
Even under normal physiologic conditions, PAO2 is not equal in all alveoli throughout the lungs.83 Because of postural differences in fluid hydrostatic pressures across the lung fields, alveolar expansion is not uniform and some alveoli inevitably have higher PAO2 than others. HPV is a protective physiologic mechanism that optimizes arterial oxygenation by regionally vasoconstricting within the pulmonary vascular bed, thereby diverting pulmonary blood flow away from alveoli with lower PAO2, and vasodilating in other regions to increase blood flow to alveoli with higher PAO2.84 (Figure 5) Optimal V/Q matching yields a PaO2 of approximately 100 mm Hg, when breathing air at atmospheric pressure. However, if the HPV response is impaired, blood flows through the lungs in an unregulated fashion, and poorly oxygenated blood with low PaO2, enters the systemic circulation85 (Figure 5).
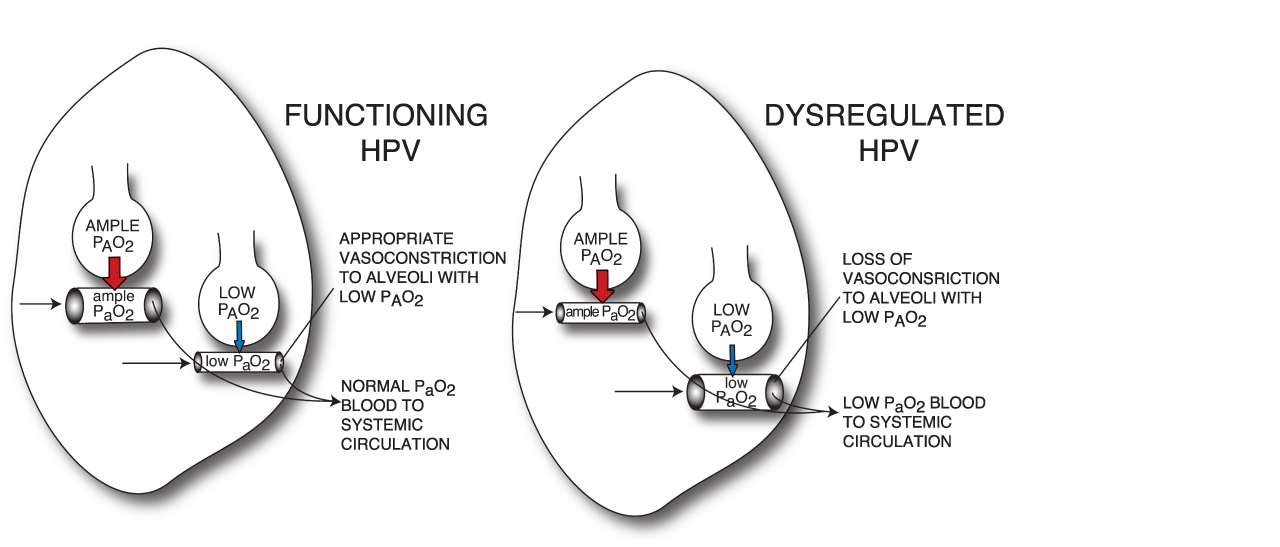 |
Figure 5 Hypoxic pulmonary vasoconstriction (HPV) and ventilation/perfusion (VQ) matching. Alveolar partial pressure of oxygen (PAO2) is the top end of an oxygen gradient, driving transfer of oxygen from the alveolar air spaces into the pulmonary blood. Thick red arrows designate a sizable gradient driving oxygen transfer. Thin blue arrows designate smaller oxygen gradients, with less potent driving force for oxygen transfer between alveolar air spaces and pulmonary blood. Functional HPV regionally vasoconstricts (designated in the figure by small blood vessel tube) within the lungs, to divert pulmonary blood flow away from poorly oxygenated alveoli, which would have a more limited ability to transfer oxygen into the pulmonary blood. Such appropriate VQ matching ensures that adequately oxygenated blood, with normal arterial partial pressure of oxygen (PaO2), is returned to the systemic circulation. When HPV fails, pulmonary flow is instead sent in excess to poorly oxygenated alveoli, such that larger volumes of poorly oxygenated blood, with low PaO2, are returned to the heart, to then enter the systemic circulation. Figure property of the authors. |
RAS plays a role in HPV that is still not precisely defined but may involve hypoxic activation of NADPH oxidase, generating ROS that close potassium channels, depolarizing pulmonary vascular smooth muscle, which in turn opens voltage-gated L-type calcium channels, activating the smooth muscle contractile apparatus.84,86 Mice lacking cytosolic NADPH oxidase had a 25% decrease in initial phase HPV.87 AII has been shown to activate pulmonary vasoconstriction in hypoxic humans,88 and ACEIs and ARBs have been shown to inhibit HPV.89,90 Recombinant ACE2 blunts pulmonary vasoconstriction in hypoxic pigs.91 Therefore, the loss of ACE2 due to SARS2 coronavirus together with the resultant ACE imbalance61 would be expected to dysregulate HPV and lead to hypoxemia.
Extreme Mortality Rates in Ventilated COVID Patients: The Final Straw
Mortality rates in ARDS hover around 45%.75 By contrast, a COVID-19 case series of 5700 patients in and around New York City reported mortality rates of 88.1% in ventilated patients, essentially doubling the ARDS rate,92 and providing further support that COVID-19 is not traditional ARDS.82
Lung tissue locally expresses almost all components of RAS.75 ACE is heavily expressed across the entire alveolar capillary network, and the lungs are the dominant site of circulating AII production.75 In contrast, ACE’s anti-inflammatory counterpart, ACE2 is more restricted within the lungs, principally to alveolar type II pneumocytes75 (Figure 3). In addition, in healthy adults, pulmonary expression of anti-inflammatory AT2 is low.75 With such broad expression of the ACE axis, and comparatively restricted expression of the protective arms of RAS, the lung is poised for more severe ACE imbalance in the event of ACE2 downregulation by the SARS2 virus.
Without ACE2, ACE actions would be essentially unchecked,61 with no means to achieve appropriate V/Q matching, amidst significant inflammatory destruction of lung tissue.75 In animal models, mechanical ventilation has been shown to upregulate ACE, promoting activation of AII.93–95 It may be the case for many severe COVID-19 patients that initiation of mechanical ventilation, and consequent further ACE axis upregulation, becomes the final straw, especially when combined with anesthetic/sedating drugs that may further impair HPV.
The Kidneys and Heart in COVID: Sword versus Shield
Emerging data show a mixed picture of COVID-19 effects on the heart and kidneys. Preexisting cardiovascular risk factors are definitively associated with increased COVID-19 severity. In an Italian retrospective case series, 49% of 1591 patients admitted to the ICU for COVID-19 had preexisting hypertension, and 21% had cardiovascular disease.96 Among a retrospective cohort of 191 COVID-19 cases hospitalized in Wuhan, China, 30% had hypertension, and 8% had coronary heart disease.97 Hypertension and atherosclerosis have well-established associations with ACE axis dysregulation.98–102 Moreover, ACE2 levels in the heart are reported to be diminished in hypertension and in diabetes-associated cardiovascular disease.103 Thus, it makes sense that these comorbidities would be heavily represented among more severe COVID-19 presentations. Studies suggest that myocardial injury worsens COVID-19 prognosis;104 nevertheless, reported rates of cardiac as well as renal injury are lower than rates of pulmonary damage, suggesting that the heart and kidneys may possess some protection.
For example, the Wuhan cohort cited that among fatal cases, 93% had an ARDS diagnosis, but only 59% had a myocardial infarction (MI), and 50% had acute kidney injury (AKI) prior to death.97 Among survivors, 7% had ARDS while only 1% had MI or AKI. Another study reported myocardial injury in 7% to 17% of hospitalized COVID-19 patients, and 22% to 31% of ICU cases.105 In contrast, the Italian ICU cohort reported 88% requiring intubation and mechanical ventilation, indicative of dire pulmonary compromise.96
Kidneys locally express all ACE axis components.29,106 Additionally, the renal glomeruli filter most circulating RAS proteins, which are then reabsorbed by the proximal tubule.106 Combined resorption and local production result in renal AII levels 100 to 1000 times higher than in plasma.29,106 Dysregulated tissue RAS, as occurs in hypertension, results in significant renal inflammatory damage, with progressive destruction of nephrons amid RAS-mediated remodeling and fibrosis.49 Similarly, the heart has significant local RAS, which mediates inflammatory remodeling in conditions such as hypertension and infarction.107
However, the counterbalancing anti-inflammatory AT2 and ACE2 arms of RAS also have an important role in the heart and kidneys. ACE2 is highly expressed in the heart,62 in particular in the coronary arteries,32 and in endothelial cells within the kidneys,32 achieving significant production of A1–7.27 Therefore, with higher baseline ACE2 levels, the heart and kidney may have a degree of shielding against the effects of ACE2 loss perpetrated by the SARS2 virus. In addition, cardiac and renal tissue RAS often bypass ACE, instead using chymase, trypsin, kallikrein, and cathepsins.32 Thus, local RAS function in these organs could potentially be less imbalanced by the loss of ACE2 due to the SARS2 virus.
Additionally, the coronary arteries52 and kidneys express comparatively higher levels of AT2 during adult life.55 The heart also has a demonstrated capacity to upregulate AT2 during pathologic states such as ischemia.55 Therefore, the effects of COVID-19-induced imbalance of RAS may be blunted somewhat by AII binding to AT2.
There are also reports of COVID-19-triggered myocarditis.108 In one study, myocarditis was deemed likely causative in 7% of COVID-19 deaths.105 AT1 has been shown to promote myocarditis in experimental models, secondary to actions on T-cell function.109 T-cells have an intrinsic RAS that regulates their proliferation and migration.109 AT1 fosters cytoskeletal changes that promote T-cell migration and trigger T-cell production of cytokines and chemokines, thereby increasing T-cell recruitment to regions of active inflammation.109 By inhibiting AT1, ARBs decrease experimental myocarditis.110 RAS-driven T-cell-mediated effects in the heart, coupled with T-cell-mediated skin effects, which are also well described,111,112 could potentially explain the pediatric Kawasaki-like condition linked to SARS2.16
Besides ischemic and inflammatory tissue destruction, another aspect of cardiac pathology in COVID-19 is the development of arrhythmias and sudden cardiac death.113,114 In addition to the arrhythmia-promoting effects of hypoxemia, arrhythmias in COVID-19 patients bear hallmarks of RAS dysregulation. Cardiac remodeling by the ACE axis is a known driver of arrhythmia events. In the atria, AII promotes fibrosis that facilitates reentry,115 and ACEIs and ARBs do provide some protection against the recurrence of atrial fibrillation after cardioversion in patients with left ventricular dysfunction.48 AII promotes ventricular arrhythmias by prolonging QRS duration, and by promoting reentry through both fibrosis and slowed ventricular conductance.48 These factors are likely playing out in COVID-19 pathology, perhaps synergizing with potential arrhythmia-promoting effects of experimental drug treatments, including hydroxychloroquine,114 which was studied earlier in the pandemic but the use of which has now been curtailed.
Hypercoagulability and Strokes in COVID: Sabotage by RAS
During the initial United States COVID-19 case escalation, stroke was reported in patients without traditional stroke risk factors.12,116 Although unanticipated in the context of a viral respiratory tract infection, stroke risk is known to be elevated in conditions of RAS overactivation such as diabetes and hypertension.117,118 RAS blockade with ACEIs and ARBs has been shown to reduce stroke rates in high-risk populations.55 With the downregulation of ACE2, the coagulation-promoting effects of the ACE axis would drive arterial thrombosis.
Healthy vascular endothelium promotes vasodilation and inhibits coagulation and inflammation,42,43 through expression of natural anticoagulants, including tissue factor pathway inhibitor (TFPI), thrombomodulin (TM), tissue plasminogen activator (tpa), and the anti-inflammatory vasodilator NO.42 Produced in endothelial cells by nitric oxide synthase (NOS),43 NO diffuses to neighboring vascular smooth muscle to activate guanylate cyclase/cyclic GMP/protein kinase G-mediated vasodilation43 (Figure 1). NO also has myriad anti-inflammatory and anti-coagulant functions, including inhibiting both platelet aggregation and monocyte adhesion.119
During injury response, AII inhibits endothelium-mediated vasodilation and anti-coagulation.42 AT1 activates NADPH oxidase, generating ROS such as superoxide anion O2−.44 (Figure 1) O2− quenches existing NO, in the process creating ONOO−, a very potent oxidant which in turn disables NOS so that it cannot manufacture new NO; hence, NO levels are severely depleted43 (Figure 1).
AT1 also promotes increased expression of adhesion molecules such as VCAM-1 and E-Selectin that recruit inflammatory cells to the injury site, and inflammation synergistically promotes coagulation.119,120 AII prompts endothelial cells to promote coagulation, by new expression of procoagulants—tissue factor (TF) to initiate the blood clotting cascade,121,122 von Willebrand factor (vWF) to enhance platelet adhesion,123 and plasminogen activator inhibitor (PAI)-1 to blunt fibrinolytic degradation of blood clots.37,42,43,119 Activation of these processes would then be the drivers of stroke in COVID-19-induced RAS imbalance, with overactive AII sabotaging the anti-coagulant actions of otherwise healthy endothelium.
Higher COVID Mortality in Obesity and Diabetes: The Map Kinase Connection
Obesity and diabetes are reported to worsen COVID-19 severity and mortality. In the case series of 5700 hospitalized COVID-19 patients, 33.8% had diabetes as a preexisting comorbidity, and 41.7% were obese.92 In a meta-analysis of 30 published studies that assessed the outcomes among more than 6400 patients, diabetes was found to more than double both the risk of severe COVID-19 disease and the risk of mortality.124 Diabetes more than quadrupled the risk of severe ARDS-like pulmonary outcomes in COVID-19.124 A study in New York City reported that patients with body mass index (BMI) equal to or above 35 had more than triple the risk of in-hospital mortality from COVID-19, compared to leaner patients [OR: 3.78; 95% CI 1.45–9.83, p=0.006].125
Multiple factors may be at work in these outcomes. Immune function has been shown to be impaired by nutritional deficiencies in obesity,126–128 and by poor glycemic control in diabetes.129–131 Obesity-compromised lung mechanics have been well described.132–134 However, both obesity and diabetes also bear the hallmarks of RAS dysregulation at baseline. In the developed world, nearly 90% of diabetes cases are the result of obesity,135 and the two conditions are mechanistically intertwined via RAS overactivation,119,135–138 which primes for worse COVID-19 outcomes.
Adipocytes locally produce a full set of RAS components, including angiotensinogen, ACE, AII, AT1.139 In obesity, this adipose ACE axis is highly upregulated,140 but with diminished counterbalancing ACE2,61 this ACE imbalance negatively impacts insulin function.140
The insulin receptor has two principal second messenger cascades.135 The first, phosphatidyl inositol-3 kinase (PI3K) promotes cell membrane localization of GLUT transporters that facilitate glucose uptake.139 PI3K also activates NOS, which manufactures NO to mediate the anti-inflammatory, anti-coagulant effects of insulin.139 The second cascade, utilizing MAP Kinase, is responsible for the growth-factor-like actions of insulin, including fibrosis, vascular smooth muscle proliferation, and cardiac myocyte hypertrophy.136,139
Under normal circumstances, PI3K-produced NO inhibits the MAP Kinase arm.139 In RAS upregulation, however, superoxide produced by AT1 eliminates NO, thus short-circuiting the PI3K function of the insulin receptor, so that glucose uptake fails, hence the connection between obesity and diabetes.141 This mechanism would also explain the correlation between COVID-19 and hyperglycemia. With ACE axis overactivity, insulin receptor function is shunted to MAP Kinase,139 and as a result of this alteration, insulin receptors and AT1 receptors—which also use MAP Kinase as a second messenger—synergize to promote inflammatory and hypertrophic damage to the cardiovascular system which is a hallmark of diabetes. Blocking RAS overactivation with ACEIs or ARBs helps restore normal insulin receptor function, improves insulin-mediated glucose uptake, and mitigates vascular damage.139
Higher COVID Mortality Among African-Americans: Visible Meets Invisible
As data accumulate showing elevated COVID-19 mortality rates in African-Americans, attention has focused on socioeconomic inequalities, including limited access to nutritious food, overcrowded housing conditions, and consequent health-related comorbidities, such as obesity, diabetes, and hypertension. These clearly evident factors undoubtedly play important roles, but less visible factors may also be at work. There is evidence of RAS dysregulation that drives hypertension disparities in African-Americans. This could also be contributing to the increased severity of COVID-19 among this population.
It has been firmly established that African-Americans have higher prevalence, earlier onset, and more severe complications of hypertension than Caucasians.142 Recent data from the US Department of Health and Human Services Office of Minority Health attest that African-Americans currently have higher rates of obesity than Caucasians.143 Obesity greater than 20% above ideal body weight doubles-to-triples hypertension risk,144 and obesity significantly increases ACE activity.145 Yet obesity alone cannot explain the unequal prevalence of hypertension. According to National Health and Nutrition Examination Survey (NHANES) data from 1988 to 1994, African-American and Caucasian males had almost identical obesity rates during that time frame, but African-Americans had 46% higher rates of hypertension.146
Research has identified critical physiologic distinctions among many African-Americans that influence response to hypertension therapies. Most African-American hypertensives have low-renin hypertension secondary to either a mutation of aldosterone synthetase (and consequent aldosterone overproduction) or of the epithelial sodium (eNaC) channels in the kidney, both of which lead to excessive renal sodium resorption.147 Although sodium retention inhibits renin release into the circulation, reduced activation of the circulating RAS does not prevent hypertension. However, in mouse experiments, renin gene deletion solely within the kidneys did confer protection against developing AII-mediated hypertension, suggesting that tissue RAS is more critical than circulating RAS for blood pressure regulation—and for the development of hypertensive nephropathy.106 In chronic kidney disease, renal tissue RAS was found to be elevated, independent of circulating RAS levels.146 African-American hypertensives have increased incidence of RAS-mediated organ damage, congruent with overactive tissue RAS despite low circulating renin.142 In keeping with this, African-Americans taking ACEIs and ARBs have comparatively poor blood pressure decrease, yet have significant protection against hypertensive end-organ damage.146
Several gene polymorphisms have been discovered to increase RAS activity. For example, increased hypertension prevalence among African-Americans has been linked to the −535T polymorphism of AT1.144 Best studied, however, is the ACE gene I/D polymorphism, distinguished by insertion (I) or deletion (D) of a nearly 300 base pair DNA segment.35 The DD genotype doubles ACE activity, in both circulation and tissue, compared to the II homozygous genotype; heterozygous ACE ID is intermediate between the two.35 In a population-based study, the ACE D polymorphism was slightly more common with African ancestry than with European or Japanese ancestry.148 African-American hypertensives in particular have been shown to have higher prevalence of the D allele.149,150` It is important to stress, however, that although those of African descent may be more likely to carry the D allele, D is found among many ethnicities. For instance, it was found to occur at increased frequency among Caucasian Australians who suffered myocardial infarction compared to the general healthy population.151
When ACE D polymorphism occurs, it has often been linked to adverse medical conditions, including increased susceptibility to ARDS.75 In children, the D allele was associated with worse outcomes in focal glomerulosclerosis152 and single ventricle congenital heart disease.153 ACE D allele correlates with ventricular hypertrophy in endurance athletes.35 Additionally, the Rotterdam Study showed an association of D allele with early mortality.35
Despite current potentially negative consequences, ethnobiological studies have hypothesized an evolutionary survival benefit during ancient times for RAS upregulation polymorphisms such as ACE D allele. In tropical areas with sodium scarcity, these polymorphisms promoted sodium and water retention, and maintenance of blood pressure.109,154 However, because of the current abundance of dietary sodium, modern descendants face increased damaging consequences from those same polymorphisms.109,154
Other ethnicities with reportedly increased COVID-19 mortality, including Latinos and South Asians, are less well represented in the hypertension literature than African-Americans. There is, nonetheless, some evidence for RAS upregulation. Low-renin hypertension is common in Caribbean Hispanics.146 The ACE-upregulating D allele has also been found with increased frequency among South Indians with hypertension.155 These observations suggest again that RAS upregulation portends the worse COVID-19 outcomes seen in some populations.
Gender Disparity in COVID Severity: The Estrogen Advantage
In a retrospective case series of 1591 consecutive patients requiring ICU level care for COVID-19 in Milan, Italy, 82% were male.96 Researchers from Beijing retrospectively analyzed mortality data from the earliest 500-plus reported cases of COVID-19 in Wuhan, China.156 They reported that, while men and women had similar COVID-19 prevalence, men were nearly 2.5 times more likely to die of COVID-19.156 Simultaneous retrospective re-analysis of mortality data collected in 2003 showed that the original SARS coronavirus also skewed toward higher mortality in men,156 in keeping with prior published reports.157
Although gender-specific differences in immune response158 may play a role in unequal outcomes of both SARS and COVID-19, there are definitive differences between the genders in the RAS system and, in keeping with Occam’s Razor, these differences likely contribute to worse male outcomes.
Sex hormones regulate the expression and function of RAS components.159 Males generally have higher levels of RAS than premenopausal females,160 perhaps explaining why male hypertensive rats show a greater blood pressure decrease with ACEIs.161 Estrogen downregulates the expression of the AT1 gene162,163 and suppresses both ROS production in vascular smooth muscle and the enzymatic activity of ACE.159 Estrogen also promotes the production of NO, the counterweight to AII in the endothelium.159
Estrogen also positively impacts the protective arms of RAS, including increasing ACE2 levels in rats.160 Female mice display higher renal AT2 expression than male counterparts, an effect that dissipates if ovaries are removed,55 and estrogen upregulates AT2 levels in the mouse kidney.55 Both the AT2 and ACE2 genes are found on the X chromosome,164,165 so it is reasonable to conclude that women would regulate and express these genes differently than men. In keeping with this, women have been reported to have higher levels of protective A1–7 than men, both in plasma166 and in urine,167 suggesting higher levels of ACE2 activity both in the circulation and at the renal tissue level. These factors likely synergize to confer comparative COVID-19 protection in females, especially pre-menopausally.
Very High COVID Mortality in Cancer Patients: Collaterals to Invasion
When assessing the elevated death rates among cancer patients who develop COVID-19,168 immune impairment evoked by therapies and nutritional depletion cannot be ignored as contributory factors. However, RAS dysregulation has also been shown to be associated with cancer.169,170
Increased ACE and AT1 expression have been reported in patients with metastatic ovarian cancer and prostate cancer, compared to those with benign neoplasms.171 AT1 is also increased in squamous cell carcinoma, and in 20% of estrogen-receptor-positive breast cancers.170
The cellular growth-promoting aspects of RAS are brought into play by tumor cells to facilitate tumor expansion and invasion. AII promotes differentiation of tumor-associated macrophages (TAMs) from immature progenitors.170 TAMs are abundant in the stroma surrounding the tumor, and they release factors that modulate tumor progression and metastatic migration.170 TAMs also inhibit the body’s anti-tumor immune mechanisms.170 TAMs express AT1, whose effect profile is proangiogenic, augmenting levels of vascular endothelial growth factor (VEGF), and VEGF type 2 receptor.171 AT1 facilitates epithelial-to-mesenchymal transition, which is crucial to metastatic progression.170
Knockout mice lacking the murine version of AT1 have decreased tumor growth compared to normal counterparts. ARBs blunt tumor size in animal models,171 and they also decrease VEGF levels, metastatic burden, and tumor vascularization.170 Similarly, ACEIs have been reported to inhibit tumor growth, angiogenesis, and metastasis.170
Among gastric cancer patients taking either ACEIs or ARBs, prolonged survival has been noted.169 Also, five-year recurrence-free survival was significantly higher among patients with non-invasive bladder cancer who were receiving RAS-blocking drugs.169 A small retrospective study of advanced non-small-cell lung cancer estimated a 3-month prolongation in survival with RAS blockers.169
Human studies of RAS blockade to limit cancer progression may be confounded by the invisible effects of RAS polymorphisms, which are only rarely assessed in trial subjects.169 In the prospective Rotterdam Study, however, ACE genotyping was performed and revealed that the high ACE-activity DD genotype had the most significant benefit in cancer risk reduction with RAS blockade.169 The ACE DD genotype has been associated with a higher risk of cancer, while the ACE II homozygote pools with a lower risk.169,170 These findings are congruent with RAS overactivation portending poorer COVID-19 prognosis.
Very High COVID Mortality in the Elderly: Unprotected from ACE
From the outset of the COVID-19 epidemic, elderly patients have accounted heavily among fatalities. A retrospective comparison of COVID-19 data from Italy and China revealed a large increase in case fatality rates with increasing age in both population groups.172 The Italian case fatality rate was 1% in patients aged 50–59, 3.5% in those aged 60–69, 12.8% in those aged 70–79, and 20.2% in persons above 80 years of age.172 Among the Chinese patients, case fatality rate was 1.3% in those 50–59, 3.6% in the 60–69 age group, 8% in those 70–79, and 14.8% above age 80.173
Elderly people often have nutritional deficiencies that can impair immune function.174 Increasing age is also associated with impairment of mucociliary clearance, which contributes to increased susceptibility to respiratory infections in the elderly,175 because mucociliary clearance is the primary barrier defense mechanism of the lungs.176 The elderly account for a higher percentage of patients having the various co-morbidities associated with poor COVID-19 prognosis, such as hypertension, diabetes, cardiovascular disease, and cancer. However, even healthy aging also leads to RAS imbalance, with elevated tissue RAS components, and lower levels of counterbalancing protective arms of RAS.58
It is well described that circulating AII levels typically decline in the elderly.55,177 Importantly, however, tissue levels of AII, ACE, AT1, and angiotensinogen increase with aging in the heart, kidney, and vasculature.55,177 Data from African-American hypertensives, including those treated with ACEIs and ARBs, revealed that tissue RAS has a more significant deleterious effect on organ function than circulating RAS.142,146 It is reasonable to expect that tissue RAS levels, rather than circulating levels, would also be the more important variable in elderly individuals, coinciding with the higher incidence of the RAS dysregulation-associated comorbidities in aged adults, such as hypertension and diabetes, which portend worse COVID-19 outcomes.
At the same time, there is evidence of lower levels of the protective counterbalancing arms of RAS,178 leaving the elderly comparatively more vulnerable to the damaging effects of the ACE axis. Animal experiments showed that the aortic vasodilatory response to A1–7 is lost in elderly female rats, but restored by exogenous estrogen replacement.178 In rat lungs of both genders, ACE2 levels have been found to decrease with aging61,179,180 This loss of ACE2 was more pronounced in elderly male rats than in similarly aged females,61,179,180 which is in keeping with the worse prognosis reported for men compared to women with COVID-19.
It has been posited that patients with baseline ACE2 deficiency would be particularly vulnerable to severe outcomes because of unchecked ACE activity with SARS2 virus downregulating ACE2 expression.61 In elderly COVID-19 patients, the higher basal tissue ACE axis components and lower counterweight ACE2 levels even at the start of infection would disadvantage these patients for poor outcome.
Unexpected Severity in Young, Otherwise Healthy: Through the Smokescreen
The COVID-19 outbreak in the west brought unexpectedly severe cases affecting younger adults. The explanation for this situation remains undefined and is probably multifactorial. Lifestyle factors may impair immune defense against COVID-19 in younger people. For instance, dehydration which may occur during strenuous exercise, impairs mucociliary clearance,181 as does aerobic exercise in polluted urban environments.182 Rising obesity rates may play a role, as well as genetic factors, including high RAS activity polymorphisms in the general population.183–185 Acquired, non-genetic RAS dysregulation may also be playing a role here, due to the resurgence of inhaled nicotine and its effects upon RAS expression.
Data reported on 191 patients with confirmed COVID-19 from two hospitals in China showed that the odds ratio of dying among current smokers, compared to nonsmokers, was 2.23 (95% CI: 0.65).97 The impact of e-cigarettes (“vaping”) has not yet been reported in the context of COVID-19. But experimental evidence has been accumulating, showing that e-cigarettes may be even more deleterious than traditional cigarettes, so it is reasonable to suppose that vaping will also influence COVID-19 outcomes.
Smoking is known to inhibit mucociliary clearance,186 an essential defense feature against airway pathogens. Recent experimental evidence suggests that high nicotine levels in vaping may be even more harmful to this critical defense against inhaled pathogens.181,187 In a sheep model, high-nicotine-containing e-cigarettes caused a 50% reduction in experimentally modelled mucociliary clearance.188
Moreover, nicotine also adversely impacts RAS levels, which would be deleterious during COVID-19 infection. Nicotine has been shown to decrease levels of Mas receptor and A1–7, while increasing ACE, AII, and AT1.189 Additionally, nicotine decreased both ACE2 and AT2 expression in several cell types, including cardiac fibroblasts.190
The rising use of e-cigarette products among young Americans has alarmed the medical community and public health officials. CDC data showed that 3.7% of US adults—more than 9 million people—used e-cigarettes in 2014.191 A 2018 Gallup poll reported that vaping had increased to more than 9% of US adults; in addition, 20% of respondents reported smoking traditional cigarettes.192 The National Youth Tobacco Survey revealed that 5 million teenagers vaped in 2019, up from 3.6 million in 2018.193 This constitutes a sizable portion of the younger US population who would be at increased risk during the current pandemic, given the evidence that baseline ACE2 deficiency predisposes to poorer COVID-19 outcomes.61
Conclusion: Taking Aim at RAS
With COVID-19 deaths now surpassing 500,000 worldwide, and further increases becoming evident in some areas as economies reopen—let alone the possibility of a second wave of infections surging late in the year—developing a therapeutic means to mitigate the severity of COVID-19 has taken on utmost urgency. With the evidence of ACE axis overactivation at the root of varied organ system pathology in COVID-19, the most obvious recourse to attempt to blunt COVID-19 progression and mortality would seem to be ACEIs and ARBs. These drugs are widely available, inexpensive, and they target the very system that is unbalanced in COVID-19. Yet the use of these drugs in COVID-19 is not clear cut.
Early in the COVID-19 pandemic, concerns were raised that ACEIs and ARBs might predispose to increased COVID-19 severity.194 In COVID-19, the role of ACE2 appears paradoxical.61 ACE2 facilitates entry of SARS2 virus into cells, but at the same time, ACE2 expression in alveolar cells protects against experimentally induced lung injury.61,64,66 After viral entry into alveolar cells, the spike protein of the SARS-CoV-2 virus downregulates further expression of ACE2,61 thereby reducing the protective effect of ACE2 on lung tissue.60
In animal models, ACEIs and ARBs elevate the expression of ACE2 in the heart.195,196 During fetal kidney development, AT1 exerts negative feedback to downregulate ACE2 levels.197 It has been suggested that ACEIs and ARBs upregulate ACE2 levels in cardiac and diabetic patients,179,198 and therefore could potentially accelerate SARS2 entry.61,198 Yet these drug classes have shown clear cardiovascular and renal protective effects,199,200 which might prove beneficial in blunting COVID-19 organ damage.201
In the midst of this “double-edged sword” debate,194 the European Society of Cardiology and the American College of Cardiology recommended that, without clear clinical evidence, already-prescribed ACEIs and ARBs should not be discontinued. A case series of 1178 patients from Wuhan, China found that ACEIs and ARBs were not associated with increased COVID-19 case severity or mortality.202 An Italian case–control study comparing records of 6272 COVID-19 positive patients to more than 30,000 registered recipients of Lombardy’s Regional Health Service came to the same conclusion.203 An observational study of Medicare Advantage health records, conducted by Yale University in partnership with United Health Group Insurance Corporation, announced that ACEIs—but not ARBs—might even be protective against more severe COVID-19 outcomes, since ACEI use correlated with a lower rate of COVID-related hospitalization in persons over 65 years of age in this study.204 Multiple ongoing prospective trials are attempting to assess whether differences in COVID-19 outcomes result from maintaining versus stopping ACEIs and ARBs (eg, study NCT04330300 in the WHO International Clinical Trials Registry Platform). Readers may access the database of international clinical studies at http://www.WHO.int.ictrp).
Independent of a potential impact on virus entry into cells, these two drug classes have been shown in human studies and animal models to lower the risk and severity of numerous problems that COVID-19 precipitates. Among their many benefits, ACEIs are lung protective in ARDS,75 and ACEIs and ARBs reduce stroke rates.55 ARBs reduce cardiovascular oxidative stress, reverse endothelial dysfunction, and decrease inflammatory and oxidative renal damage in patients with diseases associated with RAS dysregulation, such as hypertension and diabetes.205,206 ARBs have mitigated the severity of experimental myocarditis110 and have been shown to upregulate AT2 in vascular endothelial cells.52 Moreover, AT1 blockade may shunt existing AII to activate AT2, yielding anti-inflammatory effects.205,206
Therefore, ACEIs and ARBs might hold the potential to mitigate COVID-19 organ damage. But clarity may come only once the drugs have been assessed as individual agents, rather than as ACEIs and ARBs en masse. Although there are definitive class effects of ACEIs and ARBs, such as blood pressure lowering,207 the drugs themselves each have unique pharmacological characteristics. Evaluating them en masse may wash out otherwise important distinctions.
For example, not all ACEIs have equivalent tissue penetration, due to differences in lipophilicity.24 In a rat model, fosinopril had better cardiac entry than captopril, lisinopril, and ramipril.24 Similarly, quinapril had higher tissue penetration and better prevention of left ventricular hypertrophy than enalapril.24 In spontaneously hypertensive rats, moexipril more potently inhibited lung, aortic, and cardiac ACE than enalapril.208 And among ARBs, the unique structure of eprosartan seems to confer high renal affinity.209
In addition, some ACEIs, such as captopril and perindopril, cross the blood brain barrier (BBB); others, such as enalapril and imidapril, do not.109 This distinction could potentially hold importance, given that neurons express ACE2,210 and some severe COVID-19 patients have manifested CNS dysfunction, including encephalitis and a neurocognitive, dementia-like syndrome.211 Parenthetically, these reports are consistent with accumulating evidence that RAS is also involved in the amyloid pathology of Alzheimer’s dementia.212
Among the ARBs, only telmisartan has been shown to cross the BBB,185 thereby reinforcing the importance of carefully evaluating ARBs individually, rather than as whole drug classes, in COVID-19 studies.
There may be potential pitfalls deploying ACEIs and ARBs against COVID-19. ARBs have two broad categories of action: irbesartan, candesartan, and telmisartan are noncompetitive inhibitors of AT1, while losartan, valsartan, and eprosartan are competitive blockers.209,213 High concentrations of AII have been shown to overcome competitive blockade in vitro,213 possibly negatively impacting the therapeutic potential of competitive AT1 blockers in COVID-19.
Additionally, many ACEIs are prodrugs, dependent on hepatic metabolism for activation, but there is evidence that, in some cases, the prodrugs may inhibit the function of the activated counterpart.207 Lisinopril, by contrast to other class members, is orally formulated as the active drug.214 In the ARB class, losartan and candesartan are prodrugs, but activated candesartan has 10 times higher affinity for AT1 than activated losartan.213 Such pharmacological differences may impact the outcome of these therapies in COVID-19.
At least some ACEIs are dialyzable, but ARBs are not,209 and this may introduce additional difficulties using these agents in COVID-19 patients with renal failure. Because RAS blockade inhibits HPV,89,90 it is not known whether ACEIs and ARBs would worsen hypoxia in COVID-19 patients, or alternatively, push V/Q matching back toward normal.
Because ACEIs, but not ARBs, impact the metabolism of the vasodilator bradykinin (BK), the distinction between the two drug classes has the potential to differentially impact the course of treatment in COVID-19 patients. Bradykinin (BK) is the major vasoactive peptide in the kallikrein-kinin system,215–217 interfacing with two principal receptors:218,219 constitutively expressed B2, and inflammation-inducible B1.218 In the lungs, BK receptors are situated on the endothelial cells of alveolar capillaries, with B1 upregulated in patients with COVID-19.220 ACE metabolizes BK to inactive fragments; thus, ACEIs may prolong the action of BK.221–225
BK dysregulation can cause angioedema.226–228 B2 receptors inhibit the function of adherens junctions between vascular endothelial cells, leading to plasma fluid extravasation into the interstitium229 (Figure 1). Clinical data indicate that this occurs as a side effect in about one or two patients per 1000 people taking an ACEI,227,228 but the reason why only such a small percentage of patients are affected remains unknown.230–232 The concept that angioedema-like changes occur in the lungs of patients with COVID-19 is supported by both indirect (radiologic and clinical observation) evidence233,234 and by more direct evidence at autopsies.234–236,237
Other than producing AII and inactivating bradykinin, ACE also contributes to the production of A1–7, the counterbalance to AII. So, the interruption of these additional effects by ACEIs may potentially impact outcomes in COVID-19 in ways that the ARBs do not.
As of this time, a small number of prospective clinical trials have begun enrolling patients to assess the potential of individual ACEI and ARB drugs as a therapy to lessen COVID-19 severity. Among them, a study at the University of California San Diego is evaluating the ACEI Ramipril (NCT04366050). Trials at the University of Kansas Medical Center (NCT04335123) and the University of Minnesota (NCT04312009) are assessing the ARB losartan, and telmisartan is being assessed at Universidad de Buenos Aires (NCT04355936). Many other agents remain to be separately evaluated.
Aside from ACEIs and ARBs to downregulate the ACE axis, there are also potential agents to augment the activity of the protective arms of RAS. Recombinant ACE2 has shown benefit in animal models of ARDS,64 so it may prove beneficial in COVID-19 treatment. Apeiron Biologics has announced that its recombinant ACE2, called APN01, will enter Phase II trials in COVID-19 patients.238
In animal models, an AT2 agonist called C21 has proven effective at countering damaging effects of RAS. For example, C21 decreased collagen deposition fibrosis after MI and deceased arterial stiffness.53 In addition, C21 blocked the rise in AII that accompanies a high sodium diet in obese Zucker rats and blunted progression of glomerulosclerosis.54 C21 also decreased renal AT1 protein levels as measured by Western blot.54 A human trial of C21 in pulmonary fibrosis has been conducted in Europe (The European Union Registry number of this trial is EUDRACT 2017–004923-62).75 This agent could potentially be studied in COVID-19 patients.
In animal studies, the vitamin D receptor inhibits RAS activity, especially at the tissue level.106 In human studies, vitamin D supplementation did not significantly lower blood pressure, although these studies were small and had few African-American participants, who are at elevated risk from COVID-19 and who typically exhibit lower vitamin D levels than Caucasians.106 Perhaps vitamin D could have a positive impact on COVID-19 course if given to high-risk patients.
No outcome in medicine is ever guaranteed, but the evidence of RAS dysregulation in COVID-19 supports attempting to mitigate COVID-19 outcomes by modulating RAS. Such an approach would not be a shot in the dark, but rather would be guided by decades of experience modifying RAS in other diseases. Trials of RAS-modulating therapies in COVID-19 could be tailored to patients with high-RAS profiles, and would likely need initiation of medication early in the symptomatic course, before the tipping point of severe organ damage has occurred. Unfortunately, outpatient therapy opens doors to patient noncompliance, but enrollment of patients early in a hospitalization course may also be beneficial. Despite potential pitfalls, the urgency to combat COVID-19 requires tapping the full armamentarium of RAS modifier agents for possible efficacy. A recent model predicts that even with continued social distancing, COVID-19 fatalities could triple in the US alone by year’s end.239 Human beings are now the ones with no time to lose.
Acknowledgment
The authors would like to thank Ms Melinda Judson and Ms Cassandra Graybeal Brown for assistance in digitizing the manuscript figures from the authors’ hand-sketched originals.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cha AE A mysterious blood-clotting complication is killing coronavirus patients. Washington Post. 2020 April 22.
2. Belluck P 32 days on a ventilator: one covid patient’s fight to breathe again. New York Times. 2020 April 26.
3. Mays JC, Newman A Virus is twice as deadly for Black and Latino people than whites in NYC. New York Times. 2020 April 8.
4. Poston B, Barboza T, Reyes-Velarde A Younger blacks and Latinos are dying of covid-19 at higher rates in California. Los Angeles Times. April 25, 2020
5. Rabin RC In N.Y.C., the coronavirus is killing men at twice the rate of women. New York Times. 2020 April 7.
6. McGinley L Patients with certain cancers are nearly three times as likely to die of covid-19, study says. Washington Post. 2020 April 28.
7. Cotovio V, Brathwaite S Black people four times more likely to die of Covid-19 in the UK compared to white people. CNN International. 2020 May 7.
8. Bernstein L, Johnson CY, Kaplan S, McGinley L Coronavirus destroys lungs. But doctors are finding its damage in kidneys, hearts and elsewhere. Washington Post. 2020 April 15.
9. Rabin RC Obesity linked to severe coronavirus disease, especially for younger patients. New York Times. 2020 April 16.
10. Zeng JH, Liu YX, Yuan J, et al. First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection. 2020:1–5. doi:10.1007/s15010-020-01424-5.
11. Cha AE Young and middle-aged people, barely sick with covid-19, are dying of strokes. Washington Post. 2020 April 25.
12. Avula A, Nalleballe K, Narula N, et al. COVID-19 presenting as stroke. Brain Behav Immun. 2020;87:115–119. doi:10.1016/j.bbi.2020.04.077
13. LaMotte S Silent hypoxia: covid-19 patients who should be gasping for air but aren’t. CNN. 2020 May 6.
14. Aliosi S, Beasley D, Borter G, Escritt T, Kelland K Special report: as virus advances, doctors rethink rush to ventilate. Berlin: Reuters. 2020 April 23.
15. Cha AE, Janes C Children are falling ill with perplexing inflammatory syndrome thought to be linked to COVID-19. Washington Post. 2020 May 6.
16. Viner RM, Whittaker E. Kawasaki-like disease: emerging complication during the COVID-19 pandemic. Lancet. 2020;395(10239):1741–1743. doi:10.1016/S0140-6736(20)31129-6
17. Topol EJ, Whyte J. Coronavirus in context: eric topol explains the emerging research. Medscap. March 25, 2020.
18. Kim NY, Ha E, Moon JS, Lee YH, Choi EY. Acute hyperglycemic crises with coronavirus disease-19: case reports. Diabetes Metab J. 2020;44(2):349–353. doi:10.4093/dmj.2020.0091
19. Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA, Diz DI. Update on the angiotensin converting enzyme 2-angiotensin (1–7)-MAS receptor axis: fetal programing, sex differences, and intracellular pathways. Front Endocrinol (Lausanne). 2014;4:201. doi:10.3389/fendo.2013.00201
20. Brown NJ, Vaughan DE. Angiotensin-converting enzyme inhibitors. Circulation. 1998;97(14):1411–1420. doi:10.1161/01.cir.97.14.1411
21. Johnston CI. Biochemistry and pharmacology of the renin-angiotensin system. Drugs. 1990;39(Suppl 1):
22. Dzau VJ, Burt DW, Pratt RE. Molecular biology of the renin-angiotensin system. Am J Physiol. 1988;255(4 Pt 2):F563–F573. doi:10.1152/ajprenal.1988.255.4.F563
23. Okuyama A, Nonomura N, Nakamura M, Namiki M, Sonoda T. Renin-angiotensin system. Arch Androl. 1988;21(3):169–180. doi:10.3109/01485018808986739
24. White CM. Pharmacologic, pharmacokinetic, and therapeutic differences among ACE inhibitors. Pharmacotherapy. 1998;18(3):588–599.
25. Riordan JF. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003;4(8):225. doi:10.1186/gb-2003-4-8-225
26. Fournier D, Luft FC, Bader M, Ganten D, Andrade-Navarro MA. Emergence and evolution of the renin-angiotensin-aldosterone system. J Mol Med (Berl). 2012;90(5):495–508. doi:10.1007/s00109-012-0894-z
27. Zhuo JL, Ferrao FM, Zheng Y, Li XC. New frontiers in the intrarenal renin-angiotensin system: a critical review of classical and new paradigms. Front Endocrinol (Lausanne). 2013;4:166. doi:10.3389/fendo.2013.00166
28. Wynne BM, Chiao CW, Webb RC. Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin-1. J Am Soc Hypertens. 2009;3(2):84–95. doi:10.1016/j.jash.2008.09.002
29. Lee JH, Jang SJ, Rhie S. Antinatriuretic phenomena seen in children with acute pyelonephritis may be related to the activation of intrarenal RAAS. Medicine (Baltimore). 2018;97(36):e12152. doi:10.1097/MD.0000000000012152
30. Campbell DJ. Clinical relevance of local renin angiotensin systems. Front Endocrinol (Lausanne). 2014;5:113. doi:10.3389/fendo.2014.00113
31. Roman RJ, Fan F, Zhuo JL. Intrarenal renin-angiotensin system: locally synthesized or taken up via endocytosis? Hypertension. 2016;67(5):831–833. doi:10.1161/HYPERTENSIONAHA.116.07103
32. Nehme A, Zouein FA, Zayeri ZD, Zibara K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J Cardiovasc Dev Dis. 2019;6(2):14. doi:10.3390/jcdd6020014
33. De Mello WC. Beyond the circulating renin-angiotensin aldosterone system. Front Endocrinol (Lausanne). 2014;5:104. doi:10.3389/fendo.2014.00104
34. Wong MKS, Takei Y. Molecular and evolutionary perspectives of the renin- angiotensin system from lamprey. Gen Comp Endocrinol. 2018;257:137–142. doi:10.1016/j.ygcen.2017.01.031
35. Sayed-Tabatabaei FA, Oostra BA, Isaacs A, van Duijn CM, Witteman JC. ACE polymorphisms. Circ Res. 2006;98(9):1123. doi:10.1161/01.RES.0000223145.74217.e7
36. Opal SM. Phylogenetic and functional relationships between coagulation and the innate immune response. Crit Care Med. 2000;28(9 Suppl):S77–S80. doi:10.1097/00003246-200009001-00017
37. Aird WC. Coagulation. Crit Care Med. 2005;33(12 Suppl):S485–S487. doi:10.1097/01.ccm.0000186267.66402.24
38. Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131(4):417–430. doi:10.1111/j.1365-2141.2005.05753.x
39. Esmon CT. The impact of the inflammatory response on coagulation. Thromb Res. 2004;114(5–6):321–327. doi:10.1016/j.thromres.2004.06.028
40. Joyce DE, Nelson DR, Grinnell BW. Leukocyte and endothelial cell interactions in sepsis: relevance of the protein C pathway. Crit Care Med. 2004;32(5 Suppl):S280–S286. doi:10.1097/01.ccm.0000128037.72072.22
41. Pawlinski R, Mackman N. Tissue factor, coagulation proteases, and protease-activated receptors in endotoxemia and sepsis. Crit Care Med. 2004;32(5 Suppl):S293–S297. doi:10.1097/01.ccm.0000128445.95144.b8
42. Aird WC. Sepsis and coagulation. Crit Care Clin. 2005;21(3):417–431. doi:10.1016/j.ccc.2005.04.004
43. Harrison DG, Cai H. Endothelial control of vasomotion and nitric oxide production. Cardiol Clin. 2003;21(3):289–302. doi:10.1016/s0733-8651(03)00073-0
44. Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal. 2013;19(10):1110–1120. doi:10.1089/ars.2012.4641
45. Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005;1(3):183–198.
46. Funk SD, Yurdagul A
47. Wang CH, Li F, Takahashi N. The renin angiotensin system and the metabolic syndrome. Open Hypertens J. 2010;3(1):1–13. doi:10.2174/1876526203010001
48. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122(6):877–902. doi:10.1161/CIRCRESAHA.117.311401
49. Muñoz-Durango N, Fuentes CA, Castillo AE, et al. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int J Mol Sci. 2016;17(7):797. doi:10.3390/ijms17070797
50. Harvey A, Montezano AC, Lopes RA, Rios F, Touyz RM. Vascular fibrosis in aging and hypertension: molecular mechanisms and clinical implications. Can J Cardiol. 2016;32(5):659–668. doi:10.1016/j.cjca.2016.02.070
51. Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi:10.1155/2010/214074
52. Watanabe T, Barker TA, Berk BC. Angiotensin II and the endothelium: diverse signals and effects. Hypertension. 2005;45(2):163–169. doi:10.1161/01.HYP.0000153321.13792.b9
53. Kaschina E, Namsolleck P, Unger T. AT2 receptors in cardiovascular and renal diseases. Pharmacol Res. 2017;125(PtA):39–47. doi:10.1016/j.phrs.2017.07.008
54. Carey RM. Update on angiotensin AT2 receptors. Curr Opin Nephrol Hypertens. 2017;26(2):91–96. doi:10.1097/MNH.0000000000000304
55. Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: functional relevance in cardiovascular disease. Pharmacol Ther. 2008;120(3):292–316. doi:10.1016/j.pharmthera.2008.08.009
56. Clarke NE, Turner AJ. Angiotensin-converting enzyme 2: the first decade. Int J Hypertens. 2012;2012:307315. doi:10.1155/2012/307315
57. Chappel MC, Ferrario CM. ACE and ACE2: their role to balance the expression of angiotensin II and angiotensin-(1–7). Kidney Int. 2006;70(1):8–10. doi:10.1038/sj.ki.5000321
58. Jugdutt BI. Expanding saga of the renin-angiotensin system: the angiotensin II counter-regulatory AT2 receptor pathway. Circulation. 2015;131(16):1380–1383. doi:10.1161/CIRCULATIONAHA.115.016328
59. Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–590. doi:10.1007/s00134-020-05985-9
60. Glowacka I, Bertram S, Herzog P, et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. 2010;84(2):1198–1205. doi:10.1128/JVI.01248-09
61. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76:14–20. doi:10.1016/j.ejim.2020.04.037
62. Tikellis C, Thomas MC. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept. 2012;2012:256294. doi:10.1155/2012/256294
63. Zhang H, Baker A. Recombinant human ACE2: acing out angiotensin II in ARDS therapy. Crit Care. 2017;21(1):305. doi:10.1186/s13054-017-1882-z
64. Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi:10.1038/nature03712
65. Oudit GY, Imai Y, Kuba K, Scholey JW, Penninger JM. The role of ACE2 in pulmonary diseases–relevance for the nephrologist. Nephrol Dial Transplant. 2009;24(5):1362–1365. doi:10.1093/ndt/gfp065
66. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879. doi:10.1038/nm1267
67. Burki NK, Lee LY. Mechanisms of dyspnea. Chest. 2010;138(5):1196–1201. doi:10.1378/chest.10-0534
68. Buchanan GF, Richerson GB. Role of chemoreceptors in mediating dyspnea. Respir Physiol Neurobiol. 2009;167(1):9–19. doi:10.1016/j.resp.2008.12.002
69. Czick ME, Waldman JC, Gross JB. Sources of inspiration: a neurophysiologic framework for understanding anesthetic effects on ventilatory control. Curr Anesthesiol Rep. 2013. doi:10.1007/s40140-013-0042-5
70. Fitzgerald RS. Carotid body: a new target for rescuing neural control of cardiorespiratory balance in disease. Front Physiol. 2014;5:304. doi:10.3389/fphys.2014.00304
71. Patel KP, Schultz HD. Angiotensin peptides and nitric oxide in cardiovascular disease. Antioxid Redox Signal. 2013;19(10):
72. Fung ML. The role of local renin-angiotensin system in arterial chemoreceptors in sleep-breathing disorders. Front Physiol. 2014;5:336. doi:10.3389/fphys.2014.00336
73. Wang R, Zagariya A, Ibarra-Sunga O, et al. Angiotensin II induces apoptosis in human and rat alveolar epithelial cells. Am J Physiol. 1999;276(5):L885–L889. doi:10.1152/ajplung.1999.276.5.L885
74. Papp M, Li X, Zhuang J, Wang R, Uhal BD. Angiotensin receptor subtype AT(1) mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):L713–L718. doi:10.1152/ajplung.00103.2001
75. Wang D, Chai XQ, Magnussen CG, et al. Renin-angiotensin-system, a potential pharmacological candidate, in acute respiratory distress syndrome during mechanical ventilation. Pulm Pharmacol Ther. 2019;58:101833. doi:10.1016/j.pupt.2019.101833
76. De Mello WC. Renin Angiotensin system as a regulator of cell volume. Implications to myocardial ischemia. Curr Cardiol Rev. 2009;5(1):65–68. doi:10.2174/157340309787048149
77. Garcia-Garrote M, Perez-Villalba A, Garrido-Gil P, et al. Interaction between angiotensin type 1, type 2, and Mas receptors to regulate adult neurogenesis in the brain ventricular-subventricular zone. Cells. 2019;8(12):1551. doi:10.3390/cells8121551
78. Shigemura N, Takai S, Hirose F, Yoshida R, Sanematsu K, Ninomiya Y. Expression of renin-angiotensin system components in the taste organ of mice. Nutrients. 2019;11(9):2251. doi:10.3390/nu11092251
79. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. COVID-19 does not lead to a “typical” acute respiratory distress syndrome. Am J Respir Crit Care Med. 2020;201(10):1299–1300. doi:10.1164/rccm.202003-0817LE
80. Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5(1):18. doi:10.1038/s41572-019-0069-0
81. Rawal G, Yadav S, Kumar R. Acute respiratory distress syndrome: an update and review. J Transl Int Med. 2018;6(2):74–77. doi:10.1515/jtim-2016-0012
82. Gattinoni L, Chiumello D, Rossi S. COVID-19 pneumonia: ARDS or not? Crit Care. 2020;24(1):154. doi:10.1186/s13054-020-02880-z
83. Sarkar M, Niranjan N, Banyal PK. Mechanisms of hypoxemia. Lung India. 2017;34(1):47–60. doi:10.4103/0970-2113.197116
84. Sommer N, Dietrich A, Schermuly RT, et al. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J. 2008;32(6):1639–1651. doi:10.1183/09031936.00013908
85. Petersson J, Glenny RW. Gas exchange and ventilation-perfusion relationships in the lung. Eur Respir J. 2014;44(4):1023–1041. doi:10.1183/09031936.00037014
86. Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92(1):367–520. doi:10.1152/physrev.00041.2010
87. Michelakis ED, Hampl V, Nsair A, et al. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res. 2002;90(12):1307–1315. doi:10.1161/01.res.0000024689.07590.c2
88. Cargill RI, Lipworth BJ. Acute effects of hypoxaemia and angiotensin II in the human pulmonary vascular bed. Pulm Pharmacol. 1994;7(5):305–310. doi:10.1006/pulp.1994.1036
89. Cargill RI, Lipworth BJ. Lisinopril attenuates acute hypoxic pulmonary vasoconstriction in humans. Chest. 1996;109(2):424–429. doi:10.1378/chest.109.2.424
90. Kiely DG, Cargill RI, Lipworth BJ. Acute hypoxic pulmonary vasoconstriction in man is attenuated by type I angiotensin II receptor blockade. Cardiovasc Res. 1995;30(6):875–880. doi:10.1016/S0008-6363(95)00129-8
91. Kleinsasser A, Pircher I, Treml B, et al. Recombinant angiotensin-converting enzyme 2 suppresses pulmonary vasoconstriction in acute hypoxia. Wilderness Environ Med. 2012;23(1):24–30. doi:10.1016/j.wem.2011.09.002
92. Richardson S, Hirsch JS, Chappel MC, Ferrario CM. Narasimhan M et al and the northwell COVID-19 research consortium. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323(20):2053–2059. doi:10.1001/jama.2020.6775
93. Jerng JS, Hsu YC, Wu HD, et al. Role of the renin-angiotensin system in ventilator-induced lung injury: an in vivo study in a rat model. Thorax. 2007;62(6):527–535. doi:10.1136/thx.2006.061945
94. Wösten-van Asperen RM, Lutter R, Specht PA, et al. Ventilator-induced inflammatory response in lipopolysaccharide-exposed rat lung is mediated by angiotensin-converting enzyme. Am J Pathol. 2010;176(5):2219–2227. doi:10.2353/ajpath.2010.090565
95. Wösten-van Asperen RM, Lutter R, Haitsma JJ, et al. ACE mediates ventilator-induced lung injury in rats via angiotensin II but not bradykinin. Eur Respir J. 2008;31(2):363–371. doi:10.1183/09031936.00060207
96. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574–1581. doi:10.1001/jama.2020.5394
97. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi:10.1016/S0140-6736(20)30566-3
98. Corti R, Farkouh ME, Badimon JJ. The vulnerable plaque and acute coronary syndromes. Am J Med. 2002;113(8):668–680. doi:10.1016/s0002-9343(02)01344-x
99. Ridker PM, Morrow DA. C-reactive protein, inflammation, and coronary risk. Cardiol Clin. 2003;21(3):315. doi:10.1016/s0733-8651(03)00079-1
100. Stenvinkel P, Pecoits-Filho R, Lindholm B. Coronary artery disease in end- stage renal disease: no longer a simple plumbing problem. J Am Soc Nephrol. 2003;14(7):1927–1939. doi:10.1097/01.asn.0000069165.79509.42
101. Tiong AY, Brieger D. Inflammation and coronary artery disease. Am Heart J. 2005;150(1):11–18. doi:10.1016/j.ahj.2004.12.019
102. DeMarco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol. 2014;10(6):364–736. doi:10.1038/nrendo.2014.44
103. Sanchis-Gomar F, Lavie CJ, Perez-Quilis C, Henry BM, Lippi G. Angiotensin-converting enzyme 2 and antihypertensives (angiotensin receptor blockers and angiotensin-converting enzyme inhibitors) in coronavirus disease 2019. Mayo Clin Proc. 2020;95(6):1222–1230. doi:10.1016/j.mayocp.2020.03.026
104. Lippi G, Lavie CJ, Sanchis-Gomar F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): evidence from a meta-analysis [published online ahead of print, 2020 Mar 10]. Prog Cardiovasc Dis. 2020. doi:10.1016/j.pcad.2020.03.001
105. Long B, Brady WJ, Koyfman A, Gottlieb M. Cardiovascular complications in COVID-19. Am J Emerg Med. 2020;38(7):1504–1507. doi:10.1016/j.ajem.2020.04.048
106. Yang T, Xu C. Physiology and pathophysiology of the intrarenal renin-angiotensin system: an update. J Am Soc Nephrol. 2017;28(4):1040–1049. doi:10.1681/ASN.2016070734
107. Sun Y. Intracardiac renin-angiotensin system and myocardial repair/remodeling following infarction. J Mol Cell Cardiol. 2010;48(3):483–489. doi:10.1016/j.yjmcc.2009.08.002
108. Hu H, Ma F, Wei X, Fang Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur Heart J. 2020;ehaa190. doi:10.1093/eurheartj/ehaa190
109. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2(7):247–257. doi:10.1002/emmm.201000080
110. Liu X, Zhu X, Wang A, Fan H, Yuan H. Effects of angiotensin-II receptor blockers on experimental autoimmune myocarditis. Int J Cardiol. 2009;137(3):282–288. doi:10.1016/j.ijcard.2009.09.540
111. Sharma A, Saito Y, Hung SI, et al. The skin as a metabolic and immune-competent organ: implications for drug-induced skin rash. J Immunotoxicol. 2019;16(1):1–12. doi:10.1080/1547691X.2018.1514444
112. Pavlos R, Mallal S, Ostrov D, et al. T cell-mediated hypersensitivity reactions to drugs. Annu Rev Med. 2015;66(1):439–454. doi:10.1146/annurev-med-050913-022745
113. Kuck KH. Arrhythmias and sudden cardiac death in the COVID-19 pandemic. Arrhythmien und plötzlicher herztod in der COVID-19-pandemie. Herz. 2020;45(4):325–326. doi:10.1007/s00059-020-04924-0
114. Kochi AN, Tagliari AP, Forleo GB, Fassini GM, Tondo C. Cardiac and arrhythmic complications in patients with COVID-19. J Cardiovasc Electrophysiol. 2020;31(5):1003–1008. doi:10.1111/jce.14479
115. Czick ME, Shapter CL, Silverman DI. Atrial fibrillation: the science behind its defiance. Aging Dis. 2016;7(5):635–656. doi:10.14336/AD.2016.0211
116. Oxley TJ, Mocco J, Majidi S, et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. 2020;382(20):e60. doi:10.1056/NEJMc2009787
117. Tun NN, Arunagirinathan G, Munshi SK, Pappachan JM. Diabetes mellitus and stroke: a clinical update. World J Diabetes. 2017;8(6):235–248. doi:10.4239/wjd.v8.i6.235
118. Iadecola C, Gorelick PB. Hypertension, angiotensin, and stroke: beyond blood pressure. Stroke. 2004;35(2):348–350. doi:10.1161/01.STR.0000115162.16321.AA
119. Hsueh WA, Lyon CJ, Quiñones MJ. Insulin resistance and the endothelium. Am J Med. 2004;117(2):109–117. doi:10.1016/j.amjmed.2004.02.042
120. Brasier AR, Recinos A
121. Brambilla M, Gelosa P, Rossetti L, et al. Impact of angiotensin-converting enzyme inhibition on platelet tissue factor expression in stroke-prone rats. J Hypertens. 2018;36(6):1360–1371. doi:10.1097/HJH.0000000000001702
122. Roberts HR, Monroe DM, Escobar MA. Current concepts of hemostasis: implications for therapy. Anesthesiology. 2004;100(3):722–730. doi:10.1097/00000542-200403000-00036
123. Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation. 2010;17(8):641–649. doi:10.1111/j.1549-8719.2010.00060.x
124. Huang I, Lim MA, Pranata R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumonia - a systematic review, meta-analysis, and meta-regression. Diabetes Metab Syndr. 2020;14(4):395–403. doi:10.1016/j.dsx.2020.04.018
125. Palaiodimos L, Kokkinidis DG, Li W, et al. Severe obesity, increasing age and male sex are independently associated with worse in-hospital outcomes, and higher in-hospital mortality, in a cohort of patients with COVID-19 in the Bronx, New York. Metabolism. 2020;108:154262. doi:10.1016/j.metabol.2020.154262
126. Gombart AF, Pierre A, Maggini S. A review of micronutrients and the immune system-working in harmony to reduce the risk of infection. Nutrients. 2020;12(1):236. doi:10.3390/nu12010236
127. Wu D, Lewis ED, Pae M, Meydani SN. Nutritional modulation of immune function: analysis of evidence, mechanisms, and clinical relevance. Front Immunol. 2019;9:3160. doi:10.3389/fimmu.2018.03160
128. Via M. The malnutrition of obesity: micronutrient deficiencies that promote diabetes. ISRN Endocrinol. 2012;2012:103472. doi:10.5402/2012/103472
129. Kiselar JG, Wang X, Dubyak GR, et al. Modification of β-defensin-2 by dicarbonyls methylglyoxal and glyoxal inhibits antibacterial and chemotactic function in vitro. PLoS One. 2015;10(8):e0130533. doi:10.1371/journal.pone.0130533
130. Kim JH, Park K, Lee SB, et al. Relationship between natural killer cell activity and glucose control in patients with type 2 diabetes and prediabetes. J Diabetes Investig. 2019;10(5):1223–1228. doi:10.1111/jdi.13002
131. Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 diabetes and its impact on the immune system. Curr Diabetes Rev. 2020;16(5):442–449. doi:10.2174/1573399815666191024085838
132. Lotia S, Bellamy MC. Anesthesia and morbid obesity. Cont Educ Anesth Crit Care Pain. 2008;8(5):151–156. doi:10.1093/bjaceaccp/mkn030
133. Sharma S, Arora L. Anesthesia for the morbidly obese patient. Anesthesiol Clin. 2020;38(1):197–212. doi:10.1016/j.anclin.2019.10.008
134. Grassi L, Kacmarek R, Berra L. Ventilatory mechanics in the patient with obesity. Anesthesiology. 2020;132(5):1246–1256. doi:10.1097/ALN.0000000000003154
135. Govindarajan G, Alpert MA, Tejwani L. Endocrine and metabolic effects of fat: cardiovascular implications. Am J Med. 2008;121(5):366–370. doi:10.1016/j.amjmed.2008.01.032
136. Hsueh WA, Quiñones MJ. Role of endothelial dysfunction in insulin resistance. Am J Cardiol. 2003;92(4A):10J–17J. doi:10.1016/s0002-9149(03)00611-8.`
137. Sobel BE, Schneider DJ. Platelet function, coagulopathy, and impaired fibrinolysis in diabetes. Cardiol Clin. 2004;22(4):511–526. doi:10.1016/j.ccl.2004.06.009
138. Taylor AA. Pathophysiology of hypertension and endothelial dysfunction in patients with diabetes mellitus. Endocrinol Metab Clin North Am. 2001;30(4):983–997. doi:10.1016/s0889-8529(05)70223-1
139. Zhou MS, Schulman IH, Zeng Q. Link between the renin-angiotensin system and insulin resistance: implications for cardiovascular disease. Vasc Med. 2012;17(5):330–341. doi:10.1177/1358863X12450094
140. Ramalingam L, Menikdiwela K, LeMieux M, et al. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim Biophys Acta Mol Basis Dis. 2017;1863(5):1106–1114. doi:10.1016/j.bbadis.2016.07.019
141. Ceriello A. Hyperglycemia and the worse prognosis of COVID-19. Why a fast blood glucose control should be mandatory. Diabetes Res Clin Pract. 2020;163:108186. doi:10.1016/j.diabres.2020.108186
142. Ortega LM, Sedki E, Nayer A. Hypertension in the African American population: a succinct look at its epidemiology, pathogenesis, and therapy. Nefrologia. 2015;35(2):139–145. doi:10.1016/j.nefro.2015.05.014
143. U.S. Department of Health and Human Services. Office of minority health obesity and African Americans. Available from: http://www.minorityhealth.hhs.gov.
144. Lindhorst J, Alexander N, Blignaut J, Rayner B. Differences in hypertension between blacks and whites: an overview. Cardiovasc J Afr. 2007;18(4):241–247.
145. Forrester T, McFarlane-Anderson N, Bennet F, et al. Angiotensinogen and blood pressure among blacks: findings from a community survey in Jamaica. J Hypertens. 1996;14(3):315–321. doi:10.1097/00004872-199603000-00007
146. Williams SF, Nicholas SB, Vaziri ND, Norris KC. African Americans, hypertension and the renin angiotensin system. World J Cardiol. 2014;6(9):878–889. doi:10.4330/wjc.v6.i9.878
147. Cohall D, Ojeh N, Ferrario CM, Adams OP, Nunez-Smith M. Is hypertension in African-descent populations contributed to by an imbalance in the activities of the ACE2/Ang-(1–7)/Mas and the ACE/Ang II/AT1 axes? J Renin Angiotensin Aldosterone Syst. 2020;21(1):1470320320908186. doi:10.1177/1470320320908186
148. Rotimi C, Puras A, Cooper R, et al. Polymorphisms of renin-angiotensin genes among Nigerians, Jamaicans, and African Americans. Hypertension. 1996;27(3 Pt 2):558–563. doi:10.1161/01.hyp.27.3.558
149. Asamoah A, Yanamandra K, Thurmon TF, et al. A deletion in the angiotensin converting enzyme (ACE) gene is common among African Americans with essential hypertension. Clin Chim Acta. 1996;254(1):
150. Duru K, Farrow S, Wang JM, Lockette W, Kurtz T. Frequency of a deletion polymorphism in the gene for angiotensin converting enzyme is increased in African-Americans with hypertension. Am J Hypertens. 1994;7(8):759–762. doi:10.1093/ajh/7.8.759
151. Wang XL, McCredie RM, Wilcken DE. Genotype distribution of angiotensin-converting enzyme polymorphism in Australian healthy and coronary populations and relevance to myocardial infarction and coronary artery disease. Arterioscler Thromb Vasc Biol. 1996;16(1):115–119. doi:10.1161/01.atv.16.1.115
152. Frishberg Y, Becker-Cohen R, Halle D, et al. Genetic polymorphisms of the renin-angiotensin system and the outcome of focal segmental glomerulosclerosis in children. Kidney Int. 1998;54(6):1843–1849. doi:10.1046/j.1523-1755.1998.00218.x
153. Mital S, Chung WK, Colan SD, et al. Renin-angiotensin-aldosterone genotype influences ventricular remodeling in infants with single ventricle. Circulation. 2011;123(21):2353–2362. doi:10.1161/CIRCULATIONAHA.110.004341
154. Nakajima T, Wooding S, Sakagami T, et al. Natural selection and population history in the human angiotensinogen gene (AGT): 736 complete AGT sequences in chromosomes from around the world. Am J Hum Genet. 2004;74(5):898–916. doi:10.1086/420793
155. Krishnan R, Sekar D, Karunanithy S, Subramanium S. Association of angiotensin converting enzyme gene insertion/deletion polymorphism with essential hypertension in south Indian population. Genes Dis. 2016;3(2):159–163. doi:10.1016/j.gendis.2016.03.001
156. Jin JM, Bai P, He W, et al. Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health. 2020;8:152. doi:10.3389/fpubh.2020.00152
157. Karlberg J, Chong DS, Lai WY. Do men have a higher case fatality rate of severe acute respiratory syndrome than women do? Am J Epidemiol. 2004;159(3):229–231. doi:10.1093/aje/kwh056
158. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16(10):626–638. doi:10.1038/nri.2016.90
159. Fischer M, Baessler A, Schunkert H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res. 2002;53(3):672–677. doi:10.1016/s0008-6363(01)00479-5
160. Komukai K, Mochizuki S, Yoshimura M. Gender and the renin-angiotensin- aldosterone system. Fundam Clin Pharmacol. 2010;24(6):687–698. doi:10.1111/j.1472-8206.2010.00854.x
161. Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension. 2000;35(1 Pt 2):480–483. doi:10.1161/01.hyp.35.1.480
162. Nickenig G, Bäumer AT, Grohè C, et al. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation. 1998;97(22):2197–2201. doi:10.1161/01.cir.97.22.2197
163. Nickenig G, Strehlow K, Wassmann S, et al. Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation. 2000;102(15):1828–1833. doi:10.1161/01.cir.102.15.1828
164. Koike G, Horiuchi M, Yamada T, Szpirer C, Jacob HJ, Dzau VJ. Human type 2 angiotensin II receptor gene: cloned, mapped to the X chromosome, and its mRNA is expressed in the human lung. Biochem Biophys Res Commun. 1994;203(3):1842–1850. doi:10.1006/bbrc.1994.2402
165. Crackower MA, Sarao R, Oudit GY, et al. Angiotensin- converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828. doi:10.1038/nature00786
166. Sullivan JC, Rodriguez-Miguelez P, Zimmerman MA, Harris RA. Differences in angiotensin (1–7) between men and women. Am J Physiol Heart Circ Physiol. 2015;308(9):H1171–H1176. doi:10.1152/ajpheart.00897.2014
167. Cohall DH, Scantlebury-Manning T, James S, Hall K, Ferrario CM. Renin- angiotensin-aldosterone system gender differences in an Afro-Caribbean population. J Renin Angiotensin Aldosterone Syst. 2015;16(3):539–546. doi:10.1177/1470320314523659
168. Yeoh CB, Lee KJ, Rieth EF, et al. COVID-19 in the cancer patient. Anesth Analg. 2020;131(1):16–23. doi:10.1213.000000000004884
169. Wegman-Ostrosky T, Soto-Reyes E, Vidal-Millán S, Sánchez-Corona J. The renin-angiotensin system meets the hallmarks of cancer. J Renin Angiotensin Aldosterone Syst. 2015;16(2):227–233. doi:10.1177/1470320313496858
170. Munro MJ, Wickremesekera AC, Davis PF, Marsh R, Tan ST, Itinteang T. Renin-angiotensin system and cancer: a review. Integr Cancer Sci Ther. 2017;4(2):1–6.
171. Rasha F, Ramalingam L, Gollahon L, et al. Mechanisms linking the renin-angiotensin system, obesity, and breast cancer. Endocr Relat Cancer. 2019;26(12):R653–R672. doi:10.1530/ERC-19-0314
172. Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to COVID-19 in Italy. JAMA. 2020. doi:10.1001/jama.2020.4683
173. Leung C. Risk factors for predicting mortality in elderly patients with COVID-19: a review of clinical data in China [published online ahead of print, 2020 Apr 27]. Mech Ageing Dev. 2020;188:111255. doi:10.1016/j.mad.2020.111255
174. Maggini S, Pierre A, Calder PC. Immune function and micronutrient requirements change over the life course. Nutrients. 2018;10(10):1531. doi:10.3390/nu10101531
175. Ho JC, Chan KN, Hu WH, et al. The effect of aging on nasal mucociliary clearance, beat frequency, and ultrastructure of respiratory cilia. Am J Respir Crit Care Med. 2001;163(4):983–988. doi:10.1164/ajrccm.163.4.9909121
176. Bustamante-Marin XM, Ostrowski LE. Cilia and mucociliary clearance. Cold Spring Harb Perspect Biol. 2017;9(4):a028241. doi:10.1101/cshperspect.a028241
177. Abadir PM. The frail renin-angiotensin system. Clin Geriatr Med. 2011;27(1):53–65. doi:10.1016/j.cger.2010.08.004
178. Costa-Fraga FP, Goncalves GK, Souza-Neto FP, et al. Age-related changes in vascular responses to angiotensin-(1–7) in female mice. J Renin Angiotensin Aldosterone Syst. 2018;19(3):1470320318789332. doi:10.1177/1470320318789332
179. AlGhatrif M, Cingolani O, Lakatta EG. The dilemma of Coronavirus disease 2019, aging, and cardiovascular disease: insights from cardiovascular aging science [published online ahead of print, 2020 Apr 3]. JAMA Cardiol. 2020. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32242886. doi:10.1001/jamacardio.2020.1329
180. Xie X, Chen J, Wang X, Zhang F, Liu Y. Age- and gender-related difference of ACE2 expression in rat lung [published correction appears in Life Sci. 2006 Nov 25;79(26):2499.]. Life Sci. 2006;78(19):2166–2171. Available from: https://www.ncbi.nlm.nih.gov/pubmed/16303146. doi:10.1016/j.lfs.2005.09.038
181. Munkholm M, Mortensen J. Mucociliary clearance: pathophysiological aspects. Clin Physiol Funct Imaging. 2014;34(3):171–177. doi:10.1111/cpf.12085
182. Cavalcante de Sá M, Nakagawa NK, Saldiva de André CD, et al. Aerobic exercise in polluted urban environments: effects on airway defense mechanisms in young healthy amateur runners. J Breath Res. 2016;10(4):046018. doi:10.1088/1752-7163/10/4/046018
183. Benetos A, Topouchian J, Ricard S, et al. Influence of angiotensin II type 1 receptor polymorphism on aortic stiffness in never-treated hypertensive patients. Hypertension. 1995;26(1):44–47. doi:10.1161/01.hyp.26.1.44
184. Berge KE, Bakken A, Bøhn M, Erikssen J, Berg K. A DNA polymorphism at the angiotensin II type 1 receptor (AT1R) locus and myocardial infarction. Clin Genet. 1997;52(2):71–76. doi:10.1111/j.1399-0004.1997.tb02521.x
185. Singh KD, Karnik SS. Angiotensin receptors: structure, function, signaling and clinical applications. J Cell Signal. 2016;1(2):111. doi:10.4172/jcs.1000111
186. Utiyama DM, Yoshida CT, Goto DM, et al. The effects of smoking and smoking cessation on nasal mucociliary clearance, mucus properties and inflammation. Clinics (Sao Paulo). 2016;71(6):344–350. doi:10.6061/clinics/2016(06)10
187. Kesimer M. Another warning sign: high nicotine content in electronic cigarettes disrupts mucociliary clearance, the essential defense mechanism of the lung. Am J Respir Crit Care Med. 2019;200(9):1082–1084. doi:10.1164/rccm.201905-1080ED
188. Chung S, Baumlin N, Dennis JS, et al. Electronic cigarette vapor with nicotine causes airway mucociliary dysfunction preferentially via TRPA1 receptors. Am J Respir Crit Care Med. 2019;200(9):1134–1145. doi:10.1164/rccm.201811-2087OC
189. Oakes JM, Fuchs RM, Gardner JD, Lazartigues E, Yue X. Nicotine and the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2018;315(5):R895–R906. doi:10.1152/ajpregu.00099.2018
190. Yue X, Basting TM, Flanagan TW, et al. Nicotine downregulates the compensatory angiotensin-converting enzyme 2/angiotensin type 2 receptor of the renin-angiotensin system. Ann Am Thorac Soc. 2018;15(suppl2):s126–s127. doi:10.1513/AnnalsATS.201706-464MG
191. Schoenborn CA, Gindi RM. Electronic cigarette use among adults: United States, 2014. NCHS Data Brief. 2015;(217):1–8.
192. Newport F. Young people adopt vaping as their smoking rate plummets. Gallup.com. 2018 July 26. Avialable from: https://news.gallup.com/poll/237818/aspx.
193. Cullen KA, Gentzke AS, Sawdey MD, et al. E-cigarette use among youth in the United States, 2019. JAMA. 2019;322(21):2095–2103. doi:10.1001/jama.2019.18387
194. Sommerstein R, Kochen MM, Messerli FH, Gräni C. Coronavirus disease 2019 (COVID-19): do angiotensin-converting enzyme inhibitors/angiotensin receptor blockers have a biphasic effect? J Am Heart Assoc. 2020;9(7):e016509. doi:10.1161/JAHA.120.016509
195. Ferrario CM, Jessup J, Chappell MC, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):
196. Carey RM. Angiotensin type-1 receptor blockade increases ACE 2 expression in the heart. Hypertension. 2004;43(5):
197. Song R, Preston G, Yosypiv IV. Ontogeny of angiotensin-converting enzyme 2. Pediatr Res. 2012;71(1):13–19. doi:10.1038/pr.2011.7
198. Rico-Mesa JS, White A, Anderson AS. Outcomes in patients with COVID-19 infection taking ACEI/ARB. Curr Cardiol Rep. 2020;22(5):31. doi:10.1007/s11886-020-01291-4
199. Kuster GM, Pfister O, Burkard T, et al. SARS-CoV2: should inhibitors of the renin-angiotensin system be withdrawn in patients with COVID-19? Eur Heart J. 2020;41(19):1801–1803. doi:10.1093/eurheartj/ehaa235
200. Kuster GM, Osswald S. Switching antihypertensive therapy in times of COVID-19: why we should wait for the evidence. Eur Heart J. 2020;41(19):1857. doi:10.1093/eurheartj/ehaa335
201. Guo J, Huang Z, Lin L, Lv J. Coronavirus disease 2019 (COVID-19) and cardiovascular disease: a viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 infection. J Am Heart Assoc. 2020;9(7):e016219. doi:10.1161/JAHA.120.016219
202. Li J, Wang X, Chen J, Zhang H, Deng A. Association of renin-angiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) infection in Wuhan, China. JAMA Cardiol. 2020;e201624. doi:10.1001/jamacardio.2020.1624
203. Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med. 2020;382(25):2431–2440. doi:10.1056/NEJMoa2006923
204. Shelton J Seniors with COVID-19 taking ACE inhibitors have lower hospitalization risk. Yale News. 2020 May 27. Available from: http://www.news.yale.edu.
205. Dézsi CA, Szentes V. Effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers on prothrombotic processes and myocardial infarction risk. Am J Cardiovasc Drugs. 2016;16(6):399–406. doi:10.1007/s40256-016-0185-0
206. Dézsi CA. The different therapeutic choices with ARBs. Which one to give? When? Why? Am J Cardiovasc Drugs. 2016;16(4):255–266. doi:10.1007/s40256-016-0165-4
207. Reid JL. From kinetics to dynamics: are there differences between ACE inhibitors? Eur Heart J. 1997;18(SupplE):
208. Song JC, White CM. Clinical pharmacokinetics and selective pharmacodynamics of new angiotensin converting enzyme inhibitors: an update. Clin Pharmacokinet. 2002;41(3):
209. Sica DA. Pharmacology and clinical efficacy of angiotensin receptor blockers. Am J Hypertens. 2001;14(7Pt 2):
210. Parry AH, Wani AH, Yaseen M. Neurological dysfunction in Coronavirus disease-19 (COVID-19) [published online ahead of print, 2020 Jun 10]. Acad Radiol. 2020;
211. Varatharaj A, Thomas N, Ellul MA, et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study [published online ahead of print, 2020 Jun 25]. Lancet Psychiatry. 2020:
212. Kehoe PG, Perry G, Avila J, Tabaton M, Zhu X. The coming of age of the angiotensin hypothesis in Alzheimer’s disease: progress toward disease prevention and treatment? J Alzheimers Dis. 2018;62(3):1443–1466. doi:10.3233/JAD-171119
213. Oparil S. Newly emerging pharmacologic differences in angiotensin II receptor blockers. Am J Hypertens. 2000;13(1Pt 2):
214. Raia JJ
215. Hofman Z, de Maat S, Hack CE, Maas C. Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol. 2016;51(2):152–161. doi:10.1007/s12016-016-8540-0
216. Oehmcke-Hecht S, Köhler J. Interaction of the human contact system with pathogens-an update. Front Immunol. 2018;9:312. doi:10.3389/fimmu.2018.00312
217. Golias C, Charalabopoulos A, Stagikas D, Charalabopoulos K, Batistatou A. The kinin system–bradykinin: biological effects and clinical implications. Multiple role of the kinin system–bradykinin. Hippokratia. 2007;11(3):124–128.
218. Caballero T, Baeza ML, Cabañas R, et al. Consensus statement on the diagnosis, management, and treatment of angioedema mediated by bradykinin. Part I. Classification, epidemiology, pathophysiology, genetics, clinical symptoms, and diagnosis. J Investig Allergol Clin Immunol. 2011;21(5):333–347.
219. Ricciardolo FL, Folkerts G, Folino A, Mognetti B. Bradykinin in asthma: modulation of airway inflammation and remodelling. Eur J Pharmacol. 2018;827:181–188. doi:10.1016/j.ejphar.2018.03.017
220. Van de Veerdonk FL, Netea MG, van Deuren M, et al. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife. 2020;9:e57555. doi:10.7554/eLife.57555
221. Bossi F, Fischetti F, Regoli D, et al. Novel pathogenic mechanism and therapeutic approaches to angioedema associated with C1 inhibitor deficiency. J Allergy Clin Immunol. 2009;124(6):1303–1310.e4. doi:10.1016/j.jaci.2009.08.007
222. Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57(1):27–77. doi:10.1124/pr.57.1.2
223. Marceau F, Bachvarov DR. Kinin receptors. Clin Rev Allergy Immunol. 1998;16(4):385–401. doi:10.1007/BF02737658
224. Marceau F, Sabourin T, Houle S, et al. Kinin receptors: functional aspects. Int Immunopharmacol. 2002;2(13–14):1729–1739. doi:10.1016/s1567-5769(02)00189-3
225. Marceau F, Bawolak MT, Fortin JP, et al. Bifunctional ligands of the bradykinin B2 and B1 receptors: an exercise in peptide hormone plasticity. Peptides. 2018;105:37–50. doi:10.1016/j.peptides.2018.05.007
226. LoVerde D, Files DC, Krishnaswamy G. Angioedema. Crit Care Med. 2017;45(4):725–735. doi:10.1097/CCM.0000000000002281
227. Miller DR, Oliveria SA, Berlowitz DR, Fincke BG, Stang P, Lillienfeld DE. Angioedema incidence in US veterans initiating angiotensin-converting enzyme inhibitors. Hypertension. 2008;51(6):1624–1630. doi:10.1161/HYPERTENSIONAHA.108.110270
228. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165(14):1637–1642. doi:10.1001/archinte.165.14.1637
229. Busse PJ, Christiansen SC, Longo DL. Hereditary angioedema. N Engl J Med. 2020;382(12):1136–1148. doi:10.1056/NEJMra1808012
230. Ichinose M, Barnes PJ. Bradykinin-induced airway microvascular leakage and bronchoconstriction are mediated via a bradykinin B2 receptor. Am Rev Respir Dis. 1990;142(5):1104–1107. doi:10.1164/ajrccm/142.5.1104
231. Nossaman BD, Feng CJ, Kadowitz PJ. Analysis of responses to bradykinin and influence of HOE 140 in the isolated perfused rat lung. Am J Physiol. 1994;266(6 Pt 2):H2452–H2461. doi:10.1152/ajpheart.1994.266.6.H2452
232. Gama Landgraf R, Sirois P, Jancar S. Differential modulation of murine lung inflammation by bradykinin B1 and B2 selective receptor antagonists. Eur J Pharmacol. 2003;460(1):75–83. doi:10.1016/s0014-2999(02)02880-7
233. Lang M, Som A, Carey D, et al. Pulmonary vascular manifestations of COVID-19 pneumonia. Radiol Cardiothorac Imag. 2020;2(3):e200277. doi:10.1148/ryct.2020200277
234. Cohen AJ, DiFrancesco MF, Solomon SD, Vaduganathan M. Angioedema in COVID-19 [published online ahead of print, 2020 May 22]. Eur Heart J. 2020;ehaa452. doi:10.1093/eurheartj/ehaa452
235. Wichmann D, Sperhake JP, Lütgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID-19 [published online ahead of print, 2020 May 6]. Ann Intern Med. 2020:M20–2003. doi:10.7326/M20-2003.
236. Buja LM, Wolf DA, Zhao B, et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc Pathol. 2020;48:107233. doi:10.1016/j.carpath.2020.107233
237. Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID-19 autopsies, Oklahoma, USA. Am J Clin Pathol. 2020;153(6):725–733. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32374815. doi:10.1093/ajcp/aqaa062
238. Hoffman J Apeiron biologics initiates phase II clinical trials of APN01. News release, apeiron biologics; April 2, 2020. Available from: www.globalnewswire.com.
239. Basu A. Estimating the infection fatality rate among symptomatic COVID-19 cases in the United States. Health Aff (Millwood). 2020;39(7):1229–1236. doi:10.1377/hlthaff.2020.00455
240. Ghafouri-Ford S, Noroozi R, Omrani MD, et al. Angiotensin converting enzyme: a review on expression profile and its association with human disorders with special focus on SARS-CoV-2 infection. Vascul Pharmacol. 2020;130:106680. doi:10.1016/j.vph.2020.106680
241. Caruana-Montaldo B, Gleeson K, Zwillich CW. The control of breathing in clinical practice. Chest. 2000;117(1):205–225. doi:10.1378/chest.117.1.205
242. Lopez-Barneo J, Ortega-Saenz P, Pardal R, Pascual A, Piruat JI. Carotid body oxygen sensing. Eur Respir J. 2008;32(5):1386–1398. doi:10.1183/09031936.00056408
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.