")
Back to Journals » Journal of Pain Research » Volume 14
COVID-Pain: Acute and Late-Onset Painful Clinical Manifestations in COVID-19 – Molecular Mechanisms and Research Perspectives
Authors Cascella M , Del Gaudio A, Vittori A , Bimonte S , Del Prete P , Forte CA , Cuomo A, De Blasio E
Received 6 April 2021
Accepted for publication 1 June 2021
Published 10 August 2021 Volume 2021:14 Pages 2403—2412
DOI https://doi.org/10.2147/JPR.S313978
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Marco Cascella,1 Alfredo Del Gaudio,2 Alessandro Vittori,3 Sabrina Bimonte,1 Paola Del Prete,4 Cira Antonietta Forte,1 Arturo Cuomo,1 Elvio De Blasio2,5
1Division of Anesthesia and Pain Medicine, Istituto Nazionale Tumori-IRCCS-“Fondazione G. Pascale”, Naples, 80131, Italy; 2DSC Anestesia e Rianimazione 2, IRCCS Casa Sollievo Della Sofferenza, San Giovanni Rotondo, 71013, FG, Italy; 3Department of Anesthesiology and Critical Care, ARCO, Bambino Gesù Children’s Hospital, Rome, Italy; 4Direzione Scientifica, Istituto Nazionale Tumori-IRCCS-“Fondazione G. Pascale”, Naples, 80131, Italy; 5Multidisciplinary Emergency Unit for COVID-19 Campania, Naples, 80100, Italy
Correspondence: Sabrina Bimonte
Division of Anesthesia and Pain Medicine, Istituto Nazionale Tumori-IRCCS-“Fondazione G. Pascale”, Naples, 80131, Italy
Tel +39 0815903221
Email [email protected]
Abstract: Although the respiratory manifestations of COVID-19 are predominant, signs and symptoms of an extra-pulmonary involvement are usually encompassed among the clinical picture of the disease. Several painful manifestations can occur during the acute phase but also as short- or long-term complications. Myalgia, joint pain, sore throat, abdominal pain, chest pain, and headache usually accompany respiratory symptoms, but they can also occur as isolated clinical findings or can be expressed regardless of the severity of COVID-19. On these premises, given the vast spectrum of clinical manifestations and the complexity of their pathogenesis, it would be more appropriate to refer to “COVID-pain”, an umbrella term useful for encompassing all these clinical manifestations in a separate chapter of the disease. In this scenario, we addressed the topic from a molecular perspective, trying to provide explanations for the underlying pathophysiological processes. Consequently, this narrative review is aimed at dissecting the mechanisms of acute and chronic painful manifestations, summarizing fundamental concepts on the matter, controversies, current research gaps, and potential developments in this field.
Keywords: COVID-19, SARS-CoV-2, COVID-pain, COVID-19 complications
Introduction
Since the COVID-19 pandemic started, clinicians and researchers realized that, although the respiratory manifestations were predominant, signs and symptoms of extrapulmonary involvement could enrich the clinical picture of the disease. Clinical experience and literature data have shown that all organs and systems can be involved, directly or indirectly (eg, through cytokine damage), during the course of the disease. The matter, however, is very complex and in addition to the extrapulmonary manifestations occurring during the acute phase, a broad spectrum of clinical conditions can represent sequelae of the disease. These consequences can last from weeks until months after recovery from acute illness.
In this scenario, painful manifestations are included among the extrapulmonary expression of COVID-19, and they can occur during the acute phase but also as short- or long-term complications. In the acute phase of COVID-19, pain can be present in different forms. One of the first reports found that muscle pain can affect 44% of patients.1 Later on, it was showed that myalgia/arthralgia, sore throat, abdominal pain, chest pain, and headache usually accompany respiratory symptoms, but they can also occur as isolated clinical findings.2 Moreover, severe myalgia was regardless of the severity of COVID-19.3
Since the pandemic is ongoing, the chapter on post-COVID-19 complications has yet to be written. The term long COVID or post-COVID-19 syndrome refers to a range of new or ongoing symptoms experienced 4 weeks or later after acute infection with SARS-CoV-2.4 In a recent meta-analysis, the authors demonstrated more than 50 long-term effects of COVID-19.5 Interestingly, in addition to respiratory, cardiovascular, and neuropsychiatric sequelae, chronic pain must be necessarily considered among disease sequelae.6 Carfì et al,7 for instance, showed that after hospital discharge 27.3% of post-COVID-19 individuals reported joint pain, and 21.7% chest pain. Again, it was highlighted that 4 weeks after discharge from the hospital, 10–20% of patients treated in the ward suffered from myalgia, although the percentage increased to up 30% in those who needed intensive care unit (ICU) hospitalization.8 Chest pain and headache are other pain conditions observed in outpatients and inpatients until 35 days after the disease.9 These short- or long-term effects can combine with mood problems and fatigue, as well as other problems, affecting the quality of life of COVID-19-survivors.
Considering the impressive numbers of the pandemic, the problems related to the health care are manifold and can weigh on the care tasks at different levels, from the primary care to hospital management, for the most demanding cases where the pain is combined with other post-COVID-19 sequelae, such as cardiovascular and respiratory issues.
Starting from these clinical findings, it could be interesting addressing the problem from a molecular perspective, trying to provide explanations on the underlying pathophysiological processes. Consequently, this narrative review is aimed at dissecting the mechanisms of the COVID-19-associated acute and chronic painful manifestations. Although this paper summarizes fundamental concepts on the matter, controversies, current research gaps, and potential developments in the field are discussed.
For addressing the molecular mechanisms, the issues of direct viral damage, the role of macrophage activation, and the features of cytokine-induced damage are addressed. It should be emphasized that although the macrophage and cytokine involvements are strictly connected, for narrative purposes the two arguments are addressed separately.
The ACE2/RAS Pathway and the Direct Virus-Induced Damage
Structurally, spike (S) glycoprotein, envelope, membrane, and nucleocapsid proteins coat the viral genome of SARS-CoV-2. The S protein is a trimeric glycoprotein formed by an ectodomain, a single-pass transmembrane anchor, and an intracellular tail. It is composed of two subunits (S1 and S2) and exerts a fundamental role for virus binding and entry of host cells. Homotrimers of S proteins compose the spikes on the viral surface forming the typical virus crown; the S1 subunit contains the receptor-binding domain (RBD) that binds to the peptidase domain of angiotensin-converting enzyme 2 (ACE 2).10 The S2 subunit, which encompasses a fusion peptide, a transmembrane domain, and a cytoplasmic slot, is highly conserved and is responsible for the viral/host membrane fusion. Again, it is important to underline that SARS-CoV-2 S protein engages ACE2 as the crucial entry receptor and employs the host cell serine protease TMPRSS2 for S protein priming.11 This latter stage, indeed, could also play an important role in the mechanism of pain.
The ACE2 is a metalloproteinase ectoenzyme expressed mostly in the lower airway epithelial cells, such as alveolar type II cells, transient secretory cells, nasal ciliated, and secretory cells, upper olfactory neuroepithelium, and vascular endothelial cells, although it was also detected in almost all human organs.12 The enzyme was found in gastrointestinal cells, cardiac pericytes, cardiomyocytes, corneal epithelial cells, renal epithelial cells, bile duct cells, gallbladder epithelial cells, testicular Sertoli cells, and alveolar macrophages.13 It is one of the central enzymes in the renin–angiotensin system (RAS) that regulates fluids and electrolyte balance and systemic vascular resistance. Renin is the initial enzyme of the RAS cascade. It splits angiotensinogen (a serum globulin from the liver) generating angiotensin I (Ang I), a decapeptide inactive hormone. The main function of ACE is to convert Ang I to angiotensin II (Ang II). This latter is an octapeptide hormone that works by binding to two types of G-protein coupled receptors (GPCRs), Ang I type 1 (AT1) receptor (AT1R) and Ang type 2 (AT2) receptor (AT2R). AT1R is predominant in the adult organism, whereas the AT2R type is mostly found in fetal tissues and decreases after birth. Activation of AT1R induces vasoconstriction, cardiac hypertrophy, and fibrosis. On the other hand, AT2R plays a protective effect against overstimulation of AT1R; the increase of AT2R expression was observed in pathological conditions, such as vascular injury, and congestive heart failure.14
All elements that make up the RAS could play a role in the direct viral damage and, therefore, in the genesis of disease symptoms, including pain. Nevertheless, SARS-CoV-2 binds to the ACE2 isoform. Although it is a homolog of ACE, the two isoforms have important differences. The isoform type 2 removes the carboxy-terminal phenylalanine in Ang-II to generate the heptapeptide angiotensin (Ang)-(1–7). This peptide is also active and can produce the opposite effect to that of Ang-II, acting as a vasodilator and anti-inflammatory agent. These properties are due to the interaction with the GPCR, Mas. Of note, Ang-(1–7) can bind to the Mas receptor with up to 100-fold greater selectivity over the AT1R and AT2R.15 MasR is expressed throughout the central (CNS) and peripheral nervous system (PNS) including the dorsal root ganglion (DRG) and spinal cord.16 Evidence from preclinical studies showed that Ang-(1-7) may attenuate the production of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-1β, and IL-6, while increasing the expression of the anti-inflammatory cytokine IL-10 resulting in decreases of PI3K, MAPKs, and JNK signaling in multiple pain models. Forte et al17 demonstrated its antinociceptive agent in cancer-induced bone pain. Therefore, the ACE2/Ang-(1–7)/Mas axis works by producing important anti-inflammatory effects.
The search for a plausible explanation for linking SARS-CoV-2, ACE2 host receptor, and Mas-mediated Ang-(1–7) effects represents a key step to demonstrate viral direct action in the genesis of acute pain in COVID-19. Although SARS-CoV-2 binds to ACE2 for entering the host cells, theoretically, the activation of the ACE2/RAS axis should induce anti-inflammatory actions. However, the SARS-CoV-2 may cause down-regulation of ACE2 levels on cell surfaces. This phenomenon could induce Ang-II accumulation, with hyperactivation of the RAS and, in turn, enhanced oxidative stress.18 Probably, this cascade can explain, at least in part, the pathogenesis of the clinical manifestations of the disease, including those related to the altered nociception.
Thus, a possible interpretation for understanding the COVID-pain involves the activity of the ACE2/RAS pathway in key areas of nociception. Through the action of different angiotensinogen cleavage products – that act as neurotransmitters and/or neuromodulators – and their impact on particular receptors and post-receptor pathways, a complex cascade affecting the spinal transmission of nociceptive information can be postulated. Theoretically, the SARS-CoV-2 infection could directly influence these mechanisms by altering the balance between the neuromodulation systems of nociception.
Preclinical studies in mice showed that the ACE2 is expressed in neurons and microglia (but not astrocytes) in the DRG.19 The activation of AT1R leads to nociception phenomena through the phosphorylation of p38 mitogen-activated protein kinase (MAPK).20 Moreover, the octapeptide Ang II is not the only cleavage product obtained from the action of ACE2. Ang III, which is a C-terminal metabolite of Ang II; it can also bind to the AT1R, and is involved in the spinal nociceptive transmission.21 On these bases, as Su et al22 suggested, the SARS-CoV-2 infection in the human spinal dorsal horn could upset the balance between two opposing systems: the ACE/Ang II/AT1 receptor axis and the ACE2/Ang (1–7)/MAS axis.
In this complex scenario, the AT2R could play the role of protagonist/co-protagonist. It was proved that this subtype of the receptor is implicated in pain control.23 Although the precise signaling pathway remains unclear, the receptor probably works as G-protein coupled receptor and, particularly in neurons, it provides the activation of the serine/threonine protein phosphatase 2A (PP2A), generation of prostaglandin E2 from arachidonic acid by cyclooxygenase-1 (COX-1) and subsequent stimulation of the delayed rectifier K+ channel with hyperpolarization of plasma membranes. Other pathways concern the release of bradykinin and nitric oxide (NO). It was also proved that Ang II might provoke COX-2 induction.24 All these pathways are closed to the mechanisms of inflammation and nociception (Figure 1).
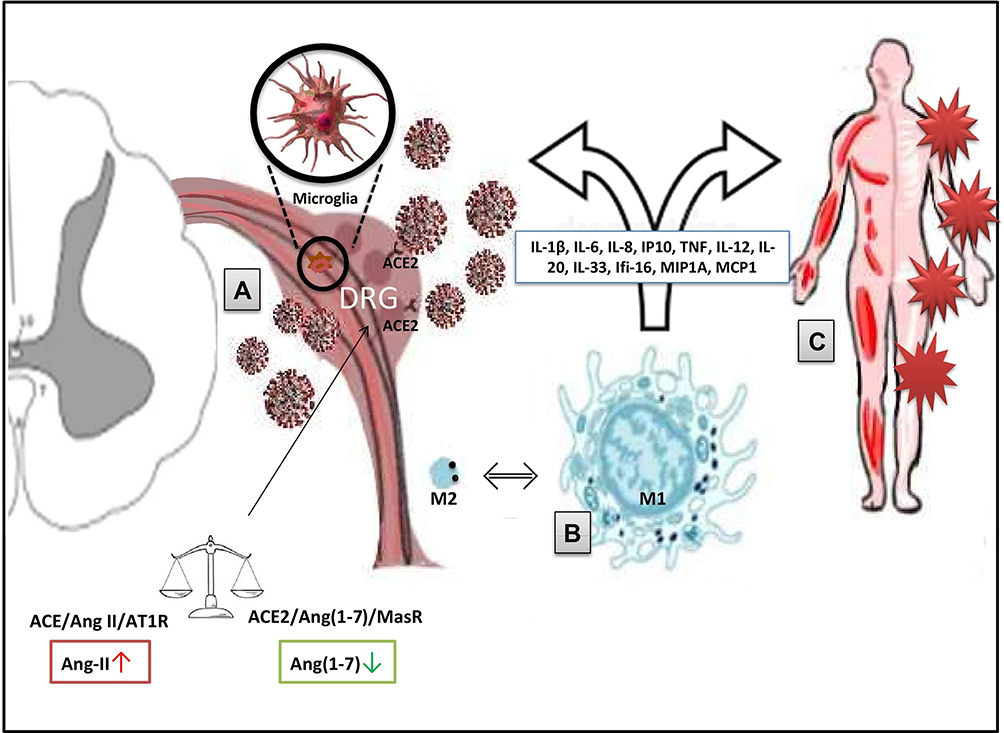 |
Figure 1 Potential mechanisms of COVID-pain (SARS-CoV-2/COVID-19-induced pain). (A) ACE2/RAS pathway and the direct virus-induced damage. Within the RAS, the virus/receptor (ACE2) interaction involves unbalance of the ACE/Ang II/AT1R and the ACE2/Ang-(1–7)/MasR axes with down-regulation of ACE2 levels on cell surfaces, Ang-II accumulation, and impairment of the anti-nociceptive Ang-(1-7) pathway. Therefore, direct damage to sensory neurons and/or glial cells is produced. (B) Macrophage activation. Macrophages and other immune cells can stimulate the production of inflammatory mediators (eg, IL-1β, TNF, and bradykinins). These processes can facilitate the sensory cells injury and can lead to chronic pain through sensitization/activation processes. (C) The exuberant immune-mediated inflammation. It is mostly responsible for systemic damage and the triggering of long-COVID problems (including widespread myalgia and joint pain) via peripheral and central mechanisms. Disease-related and predisposing factors contribute to the determinism of the damage. Abbreviations: RAS, renin–angiotensin system; ACE2, angiotensin-converting enzyme 2; Ang, angiotensin; AT1R, angiotensin 1 receptor; MasR, Mas receptor; IL, interleukin; TNF, tumour necrosis factor. |
Controversies on ACE2/RAS-Mediated Direct Viral Damage
Since the ACE2 receptor is expressed in neurons and glial cells, and SARS-CoV-2 binds to the ACE2 with a high affinity, the logical inference is that this coronavirus is particularly capable of producing direct damage to sensory neurons. Nevertheless, the matter seems to be more complex and this statement, indeed, should presuppose:
• Neurotropism of the virus
• That the pain-COVID manifestations are expressed as neuropathy phenomena with or without neuropathic pain.
Although several neurotropic viruses have pathogenic effects on the CNS and PNS, the presence of the virus and its replication in the nervous tissue must be well demonstrated. Thus, the virus/ACE2 binding and the receptor expression in the nervous tissue does not necessarily imply that SARS-CoV-2 is capable of producing direct damage to CNS and/or PNS. Again, because the chronic painful manifestations mainly manifest as diffuse myalgia and joint pain, it is more reasonable to think of nociceptive pain. From a pathophysiological point of view, nociceptive pain usually refers to the presence of tissue damage.
Previous in vitro (cultured human and rat DRG neurons) and animal studies showed that AT2R was implicated in neurite outgrowth (after nerve injury), and thus in chronic pain and hypersensitivity associated with abnormal nerve sprouting.25 In a rat model of neuropathic pain (chronic constriction injury of the sciatic nerve), and AT2R selective antagonist demonstrated important analgesic activity.26 Again, in a controlled clinical investigation, the AT2 receptor antagonist EMA401 ((S)-2-(diphenylacetyl)-1,2,3,4-tetrahydro-6-methoxy-5-(phenylmethoxy)-3-isoquinolinecarboxylic acid) was tested against post-herpetic neuropathic pain.8
In light of these pieces of evidence, the alteration of nociception may not necessarily be due to a receptor binding (virus-ACE2) within the CNS or PNS, but it can be the product of changes in the balance between the pathways of ACE2. It could be hypothesized an up-regulation of AT2 mediated (pro-nociceptive) mechanisms, caused by excessive activation of the “classic” AT1 axis. Alternatively, or additionally, a functional imbalance between cytokine systems characterized by an impairment of the Ang (1–7) pathway (anti-nociceptive) could occur. This could, at least in part, explain much of the painful manifestations in the absence of clear evidence of viral invasion into nerve tissue.
The Macrophage Involvement
Macrophages are key cells of the immune system in defense and repair processes. They represent modulators in the mechanisms of inflammation, both in the acute phase and in its chronicity.27 Their role is fundamental in inducing the inflammatory state (in the context of an innate defensive response), but also in the resolution processes. The mechanisms of pain are closely linked to these processes and involve both the peripheral inflammation site and the nociceptive processing station in the DRG. In brief, peripherally, macrophages and other immune cells stimulate the production of inflammatory mediators, such as IL-1β, TNF, and bradykinins. This effect reverberates on DRG sensory neurons, and through complex processes of sensitization/activation, it can induce pain and hyperalgesia.28 On the other hand, spontaneous or treatment-induced resolution of inflammation involves the disappearance of pain. In this context, resident macrophages in the peripheral nerve are particularly engaged in the resolution of inflammation. Nevertheless, when the resolution pathways fail, the transition from acute to chronic pain develops.29 Concerning neuropathic pain, as Yu et al30 recently demonstrated, macrophages contribute to both the initiation and maintenance of this type of pain. These cells may assume different phenotypes: an inflammatory (M1) phenotype when they infiltrate the DRG during inflammatory pain, and the resolution (M2) phenotype that probably works by restoring neuronal mitochondrial functions.31 Nevertheless, between the classically activated pro-inflammatory M1 form and the beneficial M2 phenotype, many alternatively activated/beneficial forms (M1/M2-like types) exist.32
The dynamic activity of macrophages is modulated by multiple factors. Among these factors, it was previously demonstrated that the ACE system has a role in both innate and adaptive responses by modulating macrophage and neutrophil function. Interestingly, it was demonstrated that the expression of AT2R in macrophages infiltrates the site of nerve injury but not in DRG.33 This finding could be proof of the macrophage activation in the phenomenon of COVID-19-related pain.
In the context of macrophage activity, another link with coronavirus infection could involve the interaction through elements responsible for regulating the inflammatory activity of these immune cells. The OX-2 membrane glycoprotein (CD200) and its receptor (CD200R) act as important checkpoints of the immune response by inhibiting the mitogen-activated protein kinase (MAPK) and lipid kinases (PI3K/Akt) signaling pathways.34 Thus, the CD200/CD200R inhibitory axis works for preventing the excessive inflammatory stimulus, even when secondary to infectious agents. In particular, the CD200 is expressed on myeloid lineage cells. Through a signaling network involving molecules and transcription factors, it could promote macrophage polarization toward the beneficial M2 phenotype.34 This effect does not only affect resident macrophages (closely to DRGs) as during an extensive neuro-inflammatory process, peripheral immune cells can influence the progression of nerve tissue injury.
According to this pathogenic perspective, the problem extends well beyond the issue of COVID-related pain but includes all the possible problems inherent to the direct and indirect neurotoxicity of SARS-CoV-2. Thus, from a translational perspective, these data are of great importance as modulating the CD200/CD200R inhibitory axis could attenuate many of the inflammatory responses related to infection, including but not limited to pain. Previously, Hayakawa et al35 proved that CD200 activation could limit the macrophage-induced damage to oligodendrocyte precursors. It was also demonstrated that the activation of the axis enhanced IFN production and viral clearance in mice infected by the mouse hepatitis coronavirus (MHV).36
The Exuberant Immune-Mediated Inflammation
In addition to the potential direct neurotoxic effect, SARS-CoV-2 can produce pain through the activation of an excessive immune response. This exuberant immune-mediated inflammation is a cornerstone of a cascade of events that culminates in the development of the various clinical manifestations of COVID-19. The COVID-pain can be an expression of this phenomenon.
While the whole picture of the aberrant virus-induced dysregulation of the immunologic response needs to be better elucidated, this complex chain of events is progressively enriched with new scientific findings. A key concept is dysregulation in the production of soluble immune mediators. This phenomenon involves the activation and inhibition of different immune cell subtypes and, consequently, a dysregulation between the production of mediators with anti-inflammatory action and substances that induce the recall of further cellular elements, endothelial damage, and alteration of the microcirculation. This deleterious phenomenon is termed “cytokine storm”. It expressed as high release of pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, IL-8, and IL-12 as well as IFN-γ inducible protein (IP10; also termed as motif chemokine ligand 10, CXCL10), macrophage inflammatory protein 1A (MIP1A), and monocyte chemoattractant protein 1 (MCP1).37
The processes that trigger the immune responses against the virus are, at least partly, unknown and much data has been translated from SARS research. For instance, it was shown that SARS-CoV is recognized by the toll-like receptors (TLR), especially TLR3 and TLR4. TLRs are a class of single-pass, non-catalytic transmembrane receptors, mainly expressed on the membrane of sentinel cells, such as macrophages, playing a key role in the defense of the organism, in particular in innate immunity. The binding of SARS-CoV-2 to the TLRs induces the release of pro-IL-1β which is cleaved into the active mature IL-1β mediating lung inflammation through the activation of the inflammasome.38
In addition to IL-1β, other immune mediators are aberrantly produced in response to SARS-CoV-2 and can produce neurological damage . Of those, IL-1β, IL-6, IP10, and TNFα are the pro-inflammatory cytokines with the greater ability to induce tissue injury in several organs, including the CNS. However, the list of pro-inflammatory substances triggered by the infection is particularly long and includes other ILs such as IL-12 p40, IL-20, and IL-33, and the gamma-interferon-inducible protein Ifi-16 (Ifi-16). In a mouse model, most of these pro-pro-inflammatory cytokines and the role of astrocytes and microglia were demonstrated.39 Other investigators showed that primary glial cell cultures exposed to coronavirus can secrete a great amount of several pro-inflammatory cytokines, especially IL-15, IL-6, and TNFα.40
Thus, several pathways are involved in virus-induced tissue damage. This aberrant activation of inflammatory and pro-thrombotic pathways leads to further cellular recruitment with chemo-attraction of macrophages, and cellular stress mostly resulting from activated reactive oxygen species (ROS).
Disease-Related Factors
Patients who suffered from severe forms of COVID-19 ARDS are particularly prone to develop a post-discharge complex disorder featured by persistent fatigue, depression, weakness, and limited exercise tolerance.7 This syndrome recognizes a multifactorial genesis. Among the factors involved, there is the use of medications, such as systemic corticosteroids to manage the disease and multi-organ dysfunction, for instance, during the intensive care unit (ICU) stay. Additionally, a pivotal role is played by complex psycho-affective components including psychological effects of prolonged ICU admission, sleep, and mood and affect alterations, anxiety due to separation, and isolation from family and friends. Again, other factors strictly related to systemic inflammation-like muscle atrophy due to immobility may contribute. These disorders have also been extensively described in SARS-survivors admitted to ICU.41 In the context of this clinical picture, widespread and nonspecific myalgia and joint pain are included. In a telephone follow‐up of patients post-discharge (n=100), compared to those affected by less severe forms of the disease, patients admitted to ICU showed a greater prevalence of non-specific pain (20–30% versus 10–20%), despite less affected by comorbidities.8 Results are inconclusive and, probably, also patients experienced less severe forms (eg, self-treated at home patients) may develop chronic pain and other post-COVID-19 clinical manifestations. In patients followed up for two months after hospital discharge, the persistence of joint pain, and chest pain was, respectively, 27.3% and 21.7%, although the authors failed to report precise data on the illness severity.7 Follow-up studies will be able to give us reliable data on the extent of the phenomenon.
Taken together, the data available from the literature indicate that most likely the COVID-pain phenomenon has a multifactorial genesis. Factors related to the pathogen intersect with elements produced by the response of the host to the viral attack, as well as with elements attributable to therapies for the management of the disease and other conditions related, for instance, to immobilization, and psychological stress. Regarding direct viral damage, even if scientific evidence is not strong, direct involvement in key areas of nociception cannot be excluded. On the other hand, the role of indirect damage via aberrant cytokine production and macrophage activation has more solid scientific presupposes (Table 1).
 |
Table 1 What We Know on the Pathophysiology of COVID-19-Associated Pain? |
Research Perspectives
Despite research on the pathogenesis of the COVID-19-associated pulmonary and extrapulmonary damage is advancing at unprecedented speed, multiple aspects related to pain mechanisms during and after the acute course of the disease must be well elucidated.42 It is mandatory, thus, to characterize the precise pathophysiology of these painful long-COVID clinical manifestations. From preclinical and translational research, a detailed understanding of the cascade of events leading to pain development is expected. There are many aspects to be studied. For example, the potential association of the complement system with pain in COVID should be investigated. Multiple features of severe SARS-CoV-2 infection suggest that complement activation plays a pivotal role in the pathogenesis of COVID-19, particularly during exaggerated immune responses.43 Nevertheless, little is known about the impact of this activation on chronic pain. In addition, other interesting elements in the cascade, such as the potential immune-mediated disruption of the autonomic nervous system,44 need to be better characterized.
Furthermore, research must clear up many doubts (Table 2). For example, a crucial aspect is a correlation between pain complications and the severity of the disease. In this regard, Tuzun et al3 demonstrated that, compared to muscular pain, the occurrence of arthralgia was significantly higher among the severe group of COVID-19. On the other hand, long-term symptoms can also occur in those who have suffered from less severe forms and have not been hospitalized.43
 |
Table 2 Outstanding Questions on the Pathophysiology of the Pain-COVID |
Further studies must clarify whether the post-COVID pain is produced by direct/indirect viral damage, or it is the result of other factors, such as disease-related hypoxia, consequences of more or less invasive therapies, and effects of isolation of critically ill patients or other psychological factors. Chronic pain, for instance, could represent a complication of ICU treatment and manifests itself as a form of ICU acquired weakness. Again, secondary diseases associated with the SARS-CoV-2 infection, including critical illness polyneuropathy, Guillain-Barré syndrome, polyneuritis, or central neuropathic pain due to cerebrovascular events can lead to post-COVID pain.45 Determining the factors involved and the weight of each element appears to be a challenge that will keep researchers busy for a long time after the pandemic is resolved. It is reasonable to assume that only studies with long-term follow-ups and evidence-based medicine data will provide us with adequate answers.
Probably specific groups of patients are more prone to develop COVID-pain complications. In this context, gender correlation must be elucidated, as well as the role of the age factor. Interestingly, elderly patients were more prone to suffer from myalgia, and fatigue, as compared with younger subjects who had a higher propensity to manifest symptoms related to sensorial disorders.46 Furthermore, about clinical settings, it would be interesting to verify the extent of COVID-pain in cancer patients. The translational research offers important data. For example, TMPRSS2 expression is highly increased in cancer cells, and it is also directly correlated with the degree of cancer pain.47 In a translational perspective, this aspect represents an interesting link between preclinical and clinical research.
The study of COVID-19 complications may also have other objectives. It may be possible, for example, to identify clinical-laboratory symptoms or signs suggestive of the severity of the disease, also concerning the development of the long-term sequelae. The research must finally clarify what is the weight of the patient’s general condition in the development of pain complications. Factors such as hypoxia, multi-organ damage, could override the intrinsic ability of the virus to damage the CNS.
Another important issue to be investigated concerns the role of nutrition, especially for ICU patients. Vitamin deficiency, for example, can be associated with musculoskeletal pain. This aspect was recently revised by Minelli et al.48 Moreover, as a retrospective analysis suggested, the problem seems to affect not only ICU patients.49 Epidemiological studies conducted on different populations are needed to provide more detailed data.
Finally, it is necessary to have an estimate of the duration of the symptoms and above all the effect on the quality of life. Given the clinical, social, and epidemiological impact, this is an issue of enormous importance. Probably, the acquisition of solid foundations for the pathophysiology of COVID-pain could help to better characterize the phenomenon (Table 3). Nevertheless, only follow-up studies conducted on a suitable number of samples will be able to clarify the effects of this spectrum of complications on the quality of life of the COVID-survivor, on the care needs, and on the costs associated with them. Above all, in light of the incredible numbers of the pandemic, this step is mandatory to establish the most appropriate prophylactic and therapeutic strategies. In the meantime, research must define a precise roadmap and priorities by intensifying preclinical investigations, enhancing translational research, and collecting as much as clinical data possible. In addition, the national healthcare systems must address many of the unmet needs that have emerged during the pandemic50 and organizing, in turn, dedicated paths to manage patients who report pain and other symptoms of long-COVID. These paths must involve all levels of care, identifying the tasks of general practitioners, strengthening pain unit consultations, and organizing multi-professional approaches by which scheduled follow-ups and specialist treatments can adequately be conducted.
 |
Table 3 Main Topics to Be Addressed on COVID-Pain |
Conclusions
The onset of painful manifestations is frequently seen during the acute phase of the SARS-CoV-2 infection. Additionally, COVID-19 survivors can experience chronic complications as a consequence of prolonged hospital stay, immobility, long-term mechanical ventilation, and other therapies. Pain conditions, mostly expressed as myalgia or joint pain, can frequently be found among these COVID-19 sequelae. Taken together, acute and chronic manifestations can make up a separate chapter of the disease. The pathophysiology of the pain-COVID seems to be very complex and multifactorial; it probably involves the intersection of direct viral damage, extensive injury from the aberrant production of cytokines combined with factors related to the concomitant therapies, as well as the psychological conditions of patients.
Considering the impressive numbers of the pandemic, the study of the pathophysiology of these phenomena can offer important suggestions for addressing these problems, both from a prophylactic and therapeutic point of view as well as to guide public health policy.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors declare no conflict of interest.
References
1. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi:10.1016/S0140-6736(20)30183-5
2. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708–1720. doi:10.1056/NEJMoa2002032
3. Tuzun S, Keles A, Okutan D, Yildiran T, Palamar D. Assessment of musculoskeletal pain, fatigue and grip strength in hospitalized patients with COVID-19. 2021. doi:10.23736/S1973-9087.20.06563-6
4. Venkatesan P. NICE guideline on long COVID. Lancet Respir Med. 2021;9(2):129. doi:10.1136/bmj.m4938
5. Lopez-Leon S, Wegman-Ostrosky T, Perelman C, et al. More than 50 Long-term effects of COVID-19: a systematic review and meta-analysis. medRxiv. 2021;30:21250617. doi:10.1101/2021.01.27.21250617
6. Vittori A, Lerman J, Cascella M, et al. COVID-19 pandemic acute respiratory distress syndrome survivors: pain after the storm? Anesth Analg. 2020;131:117–119. doi:10.1213/ANE.0000000000004914
7. Carfì A, Bernabei R, Landi F. Gemelli against COVID-19 post-acute care study group. persistent symptoms in patients after acute COVID-19. JAMA. 2020;324:603–605. doi:10.1001/jama.2020.12603
8. Halpin SJ, McIvor C, Whyatt G, et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: a cross-sectional evaluation. J Med Virol. 2021;93:1013–1022. doi:10.1002/jmv.26368
9. Tenforde MW, Billig Rose E, Lindsell CJ, et al. Characteristics of adult outpatients and inpatients with COVID-19–11 Academic Medical Centers, United States, March–May 2020. MMWR Morb Mortal Wkly Rep. 2020;69:841–846. doi:10.15585/mmwr.mm6926e3
10. Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 2020;41:1100–1115. doi:10.1016/j.it.2020.10.004
11. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. doi:10.1016/j.cell.2020.02.052
12. Santos RAS, Sampaio WO, Alzamora AC, et al. The ACE2/Angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev. 2018;98:505–553. doi:10.1152/physrev.00023.2016
13. Alon R, Sportiello M, Kozlovski S, et al. Leukocyte trafficking to the lungs and beyond: lessons from influenza for COVID-19. Nat Rev Immunol. 2021;21:49–64. doi:10.1038/s41577-020-00470-2
14. Steckelings UM, Unger T. Angiotensin II type 2 receptor agonists–where should they be applied? Expert Opin Investig Drugs. 2012;21:763–766. doi:10.1517/13543784.2012.681046
15. Passos-Silva DG, Verano-Braga T, Santos RA. Angiotensin-(1-7): beyond the cardio-renal actions. Clin Sci. 2013;124:443–456. doi:10.1042/CS20120461
16. Nemoto W, Ogata Y, Nakagawasai O, Yaoita F, Tadano T, Tan-No K. Angiotensin (1-7) prevents angiotensin II-induced nociceptive behaviour via inhibition of p38 MAPK phosphorylation mediated through spinal Mas receptors in mice. Eur J Pain. 2014;18:1471–1479. doi:10.1002/ejp.512
17. Forte BL, Slosky LM, Zhang H, et al. Angiotensin-(1-7)/Mas receptor as an antinociceptive agent in cancer-induced bone pain. Pain. 2016;157:2709–2721. doi:10.1097/j.pain.0000000000000690
18. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020;382:1653–1659. doi:10.1093/ehjcvp/pvaa138
19. Nemoto W, Yamagata R, Nakagawasai O, et al. Effect of spinal angiotensin-converting enzyme 2 activation on the formalin-induced nociceptive response in mice. Eur J Pharmacol. 2020;872:172950. doi:10.1016/j.ejphar.2020.172950
20. Nemoto W, Nakagawasai O, Yaoita F, et al. Angiotensin II produces nociceptive behavior through spinal AT1 receptor-mediated p38 mitogen-activated protein kinase activation in mice. Mol Pain. 2013;9:38. doi:10.1186/1744-8069-9-38
21. Nemoto W, Ogata Y, Nakagawasai O, Yaoita F, Tadano T, Tan-No K. Involvement of p38 MAPK activation mediated through AT1 receptors on spinal astrocytes and neurons in angiotensin II- and III-induced nociceptive behavior in mice. Neuropharmacology. 2015;99:221–231. doi:10.1016/j.neuropharm.2015.07.022
22. Su S, Cui H, Wang T, Shen X, Ma C. Pain: a potential new label of COVID-19. Brain Behav Immun. 2020;87:159–160. doi:10.1016/j.bbi.2020.05.025
23. Danser AH, Anand P. The angiotensin II type 2 receptor for pain control. Cell. 2014;157:1504–1506. doi:10.1016/j.cell.2014.05.030
24. Soda K, Nakada Y, Iwanari H, Hamakubo T. AT2 receptor interacting protein 1 (ATIP1) mediates COX-2 induction by an AT2 receptor agonist in endothelial cells. Biochem Biophys Rep. 2020;24:100850. doi:10.1016/j.bbrep.2020.100850
25. Moldofsky H, Patcai J. Chronic widespread musculoskeletal pain, fatigue, depression and disordered sleep in chronic post-SARS syndrome; a case-controlled study. BMC Neurol. 2011;11:37. doi:10.1186/1471-2377-11-37
26. Smith MT, Woodruff TM, Wyse BD, Muralidharan A, Walther T. A small molecule angiotensin II type 2 receptor (AT2R) antagonist produces analgesia in a rat model of neuropathic pain by inhibition of p38 mitogen-activated protein kinase (MAPK) and p44/p42 MAPK activation in the dorsal root ganglia. Pain Med. 2013;14:1557–1568. doi:10.1111/pme.12157
27. Kinet JP. The essential role of mast cells in orchestrating inflammation. Immunol Rev. 2007;217:5–7. doi:10.1111/j.1600-065X.2007.00528.x
28. Krames ES. The dorsal root ganglion in chronic pain and as a target for neuromodulation: a review. Neuromodulation. 2015;18:24–32. doi:10.1111/ner.12247
29. Peng J, Gu N, Zhou L, et al. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun. 2016;7:12029. doi:10.1038/ncomms12029
30. Yu X, Liu H, Hamel KA, et al. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat Commun. 2020;11:264. doi:10.1038/s41467-019-13839-2
31. Hayakawa K, Esposito E, Wang X, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–555. doi:10.1038/nature18928
32. Hu X, Leak RK, Shi Y, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. 2015;11:56–64. doi:10.1038/nrneurol.2014.207
33. Griffiths MR, Gasque P, Neal JW. The regulation of the CNS innate immune response is vital for the restoration of tissue homeostasis (repair) after acute brain injury: a brief review. Int J Inflam. 2010;. doi:10.4061/2010/151097
34. Hayakawa K, Wang X, Lo EH. CD200 increases alternatively activated macrophages through cAMP-response element binding protein - C/EBP-beta signaling. J Neurochem. 2016;136:900–906. doi:10.1111/jnc.13492
35. Hayakawa K;, Pham L-D-D, Seo JH, Miyamoto N, Maki T, Terasaki Y. CD200 restrains macrophage attack on oligodendrocyte precursors via toll-like receptor 4 downregulation. J Cerebr Blood Flow Metabol. 2016;36:781–793. doi:10.1177/0271678X15606148
36. Karnam G, Rygiel TP, Raaben M, et al. CD200 receptor controls sex-specific TLR7 responses to viral infection. PLoS Pathog. 2012;8:e1002710. doi:10.1371/journal.ppat.1002710
37. Lau SKP, Lau CCY, Chan KH, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel middle east respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol. 2013;94:2679–2690. doi:10.1177/0271678X15606148
38. Conti P, Ronconi G, Caraffa A, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. 2020;34:327–331. doi:10.23812/20-1-E
39. Li Y, Fu L, Gonzales DM, Lavi E. LIcorrelates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J Virol. 2004;78:3398–3406. doi:10.1128/jvi.78.7.3398-3406.2004
40. Bohmwald K, Gálvez NMS, Ríos M, Kalergis AM. Neurologic alterations due to respiratory virus infections. Front Cell Neurosci. 2018;12:386. doi:10.3389/fncel.2018.00386
41. Huang J, Zheng M, Tang X, Chen Y, Tong A, Zhou L. Potential of SARS-CoV-2 to cause CNS infection: biologic fundamental and clinical experience. Front Neurol. 2020;11:659. doi:10.3389/fneur.2020.00659
42. Logue JK, Franko NM, McCulloch DJ, et al. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw Open. 2021;4(2):e210830. doi:10.1001/jamanetworkopen.2021.0830
43. Wang X, Sahu KK, Cerny J. Coagulopathy, endothelial dysfunction, thrombotic microangiopathy and complement activation: potential role of complement system inhibition in COVID-19. J Thromb Thrombolysis. 2021;51(3):657–662. doi:10.1007/s11239-020-02297-z
44. Dani M, Dirksen A, Taraborrelli P, et al. Autonomic dysfunction in ‘long COVID’: rationale, physiology and management strategies. Clin Med. 2021;21(1):e63–e67. doi:10.7861/clinmed.2020-0896
45. Meyer-Frießem CH, Gierthmühlen J, Baron R, Sommer C, Üçeyler N, Enax-Krumova EK. Pain during and after COVID-19 in Germany and worldwide: a narrative review of current knowledge. Pain Rep. 2021;6(1):e893. doi:10.1097/PR9.0000000000000893
46. Lechien JR, Chiesa-Estomba CM, Place S, et al. Clinical and epidemiological characteristics of 1420 European patients with mild-to-moderate coronavirus disease 2019. J Intern Med. 2020;288:335–344. doi:10.1111/joim.13089
47. Lam DK, Dang D, Flynn AN, Hardt M, Schmidt BL. TMPRSS2, a novel membrane-anchored mediator in cancer pain. Pain. 2015;156(5):923–930. doi:10.1097/j.pain.0000000000000130
48. Minnelli N, Gibbs L, Larrivee J, Sahu KK. Challenges of maintaining optimal nutrition status in COVID-19 patients in intensive care settings. J Parenter Enteral Nutr. 2020;44(8):1439–1446. doi:10.1002/jpen.1996
49. Dhatt SS, Kumar V, Neradi D, Sodavarapu P, Meetei TT, Goni V. Need for testing and supplementation of vitamin D3 after release of COVID-19 lockdown in patients with increased musculoskeletal pain. Indian J Orthop. 2021;6:1–4. doi:10.1007/s43465-021-00376-8
50. Crispo A, Montagnese C, Perri F, et al. COVID-19 emergency and post-emergency in Italian cancer patients: how can patients be assisted? Front Oncol. 2020;10:1571. doi:10.3389/fonc.2020.01571
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.