")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Corosolic Acid Protects Rat Chondrocytes Against IL-1β-Induced ECM Degradation by Activating Autophagy via PI3K/AKT/mTOR Pathway and Ameliorates Rat Osteoarthritis
Authors Han H, Chen M, Li Z, Zhou S, Wu Y, Wei J
Received 6 March 2022
Accepted for publication 11 July 2022
Published 6 August 2022 Volume 2022:16 Pages 2627—2637
DOI https://doi.org/10.2147/DDDT.S365279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Hui Han,1– 3,* Ming Chen,2,* Zhenyu Li,2,* Siqi Zhou,2 Yingbin Wu,3 Jian Wei1
1Department of Sports Medicine and Joint Orthopedics, Liuzhou People’s Hospital, Liuzhou, Guangxi, People’s Republic of China; 2Department of Orthopedic Surgery, Zhongnan Hospital of Wuhan University, Wuhan, People’s Republic of China; 3Department of Orthopedic Surgery, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Wei, Department of Sports Medicine and Joint Orthopedics, Liuzhou People’s Hospital, Liuzhou, Guangxi, People’s Republic of China, Tel +86-13669663233, Email [email protected]
Purpose: Osteoarthritis (OA) is an age-related degenerative disease associated with enhanced degradation of extracellular matrix (ECM) and decreased autophagy. Our study is aimed to explore how corosolic acid (CRA) affect cartilage ECM metabolism and the potential mechanism.
Methods: Rat chondrocytes were pretreated with different concentrations of CRA (0, 2.5, 5, and 10 μM), and were stimulated with IL-1β (10ng/mL) for 24 h, subsequently. RT-qPCR, Western blot, and immunofluorescence were used to detect the expression of genes related to ECM metabolism and explore the potential molecular mechanism. The effect of CRA on articular cartilage was observed in the surgically induced OA rat model with the method of Safranin O/Fast green and immunohistochemical staining.
Results: Results showed that CRA reversed the IL-1β-induced degradation of aggrecan and type II collagen and the high expression of MMP13 and ADAMTS5. Mechanistically, CRA enhanced autophagy through inhibiting the classical PI3K/AKT/mTOR signaling pathway. Furthermore, inhibition of autophagy partly abolished the protective effects of CRA on ECM synthesis in IL-1β-treated chondrocytes. Correspondingly, the protective effect of CRA was also confirmed in a rat OA model.
Conclusion: Herein, we demonstrate that CRA can enhance autophagy by inhibiting PI3K/AKT/mTOR signaling pathway, prevent IL-1β-induced cartilage ECM degradation, and may be a potentially applicable candidate for the treatment of OA.
Keywords: osteoarthritis, corosolic acid, autophagy, extracellular matrix, PI3K/AKT/mTOR signaling
Introduction
Osteoarthritis (OA) is a common degenerative joint disease associated with joint pain and dysfunction, which can lead to disability in the elderly and bring substantial social burden.1,2 It is characterized by synovial inflammation, subchondral bone structure changes, and articular cartilage degeneration.3 IL-1β is thought to promote the occurrence and progression of OA via inducing apoptosis and inhibiting autophagy of chondrocyte.4,5 It also promotes the expression of inflammatory-related proteins such as INOS and COX-2 in chondrocytes, thereby causing cartilage extracellular matrix (ECM) degradation.6,7 The cartilage ECM is considered to be an important component for maintaining cartilage function and structure.8,9 Therefore, inhibiting IL-1β-induced ECM degradation may be an effective strategy to ameliorate OA progression. Although many drugs are used to treat OA in the clinic, none of them can effectively reverse the progression of OA. Besides, serious side effects limit their clinical use.10 Thus, it is imminent to discover a new and effective drug for OA treatment.
Autophagy is a catabolic process that degrades cellular components through lysosomal mechanisms to achieve cell homeostasis.11 LC3 and P62 are marker proteins that respond to autophagosome formation and degradation. LC3-II is a structural protein of the autophagosome. LC3/Atg8 is cleaved by Atg4 to generate LC3-I, which undergoes a ubiquitin-like reaction to generate LC3-II and attaches to the autophagosome membrane. P62 is an autophagy substrate protein. When lysosomes degrade, the P62 protein bound to the substrate is also hydrolyzed by proteolytic enzymes.12 In the development of OA, autophagy not only involves articular cartilage morphogenesis, but also plays an important role in ECM homeostasis.13,14 In the initial degeneration stage of articular cartilage, autophagy increases to protect chondrocytes from various environmental changes. Subsequently, with the gradual degradation of articular cartilage, autophagy decreases in chondrocytes.15 In addition, autophagy increased the expression of type II collagen (COL2A1) and aggrecan (ACAN) in IL-1β-treated chondrocytes, but decreased the levels of matrix metallopeptidase 13 (MMP13) and a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS5).16 The previous study has also shown that the loss of autophagy associated gene 5 (Atg5) in chondrocytes can lead to age-related osteoarthritis in mice.17 These studies suggest that activating chondrocyte autophagy can protect cells from stress and alleviate the progression of OA.
Corosolic acid (CRA) is a natural pentacyclic triterpenoid discovered in many medicinal herbs, especially Lagerstroemia speciosa. CRA initially attracted the attention of researchers due to its anti-diabetic function.18 However, some current studies have shown that it also has significant effects on anti-inflammatory, anti-tumor, anti-fibrosis, etc.19–22 Moreover, it has been reported that CRA is closely related to autophagy. It can improve mice’s myocardial hypertrophy and protect hepatocytes from ethanol-induced injury by regulating autophagy.23,24 However, its effect on chondrocyte autophagy and its potential therapeutic effect on OA remains unclear.
In this study, we explored the effects of CRA on chondrocyte autophagy and ECM metabolism and further analyzed its molecular mechanism. In addition, we evaluated the potential therapeutic effect of CRA in a rat OA model.
Materials and Methods
Reagents
CRA (purity ≥ 98%) was purchased from Mansite Biotechnology Co., Ltd., (Chengdu, China). Rat IL-1β and Cell Counting Kit-8 were purchased from Beyotime Institute of Biotechnology (Shanghai, China). 3-methyladenine (3-MA) was purchased from Meilun Biotechnology Co., Ltd., (Dalian, China). 740Y-P (PI3K activator) was purchased from MCE (Shanghai, China). The primary antibodies against LC3 and P62 were obtained from Sigma-Aldrich (St. Louis, MO, USA); antibodies against AKT, p-AKT, p-mTOR, and mTOR were purchased from Cell Signaling Technology (Danvers, MA, USA); antibodies against PI3K and p-PI3K was acquired from ABclonal Technology Co., Ltd. (Wuhan, China); antibodies against COL2A1, ACAN, ADAMTS5, and MMP13 were obtained from Proteintech (Wuhan, China). The TRIzol reagent kit was obtained from Invitrogen (Carlsbad, CA, USA). The mRNA reverse transcription and real-time RT-PCR kits were purchased from Vazyme Biotechnology Co., Ltd. (Nanjing, China).
Isolation, Culture, and Treatment of Chondrocytes
The articular cartilage was isolated from the femoral grooves and femoral condyles of 4-week-old Wistar rats. After shredded, the cartilage was fully digested with 0.3% collagenase II and then filtered by a cell screen to prepare a single-cell suspension. Finally, the cells were transferred to a culture flask and incubated with DMEM (containing 10% fetal bovine serum, 100 μg/mL streptomycin, and 100 U/mL penicillin) in a 5% CO2 incubator at 37°C. Cells from the second and third generations were used for subsequent experiments. For in vitro experiments, chondrocytes were either treated with 10 ng/mL IL-1β for 24 h or pretreated with CRA for 5 h and then co-treated with IL-1β (10 ng/mL) for 24 h. For autophagy studies, the chondrocytes were pretreated with 3-MA (5 mM) for 3 h before treatment with CRA or IL-1β. For signaling pathway analysis, the chondrocytes were pretreated with 740Y-P (30 μM) for 1 h before treatment with CRA or IL-1β.
Cell Viability Assay
CCK8 kit was used to detect cell viability. The chondrocytes were seeded into 96-well plates at a density of 5000 cells per well and incubated for 24 h. CCK-8 solution was added after CRA treatment and incubated at 37°C for 1 h in the dark. The absorbance at 450 nm was determined with a microplate reader.
Quantitative RT-PCR
Total RNA was extracted from rat chondrocytes using TRIzol reagent (Invitrogen, USA), and the RNA was reverse transcripted into cDNA using HiScript III RT SuperMix for qPCR (+ gDNA wiper) (Vazyme) according to the manufacturer’s instructions. Real-time PCR was carried out using AceQ Universal SYBR qPCR Master Mix (Vazyme). The gene expression was calculated in accordance with the 2 −ΔΔCt method. The results were analyzed after target gene expression was normalized to GAPDH expression. The sequences of the primers are listed in Table 1.
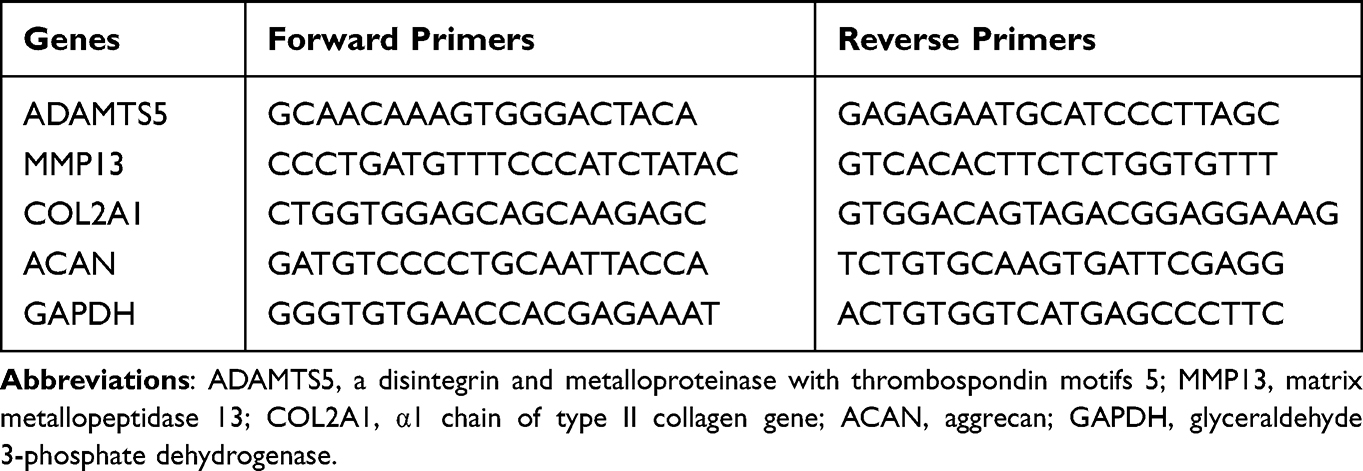 |
Table 1 Primers Used for the Real-Time Quantitative Polymerase Chain Reaction |
Western Blot
Total chondrocyte protein was extracted according to the kit instructions (Beyotime, Shanghai, China). Total 30 μg of proteins were loaded to each lane, isolated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membrane was blocked with 5% skim milk and incubated overnight with primary antibodies directed against COL2A1 (1:500), ACAN (1:500), MMP13 (1:500), ADAMTS5 (1:500), LC3B (1:1000), P62 (1:1000), PI3K (1:1000), P-PI3K (1:1000), AKT (1:1000), P-AKT (1:1000), mTOR (1:1000), P-mTOR (1:1000), GAPDH (1:5000) at 4°C. After washing with TBST, the membrane and HRP-conjugated secondary antibodies were incubated at room temperature for 1 h. The protein bands were examined with an enhanced ECL kit. Image J was used to measure the density of the membrane quantitatively.
Cellular Immunofluorescence Staining
Chondrocytes were seeded on the cover glass of the 6-well plate and treated with IL-1β or CRA. Then, they were washed with PBS, fixed in 4% paraformaldehyde, and permeated in 0.5% Triton X-100 for 15 minutes. Next, cells were blocked with 3% bovine serum albumin (BSA) and incubated with primary antibodies at 4°C overnight. On the second day, the cells were incubated with anti-rabbit CY3 and anti-rabbit FITC conjugated secondary antibody for 1 h. After labeling with DAPI, images were captured using Olympus fluorescence microscopy (Tokyo, Japan).
Transmission Electron Microscopy
After treating chondrocytes with IL-1β or CRA, the culture medium was removed and washed with PBS three times. Then, the cells were fixed with 2.5% glutaraldehyde in 0.1 M of phosphate buffer for 10 min and post-fixed with 1% osmium tetroxide/1.5% potassium ferrocyanide solution for 2 h. The chondrocytes were dehydrated through a graded series of ethanol and embedded in Epon 618. Finally, Epon 618 was embedded after ethanol grading dehydration. Ultrathin sections (50 nm) were cut with LKB-Vultra microtome (Bromma, Sweden), stained with uranyl acetate and lead citrate, and examined with a Hitachi H600 transmission electron microscope (Hitachi, Tokyo, Japan).
Animal Model of OA
Specific pathogen-free male Wistar rats [aged 8 weeks and weighing 170–230 g; license number: SCXK (Hubei), NO.2020-0018, n=10] were purchased from the Experimental Center of the Hubei Medical Scientific Academy. All the animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Animal Ethics Committee of Wuhan University (Licence number: 14016). After one week of adaptive feeding, these rats were randomly divided into three groups: Sham-operated group, OA group, and OA + CRA group. The control rats in Sham group underwent simply opening joint cavity surgery while the OA group underwent the transection of Anterior cruciate ligament (ACLT) to induce OA, as previously reported.25 Postoperatively, rats in OA + CRA group were given CRA (20 mg/kg/d) by gavage for 8 weeks. The remaining two groups were given the same amount of normal saline. The rats were sacrificed at postoperative week 8. The knee specimens of the right side were collected and fixed with 4% paraformaldehyde for 48 h for further analysis.
Histopathology and Immunohistochemical Staining
After a series of processing, the joint sections were stained with Safranin O/Fast green staining. The Osteoarthritis Research Society International (OARSI) guidelines were used to quantify the degeneration of cartilage.26
For immunohistochemical staining, samples were cut through the medial knee joints and 5 μm sections were used. Paraffin sections were dewaxed, rehydrated and antigen retrieval, then treated with EDTA antigen-repaired buffer. Sections were then blocked with 5% BSA at room temperature for 2 h, followed by overnight incubation at 4°C with primary antibodies against LC3B, COL2A1 and MMP13. Finally, the sections were visualized after interacting with HRP-conjugated secondary antibodies and DAB. All of the images were captured and then analyzed using a Nikon NIS Elements BR light microscope (Nikon, Tokyo, Japan).
Statistical Analysis
The data were analyzed by using GraphPad Prism7 software (GraphPad Software Inc.). All of the numerical results are presented as mean ± standard error of the mean (S.E.M.). Significant differences between control and treatment groups were identified using Student’s t-tests. The differences among more than two groups were determined using the one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant.
Results
Effect of CRA on Rat Chondrocytes Viability
To evaluate the toxicity of CRA (Figure 1A) to rat chondrocytes, we used CCK-8 kit to detect cell viability. Firstly, we examined the cytotoxicity of chondrocytes treated with CRA at different concentrations (0, 2.5, 5, 10, and 20 μM) for 24 h. As shown in Figure 1B, the survival rate of chondrocytes was significantly affected when the concentration of CRA increased to 20 μM. Moreover, we also detected the cytotoxicity of CRA at different time points (0, 12, 24, 36, and 48 h) at the concentration of 10 μM, and the results showed that there was no obvious cytotoxicity from the beginning time to the end time (Figure 1C). Thus, we chose 2.5–10 μM CRA for the subsequent experiments.
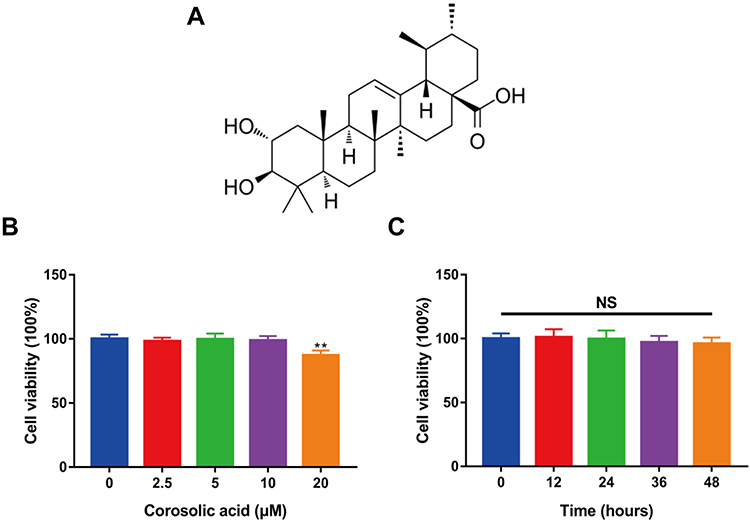 |
Figure 1 Effect of corosolic acid on rat chondrocytes viability. (A) Chemical structure of corosolic acid. (B) The chondrocytes were treated with different concentrations of corosolic acid (0, 2.5, 5, 10, and 20 μM) for 24 h, and the cell viability was detected through CCK-8 assay. (C) Chondrocytes were treated with 10 μM corosolic acid for 0, 12, 24, 36, and 48 h, and the cell viability was detected by CCK-8 assay. The data were expressed as Mean ± S.E.M., n=3, **P < 0.01 compared with control group. Abbreviation: NS, no significance. |
Effect of CRA on ECM Metabolism of Chondrocytes Treated with IL-1β
COL2A1 and ACAN were previously reported as primary components of ECM.27,28 MMP13 and ADAMTS5 are proteolytic enzymes that mediate ECM destruction during OA.29 To evaluate the effect of CRA on cartilage ECM metabolism, we treated rat chondrocytes with IL-1β alone or co-treated with different concentrations of CRA (0, 2.5, 5, and 10 μM). Finally, the expression of genes related to chondrocyte extracellular matrix metabolism (COL2A1, ACAN, MMP13, and ADAMTS5) was detected. We found that IL-1β could significantly reduce the mRNA expression of ECM synthesis-related genes COL2A1 and ACAN, and increase the mRNA expression of ECM degradation-related genes MMP13 and ADAMTS5. Nevertheless, the effects of IL-1β on the above genes were partially reversed in a concentration-dependent manner after treatment with different concentrations of CRA (Figure 2A). Then, we further detected the protein expression of the above genes by Western blot and immunofluorescence, and similar results were obtained (Figure 2B–E). When chondrocytes were treated with CRA alone, the mRNA expression levels of COL2A1 and MMP13 did not change significantly (Supplementary Figure 1A). In summary, CRA protects against ECM degradation in IL-1β-induced chondrocytes, but not in normal chondrocytes.
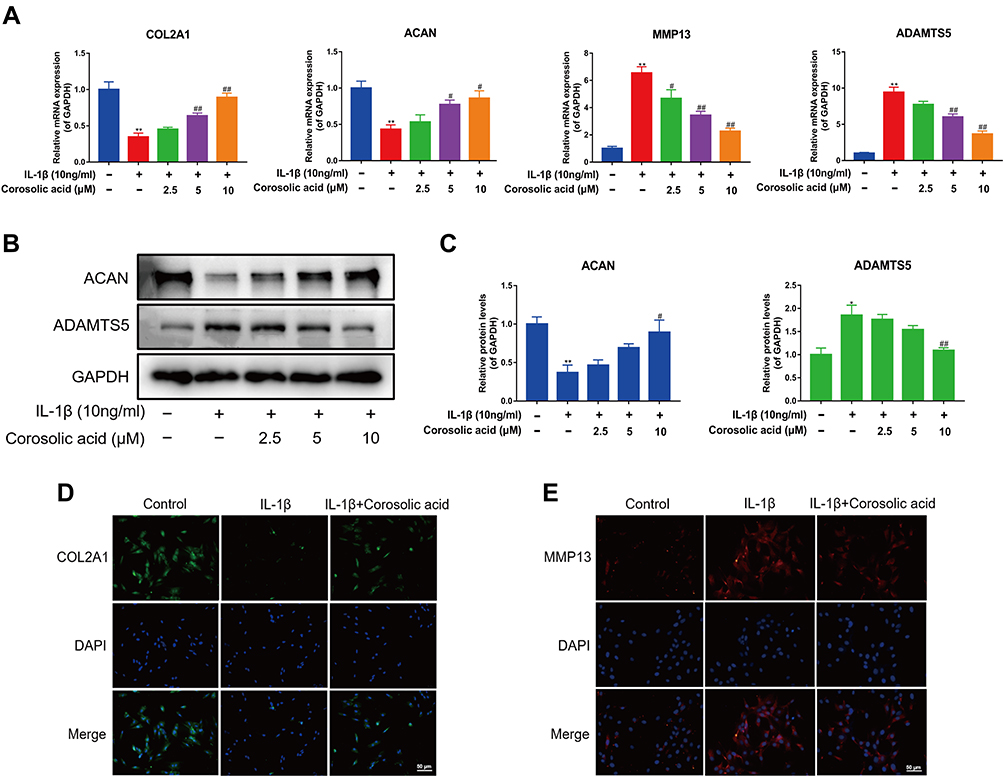 |
Figure 2 Effect of corosolic acid on ECM metabolism of chondrocytes treated with IL-1β. (A) RT-qPCR was used to detect the effect of corosolic acid on the mRNA expression of COL2A1, ACAN, MMP13, and ADAMTS5. (B) Western blot analysis of ACAN and ADAMTS5 protein expression level. (C) Quantitative analysis of ACAN and ADAMTS5 protein expression. (D and E) The protein expressions of COL2A1 and MMP13 were detected by immunofluorescence staining (scale bar = 50 μm). Mean ± S.E.M., n=3, **P < 0.01 compared with control group; #P<0.05 and ##P < 0.01 compared with IL-1β treated alone. Abbreviations: COL2A1, α1 chain of type II collagen gene; ACAN, aggrecan; MMP13, matrix metallopeptidase 13; ADAMTS5, a disintegrin and metalloproteinase with thrombospondin motifs 5; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
Effects of CRA on Autophagy of Chondrocytes Treated with IL-1β
Autophagy is often one of the important mechanisms for cells to protect themselves from damage and degeneration and maintain homeostasis.14,30 Therefore, we examined the protein expression of autophagy-related genes (LC3II/LC3I ratio and P62) to evaluate the effect of CRA on chondrocyte autophagy. Western blot analysis showed that CRA increased LC3II/LC3I ratio and decreased P62 expression in a concentration-dependent manner (Figure 3A). The data from the transmission electron microscope revealed that CRA treatment increased the number of autophagosomes in chondrocytes compared to the treatment of IL-1β alone (Figure 3B). When we treated CRA alone, the expression level of autophagy-related proteins did not change significantly (Supplementary Figure 1B and C). These findings indicated that CRA exclusively enhanced autophagy of OA chondrocytes, but for the normal chondrocytes.
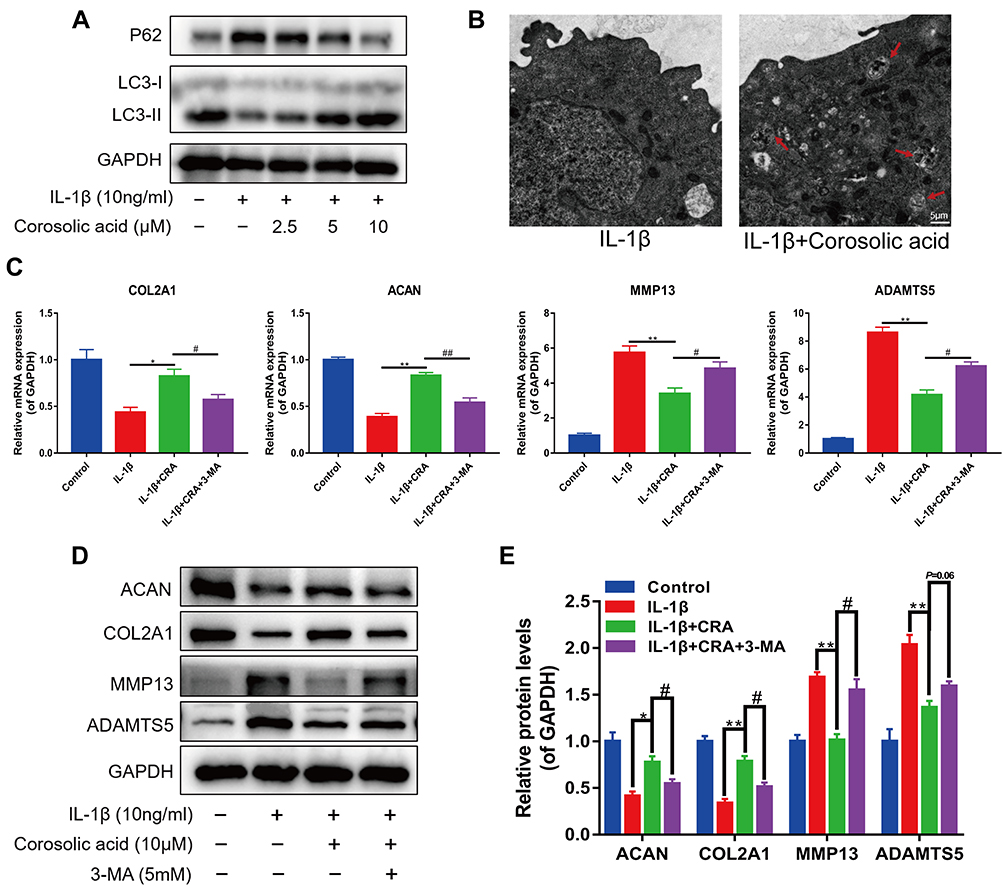 |
Figure 3 Corosolic acid participates in the regulation of cartilage ECM metabolism by enhancing autophagy. (A) Western blot analysis of autophagy-related genes LC3-II/I and P62 protein expression level. (B) Autophagosomes in chondrocytes were observed by using transmission electron microscope. The red arrowheads indicate autophagosomes. (scale bar = 5 μm). (C) RT-qPCR was used to detect the mRNA expression of COL2A1, ACAN, MMP13, and ADAMTS5 after administering the autophagy inhibitor 3-MA. (D and E) Western blot was performed to analyze the protein level of COL2A1, ACAN, MMP13, and ADAMTS5 after administering the autophagy inhibitor 3-MA and quantitative analysis. Mean ± S.E.M., n=3, *P<0.05, **P<0.01 compared with respective controls; #P<0.05, ##P < 0.01 compared with the appropriate controls. Abbreviations: CRA, Corosolic acid; 3-MA, 3-methyladenine; COL2A1, α1 chain of type II collagen gene; ACAN, aggrecan; MMP13, matrix metallopeptidase 13; ADAMTS5, a disintegrin and metalloproteinase with thrombospondin motifs 5; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
The activation of autophagy may affect the anabolism and catabolism of ECM.11 To further confirm the role of CRA-induced autophagy in chondrocyte ECM metabolism, we detected the expression of genes related to ECM metabolism in the presence of an autophagy inhibitor 3-MA. We found that CRA increased the mRNA expressions of COL2A1 and ACAN, and decreased the mRNA expressions of MMP13 and ADAMTS5 compared to the treatment of IL-1β alone. In contrast, 3-MA treatment partially eliminated the effect of CRA on the expression of these genes (Figure 3C). Then, the protein expressions of COL2A1, ACAN MMP13, and ADAMTS5 were detected by Western blot, and similar results were obtained (Figure 3D and E). These results suggested that CRA could inhibit cartilage ECM degradation by enhancing autophagy.
Effects of CRA on PI3K/AKT/mTOR Signaling Pathway in IL-1β-Treated Chondrocytes
Previous studies have shown that PI3K/AKT/mTOR signaling pathway plays an important role in regulating cartilage ECM metabolism and autophagy.31,32 Therefore, the activation of PI3K, AKT, and mTOR was examined by Western blot assay. As shown in Figure 4A and B, IL-1β significantly activated PI3K, AKT, and mTOR, while the increased phosphorylation levels of PI3K, AKT, and mTOR were inhibited with the pretreatment with different concentrations of CRA. The above data suggested that CRA could affect the PI3K/AKT/mTOR signaling pathway in IL-1β-induced OA chondrocytes. To further assess the role of the PI3K/AKT/mTOR pathway in the anti-ECM degradation of CRA, we administered 740Y-P (a PI3K agonist) treatment. Our results demonstrated that CRA’s enhanced autophagy and anti-ECM degradation effects were reversed by 740Y-P (Figure 4C and D). Taken together, our findings demonstrated that CRA could enhance autophagy by inhibiting the PI3K/AKT/mTOR signaling pathway and then play a regulatory role in cartilage ECM metabolism.
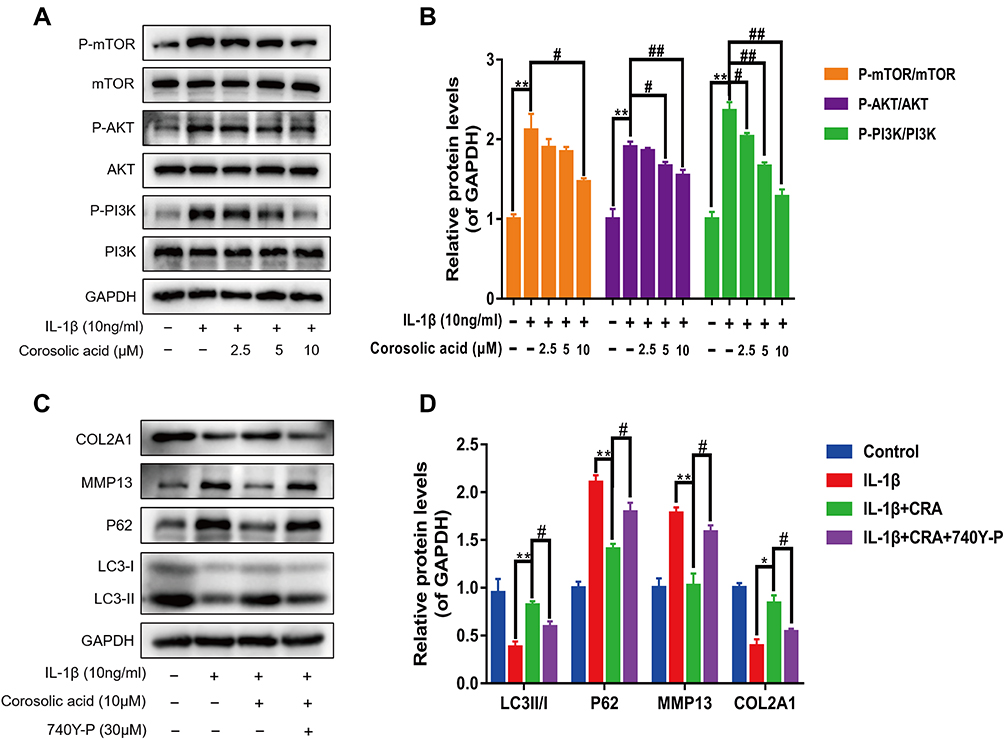 |
Figure 4 Corosolic acid inhibits the activation of PI3K/AKT/mTOR signaling. (A and B) The protein levels of PI3K, P-PI3K, AKT, P-AKT, mTOR, and P-mTOR were determined by Western blot and quantitative analysis. (C and D) The protein levels of LC3, P62, MMP13, and COL2A1 were detected by Western blot after 740Y-P (a PI3K agonist) treatment for 24 h and quantitative analysis. Mean ± S.E.M., n=3, *P<0.05, **P<0.01 compared with respective controls; #P<0.05, ##P < 0.01 compared with the appropriate controls. |
Effects of CRA on OA Progression and Autophagy in ACLT Rat Model
Since the in vitro data revealed a protective effect of CRA on the ECM metabolism associated with OA pathology, a surgically induced ACLT model was established to verify whether CRA protects against the development of OA in vivo. The pathological changes of articular cartilage were detected by Safranin O staining. We found that pathological changes such as cartilage erosion, ECM loss, and superficial cartilage destruction were reduced in the CRA-treated group compared with the OA group (Figure 5A). In addition, the CRA group also showed a lower OARSI score (Figure 5B). Immunohistochemical results showed that CRA treatment could up-regulate the expression of COL2A1 and down-regulate the expression of MMP13 in articular cartilage. At the same time, it also increased the level of autophagy-related gene LC3B (Figure 5C and D). These results suggested that CRA can delay the progression of OA and enhance autophagy in vivo.
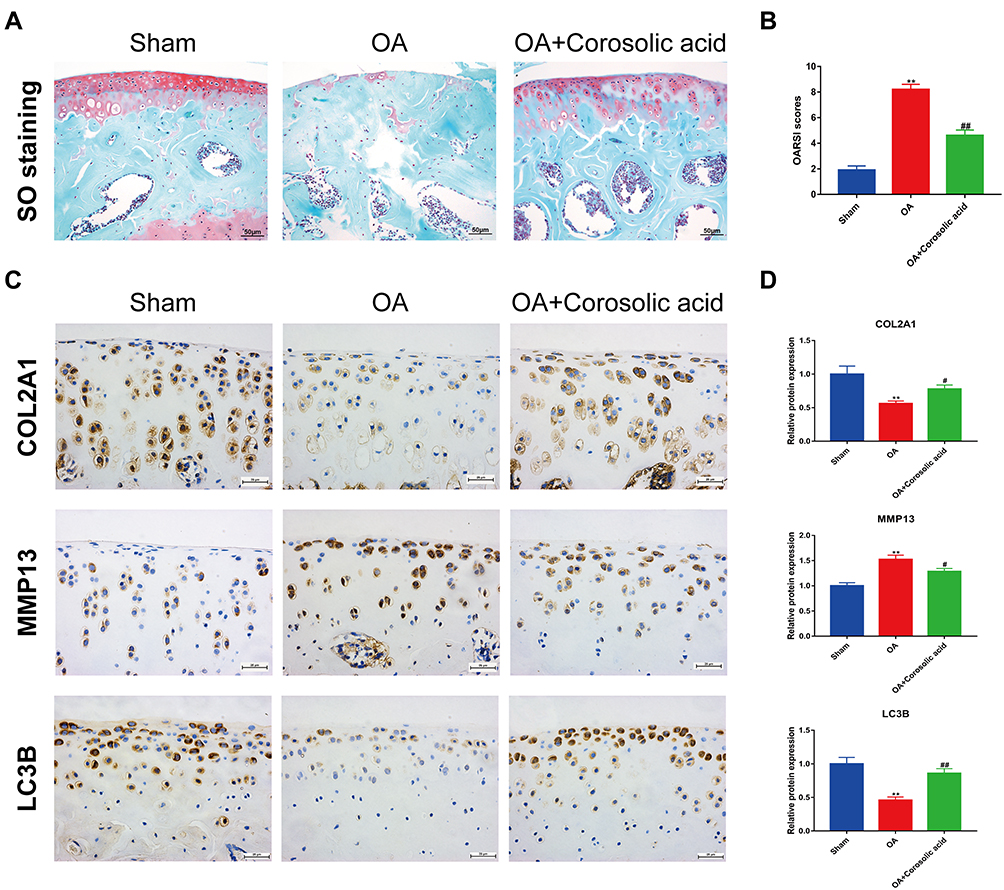 |
Figure 5 Corosolic acid retarded OA progression and enhanced autophagy in vivo. (A) Pathological changes of articular cartilage were detected by safranin O staining (scale bar = 50 μm). (B) Degree of joint damage was evaluated using the Osteoarthritis Research Society International (OARSI) scores. (C) The protein expressions of COL2A1, MMP13, and LC3B in joint tissues were detected by immunohistochemistry staining (scale bar = 25 μm). (D) Quantitative analysis was made on the protein expressions of COL2A1, MMP13, and LC3B in joint tissues. Mean ± S.E.M., n=8 per group, **P < 0.01 represents the comparison with the Sham group; #P < 0.05, ##P < 0.01 represent the comparison with OA group. |
Discussion
OA, dominated by articular cartilage degeneration and dysfunction, is one of the most important global causes of disability.3,33 ECM is the main component in cartilage, and its metabolic abnormality is one of the critical factors leading to the occurrence and progression of OA.8 Besides, many studies have shown that the promotion of cartilage ECM anabolism and inhibition of catabolism can effectively prevent the occurrence and slow down the development of OA.9,34,35 CRA, isolated from Lagerstroemia speciosa, shows anti-inflammatory, anti-tumor, and anti-fibrosis biological activities.19–22 In this study, we investigated the effect of CRA on the ECM metabolism of OA chondrocytes induced by IL-1β and explored its potential mechanism. Meanwhile, we also found that treatment with CRA reduced joint degeneration in a rat OA model in vivo.
In view of the key role of ECM in OA progress, we first studied the effect of CRA on the ECM metabolism of OA chondrocytes. The IL-1β-induced chondrocyte inflammatory response of rat chondrocytes was applied to simulate the inflammatory injury of OA in vivo. Consistent with expectations, the use of IL-1β significantly increased the expression of chondrocyte matrix-degrading enzymes and decreased ECM synthesis. However, pretreatment with different concentrations of CRA could partially reverse the ECM damage induced by IL-1β. When we treated chondrocytes with CRA alone, there was no significant effect on extracellular matrix metabolism. These results indicated that CRA could promote the anabolism of OA chondrocytes and inhibit their catabolism, but had no significant effect on normal chondrocytes.
Autophagy is a cell homeostasis mechanism in various pathological events.36 The weakening of autophagy is also one of the main characteristics of cartilage degeneration caused by aging. Due to cartilage degeneration and reduced autophagy, the adaptability of chondrocytes to the external environment is significantly reduced, which can eventually lead to cell damage and metabolic abnormalities.37,38 Previous studies have shown that CRA has a close relationship with autophagy. CRA not only antagonize pressure overload-induced cardiac hypertrophy through enhancing autophagy, but also protect hepatocytes from ethanol-induced damage via activating autophagy.23,24 Therefore, we further studied the effect of CRA on chondrocyte autophagy. Our data showed that IL-1β significantly decreased the autophagy flux marker LC3-II/LC3-I ratio and increased the expression of P62, indicating that IL-1β played a negative role in the regulation of chondrocyte autophagy, which was consistent with the results of previous studies.39,40 However, the administration of CRA could partly reverse the IL-1β–induced autophagy downregulation. Chondrocyte autophagy was not affected when CRA was administered alone. These suggested that CRA could only enhance autophagy in the presence of IL-1β. Besides, the autophagy inhibitor 3-MA can partially block the beneficial effects of CRA on cartilage ECM synthesis. Taken together, our results suggested that CRA exerted a protective effect on ECM by enhancing OA chondrocyte autophagy.
The PI3K/AKT/mTOR signaling pathway plays a crucial role in regulating cell proliferation, apoptosis, metabolism, differentiation, and cell cycle.41–43 At the same time, this signaling pathway is also an important regulator of chondrocyte autophagy flux. The promoting effects of its silencing on autophagy have been widely reported as an effective strategy to alleviate OA progression.31,44,45 Therefore, we further explored whether the enhancement of chondrocyte autophagy caused by CRA was related to the inhibition of PI3K/AKT/mTOR signaling pathway. We found that CRA can decrease IL-1β-activated phosphorylation levels of PI3K, AKT, and mTOR in a concentration-dependent manner. Besides, the PI3K activator 740Y-P can partially reverse the effects of CRA on autophagy and ECM metabolism. These indicated that CRA could enhance autophagy and inhibit ECM degradation by inhibiting PI3K/AKT/mTOR signaling pathway.
We further studied the effects of CRA on articular cartilage in a rat OA model. In this study, we established the OA model by ACLT, which is a classic model of OA.25 The results of Safranin O‐fast green staining showed that CRA could alleviate the progression of OA and reduce the OARSI histological score. At the same time, we also observed that CRA could significantly up-regulate the expression of COL2A1 and autophagy-related gene LC3-II, and reduce the expression of MMP13 in articular cartilage. In brief, all these data suggested that CRA could enhance autophagy and inhibit cartilage ECM degradation and had a potential application value in OA treatment.
Conclusions
In summary, our results indicated that CRA could enhance autophagy and alleviate IL-1β -induced cartilage ECM degradation by inhibiting PI3K/AKT/mTOR signaling pathway. Our in vivo experiments also confirmed the therapeutic effect of CRA on OA. This study provides a potential therapeutic strategy for OA, but it still needs further research before its clinical application.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kraus VB, Karsdal MA. Osteoarthritis: current molecular biomarkers and the way forward. Calcif Tissue Int. 2021;109(3):329–338. doi:10.1007/s00223-020-00701-7
2. van den Bosch MHJ. Osteoarthritis year in review 2020: biology. Osteoarthritis Cartilage. 2021;29(2):143–150. doi:10.1016/j.joca.2020.10.006
3. Abramoff B, Caldera FE. Osteoarthritis: pathology, diagnosis, and treatment options. Med Clin North Am. 2020;104(2):293–311. doi:10.1016/j.mcna.2019.10.007
4. Wang BW, Jiang Y, Yao ZL, Chen PS, Yu B, Wang SN. Aucubin protects chondrocytes against IL-1beta-induced apoptosis in vitro and inhibits osteoarthritis in mice model. Drug Des Devel Ther. 2019;13:3529–3538. doi:10.2147/DDDT.S210220
5. Lin Z, Miao J, Zhang T, et al. d-Mannose suppresses osteoarthritis development in vivo and delays IL-1beta-induced degeneration in vitro by enhancing autophagy activated via the AMPK pathway. Biomed Pharmacother. 2021;135:111199. doi:10.1016/j.biopha.2020.111199
6. Wang M, Shen J, Jin H, Im HJ, Sandy J, Chen D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann N Y Acad Sci. 2011;1240:61–69. doi:10.1111/j.1749-6632.2011.06258.x
7. Jin J, Lv X, Wang B, et al. Limonin inhibits IL-1beta-induced inflammation and catabolism in chondrocytes and ameliorates osteoarthritis by activating Nrf2. Oxid Med Cell Longev. 2021;2021:7292512. doi:10.1155/2021/7292512
8. Rahmati M, Nalesso G, Mobasheri A, Mozafari M. Aging and osteoarthritis: central role of the extracellular matrix. Ageing Res Rev. 2017;40:20–30. doi:10.1016/j.arr.2017.07.004
9. Shi Y, Hu X, Cheng J, et al. A small molecule promotes cartilage extracellular matrix generation and inhibits osteoarthritis development. Nat Commun. 2019;10(1):1914. doi:10.1038/s41467-019-09839-x
10. Taruc-Uy RL, Lynch SA. Diagnosis and treatment of osteoarthritis. Prim Care. 2013;40(4):821–836. doi:10.1016/j.pop.2013.08.003
11. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16(8):461–472. doi:10.1038/nrm4024
12. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. doi:10.1002/path.2697
13. Carames B, Olmer M, Kiosses WB, Lotz MK. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015;67(6):1568–1576. doi:10.1002/art.39073
14. Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801. doi:10.1002/art.27305
15. Li YS, Zhang FJ, Zeng C, et al. Autophagy in osteoarthritis. Joint Bone Spine. 2016;83(2):143–148. doi:10.1016/j.jbspin.2015.06.009
16. Sasaki H, Takayama K, Matsushita T, et al. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64(6):1920–1928. doi:10.1002/art.34323
17. Bouderlique T, Vuppalapati KK, Newton PT, Li L, Barenius B, Chagin AS. Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann Rheum Dis. 2016;75(3):627–631. doi:10.1136/annrheumdis-2015-207742
18. Miura T, Takagi S, Ishida T. Management of diabetes and its complications with Banaba (Lagerstroemia speciosa L.) and corosolic acid. Evid Based Complement Alternat Med. 2012;2012:871495. doi:10.1155/2012/871495
19. Yang J, Leng J, Li JJ, et al. Corosolic acid inhibits adipose tissue inflammation and ameliorates insulin resistance via AMPK activation in high-fat fed mice. Phytomedicine. 2016;23(2):181–190. doi:10.1016/j.phymed.2015.12.018
20. Jia M, Xiong Y, Li M, Mao Q. Corosolic acid inhibits cancer progress through inactivating YAP in hepatocellular carcinoma. Oncol Res. 2020;28(4):371–383. doi:10.3727/096504020X15853075736554
21. Wang ZP, Che Y, Zhou H, et al. Corosolic acid attenuates cardiac fibrosis following myocardial infarction in mice. Int J Mol Med. 2020;45(5):1425–1435. doi:10.3892/ijmm.2020.4531
22. Zhao J, Zhou H, An Y, Shen K, Yu L. Biological effects of corosolic acid as an anti-inflammatory, anti-metabolic syndrome and anti-neoplasic natural compound. Oncol Lett. 2021;21(2):84. doi:10.3892/ol.2020.12345
23. Wang ZP, Shen D, Che Y, et al. Corosolic acid ameliorates cardiac hypertrophy via regulating autophagy. Biosci Rep. 2019;39(12). doi:10.1042/BSR20191860
24. Guo X, Cui R, Zhao J, Mo R, Peng L, Yan M. Corosolic acid protects hepatocytes against ethanol-induced damage by modulating mitogen-activated protein kinases and activating autophagy. Eur J Pharmacol. 2016;791:578–588. doi:10.1016/j.ejphar.2016.09.031
25. Cope PJ, Ourradi K, Li Y, Sharif M. Models of osteoarthritis: the good, the bad and the promising. Osteoarthritis Cartilage. 2019;27(2):230–239. doi:10.1016/j.joca.2018.09.016
26. Gerwin N, Bendele AM, Glasson S, Carlson CS. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rat. Osteoarthritis Cartilage. 2010;18(Suppl 3):S24–34. doi:10.1016/j.joca.2010.05.030
27. Poole AR, Kobayashi M, Yasuda T, et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann Rheum Dis. 2002;61(Suppl 2):ii78–81. doi:10.1136/ard.61.suppl_2.ii78
28. Roughley PJ, Mort JS. The role of aggrecan in normal and osteoarthritic cartilage. J Exp Orthop. 2014;1(1):8. doi:10.1186/s40634-014-0008-7
29. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529–543. doi:10.2741/1817
30. Kim KH, Lee MS. Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10(6):322–337. doi:10.1038/nrendo.2014.35
31. Xue JF, Shi ZM, Zou J, Li XL. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed Pharmacother. 2017;89:1252–1261. doi:10.1016/j.biopha.2017.01.130
32. Xu K, He Y, Moqbel SAA, Zhou X, Wu L, Bao J. SIRT3 ameliorates osteoarthritis via regulating chondrocyte autophagy and apoptosis through the PI3K/Akt/mTOR pathway. Int J Biol Macromol. 2021;175:351–360. doi:10.1016/j.ijbiomac.2021.02.029
33. Hiligsmann M, Cooper C, Arden N, et al. Health economics in the field of osteoarthritis: an expert’s consensus paper from the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Semin Arthritis Rheum. 2013;43(3):303–313. doi:10.1016/j.semarthrit.2013.07.003
34. Wang Y, Yu D, Liu Z, et al. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res Ther. 2017;8(1):189. doi:10.1186/s13287-017-0632-0
35. Li Z, Cheng J, Liu J. Baicalin protects human OA chondrocytes against IL-1beta-induced apoptosis and ECM degradation by activating autophagy via miR-766-3p/AIFM1 axis. Drug Des Devel Ther. 2020;14:2645–2655. doi:10.2147/DDDT.S255823
36. Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. doi:10.1007/978-3-642-00302-8_3
37. Luo P, Gao F, Niu D, et al. The role of autophagy in chondrocyte metabolism and osteoarthritis: a comprehensive research review. Biomed Res Int. 2019;2019:5171602. doi:10.1155/2019/5171602
38. Castrogiovanni P, Ravalli S, Musumeci G. Apoptosis and autophagy in the pathogenesis of osteoarthritis. J Invest Surg. 2020;33(9):874–875. doi:10.1080/08941939.2019.1576811
39. Zhou X, Li J, Zhou Y, et al. Down-regulated ciRS-7/up-regulated miR-7 axis aggravated cartilage degradation and autophagy defection by PI3K/AKT/mTOR activation mediated by IL-17A in osteoarthritis. Aging. 2020;12(20):20163–20183. doi:10.18632/aging.103731
40. Chen X, Wang Y, Qu N, Zhang B, Xia C. PLCgamma1 inhibition-driven autophagy of IL-1beta-treated chondrocyte confers cartilage protection against osteoarthritis, involving AMPK, Erk and Akt. J Cell Mol Med. 2021;25(3):1531–1545. doi:10.1111/jcmm.16245
41. Marquard FE, Jucker M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem Pharmacol. 2020;172:113729. doi:10.1016/j.bcp.2019.113729
42. Malemud CJ. The PI3K/Akt/PTEN/mTOR pathway: a fruitful target for inducing cell death in rheumatoid arthritis? Future Med Chem. 2015;7(9):1137–1147. doi:10.4155/fmc.15.55
43. Xu F, Na L, Li Y, Chen L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020;10:54. doi:10.1186/s13578-020-00416-0
44. Cai C, Min S, Yan B, et al. MiR-27a promotes the autophagy and apoptosis of IL-1beta treated-articular chondrocytes in osteoarthritis through PI3K/AKT/mTOR signaling. Aging. 2019;11(16):6371–6384. doi:10.18632/aging.102194
45. Sun W, Li Y, Wei S. miR-4262 regulates chondrocyte viability, apoptosis, autophagy by targeting SIRT1 and activating PI3K/AKT/mTOR signaling pathway in rats with osteoarthritis. Exp Ther Med. 2018;15(1):1119–1128. doi:10.3892/etm.2017.5444
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.