")
Back to Journals » International Journal of Nanomedicine » Volume 12
Corneal permeation properties of a charged lipid nanoparticle carrier containing dexamethasone
Authors Ban J , Zhang Y, Huang X, Deng GH, Hou DZ, Chen YZ, Lu ZF
Received 31 October 2016
Accepted for publication 9 January 2017
Published 16 February 2017 Volume 2017:12 Pages 1329—1339
DOI https://doi.org/10.2147/IJN.S126199
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Junfeng Ban, Yan Zhang, Xin Huang, Guanghan Deng, Dongzhi Hou, Yanzhong Chen, Zhufen Lu
Guangdong Provincial Key Laboratory of Advanced Drug Delivery Systems, Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China
Abstract: Drug delivery carriers can maintain effective therapeutic concentrations in the eye. To this end, we developed lipid nanoparticles (L/NPs) in which the surface was modified with positively charged chitosan, which engaged in hydrogen bonding with the phospholipid membrane. We evaluated in vitro corneal permeability and release characteristics, ocular irritation, and drug dynamics of modified and unmodified L/NPs in aqueous humor. The size of L/NPs was uniform and showed a narrow distribution. Corneal permeation was altered by the presence of chitosan and was dependent on particle size; the apparent permeability coefficient of dexamethasone increased by 2.7 and 1.8 times for chitosan-modified and unmodified L/NPs, respectively. In conclusion, a chitosan-modified system could be a promising method for increasing the ocular bioavailability of unmodified L/NPs by enhancing their retention time and permeation into the cornea. These findings provide a theoretical basis for the development of effective drug delivery systems in the treatment of ocular disease.
Keywords: ocular drug delivery system, local bioavailability, dexamethasone, eye drop administration
Introduction
A major challenge in the treatment of the ocular posterior segment in fundus disease is that effective therapeutic concentrations of a drug delivered through the cornea cannot be maintained in the posterior segment of the eye. In addition, low bioavailability and adverse drug reactions occur with the absorption of high drug doses via systemic circulation. Three methods are used for drug delivery to the posterior segment and fundus in clinical settings (Figure 1).1–7 The first method has been applied to the treatment of posterior segment disease and involves injection or implantation of the drug into the vitreous humor via the flat part of the ciliary body. The second method targets the subconjunctival fascia by implantation or injection, with the drug eventually becoming concentrated in the choroid, retinal pigment epithelium, retina, etc. The third approach involves increasing the drug concentration in the posterior segment of the eye by ocular iontophoresis and insertion of microneedles via the cornea or sclera. Although these methods can achieve high drug concentrations in the posterior segment of the vitreous humor, lens, retina, and other ocular tissues, their broad clinical application is limited by potential complications such as tissue damage, infection and injury, and retinal hemorrhage.8–11 Drug administration into the anterior segment has been proposed to circumvent these problems. In intraocular administration, drug molecules must diffuse over a long distance from the site of corneal penetration and penetrate the ocular barrier – which consists of the convection flow of the aqueous humor from the ciliary body to Schlemm’s canal, together with lacrimal fluid, blood-aqueous humor, retinal blood, and a corneal avascular component – and be specifically absorbed by the retinal pigment epithelium. Increasing effective drug concentrations in the eye can enhance drug absorption in the posterior segment and fundus.
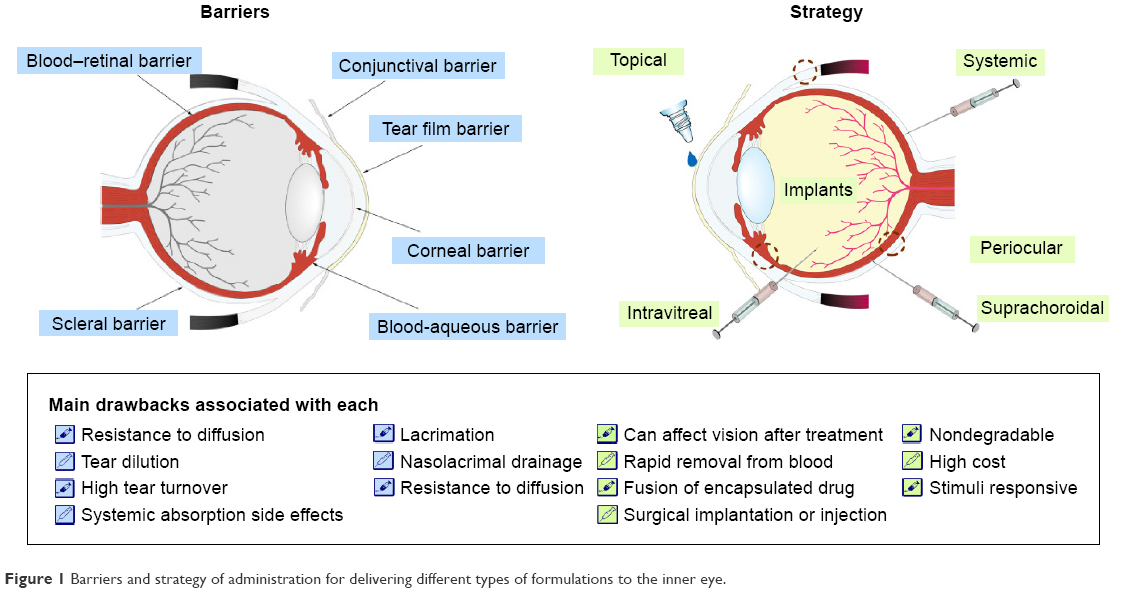 | Figure 1 Barriers and strategy of administration for delivering different types of formulations to the inner eye. |
Studies on prolonging precorneal retention and increasing contact time with the ocular surface have focused on sub-microemulsion drug delivery systems, which can transform molecules into liquid crystals in the eye;12 this was shown to increase precorneal retention time by up to 1.42-fold as compared to direct delivery of the solution. In addition, this method has been shown to achieve drug concentrations of up to 8.64 and 15.95 ng/mL in the cornea after 0.5 and 4 h, respectively.13 Ethosomes, a type of carrier molecule, have many advantages for the treatment of eye irritation, including high stability; however, very few types are available for clinical application, and high concentrations can be toxic.14,15 Cyclodextrin has been widely studied as a carrier in ophthalmic drug delivery systems and has been shown to prolong ocular retention time and increase drug solubility and bioavailability, thereby improving the drug’s effectiveness.11,16,17 Eye drops consisting of methazolamide in cyclodextrin can also reduce intraocular pressure, and are released over 6 h. The combination of acetazolamide, cyclodextrin, and triethanolamine was shown to increase permeation through the rabbit cornea by over ten-fold and to reduce intraocular pressure by >30%.18 For diseases requiring long-term drug administration such as those of the retina and vitreous body, a dexamethasone/cyclodextrin mixture applied to the ocular surface increased retinal penetration two-fold relative to systemic administration.19
In situ gels are a novel type of ophthalmic drug delivery system with a longer retention time than that of existing approaches, and they reduce administration frequency and improve bioavailability. In vivo studies have shown that a temperature-sensitive chitosan hydrogel administered as ocular drops had high penetration and sustained release over 12 h while reducing intraocular pressure.20 Although it was shown that particles in the nanometer range have longer corneal retention time, there are still no drug delivery systems that adequately target the posterior segment.21,22
Recently, drug carriers have been developed that combine two or more molecules, although effective delivery to the posterior segment by anterior corneal administration remains problematic.23–27 Lipid nanoparticles (L/NPs) can be used to encapsulate and deliver poorly soluble drugs and can enable greater drug permeation into tissues via lipid membrane fusion. We hypothesized that combining L/NPs with the water-soluble cationic polysaccharide chitosan – which is biodegradable, adhesive, and biocompatible and has superior penetration – can improve L/NP drug loading and release (Figure 2).
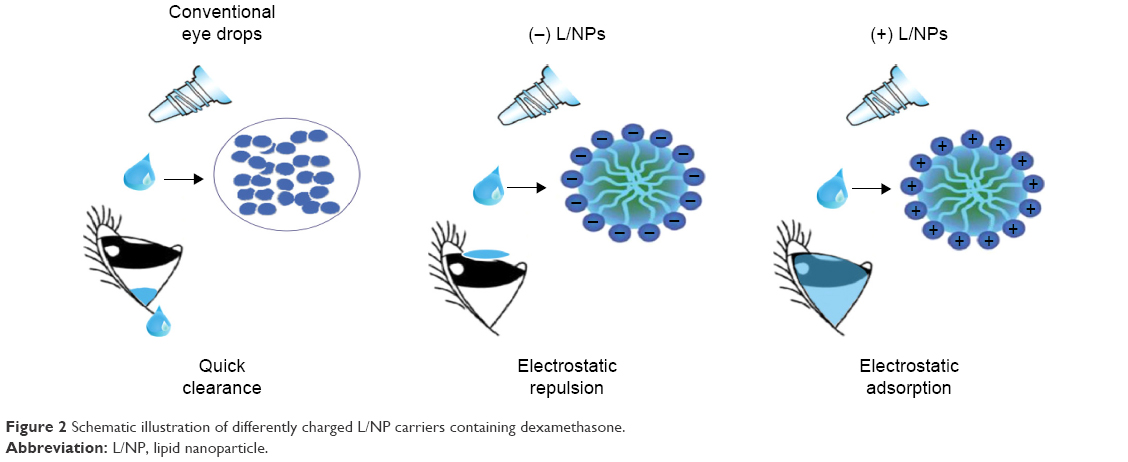 | Figure 2 Schematic illustration of differently charged L/NP carriers containing dexamethasone. |
In a previous study, we reported the potential of NPs as a novel system for delivering ocular drugs to the posterior segment of the eye, including the aqueous humor and vitreous fluid in the eyes. Briefly, fluorescence emission of coumarin-6 formulated into NPs was detected in the posterior segment of rabbit eyes after eyedrop administration.7 The rigidity of NPs is an important factor in considering drug delivery efficiency. Several factors would affect the properties in lipid drug delivery systems: particle rigidity, particles surface characteristics, components of the particles system, particle sizes, and zeta potential. Crucial findings may be obtained by investigating the relationship among these factors and drug delivery efficiency to the ocular posterior segment.
The main goal of this study was to develop a lipid drug delivery system for dexamethasone and evaluate its potential for drop delivery to the ocular posterior segment. For this purpose, L/NPs containing various agents were prepared by a film-dispersion high-pressure homogenization method. In vitro evaluation of L/NPs and the pharmacokinetics in rabbit were investigated. The effect of L/NPs on rabbit cornea was assessed, and preliminary studies on the pharmacokinetics of drop administration were performed.
Materials and methods
Materials
Soy and egg yolk lecithins were purchased from Advanced Vehicle Technology Pharmaceutical Co., Ltd. (Shanghai, China). Soybean oil and Pluronic F68 were gifts from Yipusheng Pharmaceutical Co. (Jiangxi, China) and BASF (Ludwigshafen, Germany), respectively. Dexamethasone was supplied by Tianjin Tianyao Pharmaceutical Co., Ltd. (Tianjin, China). Bitter capric acid triglyceride and medium-chain triglycerides were provided by Fengli Jingqiu Pharmaceutical Co., Ltd. (Beijing, China). All other chemicals and reagents were commercially available and of reagent grade.
Preparation of L/NPs
L/NPs were prepared using a film-dispersion high-pressure homogenization system.28,29 Briefly, soy lecithin and dexamethasone were dissolved in soybean oil under low heat, followed by organic solvent dissolution and film dispersion. Glycerol and Pluronic F68 were dissolved in deionized water as the aqueous phase. To achieve a coarse emulsion system, the two phases were separately heated to 45°C and then emulsified using an Ultra-Turrax T25 high-shear mixer (IKA, Guangzhou, China) at a speed of 14,000 rpm for 10 min. The preparation was cooled to room temperature; pH was adjusted to 6.5–7.8, and the desired volume was obtained by adding purified water. L/NPs were homogenized into a fine monodispersed emulsion with a high-pressure homogenizer (NS 1001L; NiroSoavi Co., Parma, Italy) at 500–600 bar for 10 min.
Charge modification of (+) L/NPs was carried out by mixing unmodified (−) L/NPs with specific concentrations of chitosan. After the functional polymer was added to the aqueous phase, the solution containing charge-modified (+) L/NPs was adjusted to an appropriate final volume. The solution was homogenized for 15 min; the final chitosan concentration was 1%.
Particle size and zeta potential analysis
Mean particle size and size distribution of L/NPs were measured by laser diffraction (Delsa Nano C Particle Size and Zeta Potential Analyzer; Beckman Coulter, Brea, CA, USA). Briefly, 1 mg/mL dexamethasone (±) L/NPs were prepared in distilled water at room temperature before size measurements, which were performed in triplicate following the dilution of L/NPs at 25°C. The zeta potential was determined with the same instrument under the same conditions.
Analysis of L/NP morphology by transmission electron microscopy (TEM)
L/NP morphology was visualized by TEM (JEM-1400; JEOL, Tokyo, Japan). Samples were prepared using a negative staining method at room temperature by placing a drop of each formulation onto a copper grid coated with carbon film and staining with 2% phosphotungstic acid solution.30
Analysis of drug-loading efficiency and capacity
Free drug encapsulated in L/NPs was separated by size exclusion. Briefly, Sephadex G-50 medium was loaded into a 2 mL syringe, which was centrifuged at 2,500 rpm for 2 min to obtain a dehydrated column. L/NPs (0.2 mL) were applied to the column, followed by centrifugation at 2,500 rpm for 2 min. Deionized water (0.2 mL) was added to the column as an eluent, followed by centrifugation at 2,500 rpm for 2 min. This step was repeated thrice, and resultant L/NPs were dissolved in acetonitrile and passed through a 0.22 μm membrane filter.
The solution was diluted with an appropriate amount of mobile phase, and L/NPs were homogenized to determine their total content. The solution was analyzed by high-performance liquid chromatography (HPLC) using a C18 column (5 μm, 25 cm ×4.6 mm; Kromasil, Bohus, Sweden) with a mobile phase consisting of an acetonitrile/water mixture (28:72). The flow rate was 1.0 mL/min at room temperature, and the encapsulation efficiency of L/NPs was calculated with the following equation:
|
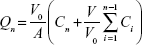
|
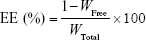
where WTotal and WFree represent the weights of total and nonencapsulated drug, respectively; and EE represents the drug encapsulation efficiency and DL, the drug loading.
In vitro evaluation of corneal penetration
New Zealand albino rabbits (Laboratory Animal Service Center, Southern Medical University, Guangzhou) were randomly divided into three groups (dexamethasone/[+] L/NPs, dexamethasone/[−] L/NPs, and dexamethasone only; n=3 each). The penetration-enhancing effect of L/NPs in aqueous solution (dexamethasone concentration =1 mg/mL) was evaluated in isolated rabbit corneas (available area =0.5024 cm2) using a Franz diffusion apparatus (Shanghai Kai Kai Technology Trade Co. Ltd., China). Briefly, 1 mL of each sample and 6 mL Ringer’s solution were placed on the epithelial (donor) and endothelial (receptor) sides of the cornea, respectively, with the system maintained at 34°C. A 1 mL volume of sample was drawn from the epithelial compartment after 0.25, 0.5, 1, 1.5, 3, 4, 5, and 6 h and immediately replaced with an equal volume of preheated Ringer’s solution. Each sample was prepared in triplicate. The amount of drug in the sample was determined by the HPLC method as described in the “Analysis of drug-loading efficiency and capacity” section. Cumulative penetration per area (Qn, μg/cm2) at different intervals was used to evaluate the efficiency of penetration according to the following formula:
|
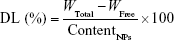
where Cn is the drug concentration in the endothelial compartment at different times (mg/mL), Ci is the drug concentration in samples at the final time point (mg/mL), V0 is the volume of the endothelial compartment (6 mL), V is the sample volume (1 mL), and A is the effective area of permeation (0.5024 cm2).
We also determined the apparent permeability coefficient (Papp, cm/min) and steady-state flux (JSS, μg/mL), where ΔQ/Δt is the slope of the straight-line portion of the Qn-t plot and A is the effective area of permeation (0.5024 cm2).31
In vitro drug release test
In vitro drug release was evaluated using the dynamic dialysis bag method.32 Briefly, 2 mL each of the (±) L/NP formulations was separately loaded into dialysis bags immersed in 40 mL freshly prepared Ringer’s solution at 34°C. The magnetic stirring speed was 100 rpm. At predetermined time points (2, 4, 6, 8, 10, 12, 24, and 36 h), 1 mL of each sample was withdrawn and immediately replaced with an equal volume of fresh Ringer’s solution. The amount of drug released by L/NPs was determined by the HPLC method as described in the “Analysis of drug-loading efficiency and capacity” section.
Evaluation of eye irritancy
All animal protocols complied with the Guide for the Care and Use of Laboratory Animals and Institute of Laboratory Animal Resources and were approved by the Institutional Animal Care and Use Committee of Guangdong Pharmaceutical University. Each rabbit was treated with 0.1 mL of L/NP formulation, which was applied to the lower conjunctival sac of the right eye, while the left eye was left untreated as a control. The ocular tissue was examined thrice daily for 7 days. The degree of eye irritation was scored using the modified Draize test.33 Rabbits were euthanized by air embolism, and eye tissue was fixed with 4% formaldehyde and embedded in paraffin for histopathological assessment.
Pharmacokinetics of L/NPs in aqueous humor
Adult New Zealand albino rabbits (n=6, weighing 2.0–3.0 kg) were used for the analysis of pharmacokinetics in the aqueous humor. A single dose (0.1 mL) of L/NPs (1 mg/mL) was applied to the lower conjunctival sac of one eye of each rabbit immediately after preparation. At predetermined time points (0.5, 1, 3, 6, 9, 18, and 24 h after L/NP application), 20 μL samples of aqueous humor were collected using a syringe fitted with a 26 G needle. Samples were mixed with 80 μL of HPLC mobile phase and centrifuged at 6,000 rpm for 30 min. A 60 μL volume of the supernatant was used for HPLC analysis as described in the “Analysis of drug-loading efficiency and capacity” section.
Statistical analysis
Data are expressed as mean ± SD. Differences between groups were assessed by analysis of variance and the independent samples test using SPSS software (SAS Institute, Cary, NC, USA). P<0.05 was considered statistically significant.
Results and discussion
Preparation and characterization of L/NPs
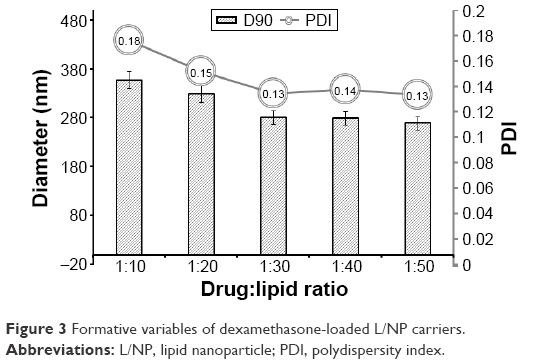
Particles with sizes on a nanometer scale not only show high solubility in water but also prolong the time that a drug remains in the ocular circulation.34–36 Various strategies such as emulsion solvent evaporation, solvent dispersion, and nano-crystallization have been used to improve the bioavailability of poorly soluble drugs. Lipids, including phospholipids, can serve as drug carriers in eye drops owing to their excellent capacity for encapsulation, safety, and biocompatibility.37–40 In this study, we prepared dexamethasone L/NPs with different surface properties by high-pressure homogenization. L/NP size has an important effect on the dispersibility of ophthalmic formulations. The particle size of D90 (the cumulative particle size distribution number reaching 90%) decreased from 356.7 to 269 nm when the drug/lipid ratio increased from 1:10 to 1:50 (Figure 3, D90), indicating that NP size is affected by lipid content. There was also a slight improvement in the stability of the system with increased drug/lipid ratio (Figure 3, polydispersity index [PDI]); we therefore prepared particles <200 nm in size by increasing the pressure in the homogenizer to achieve a drug/lipid ratio of 1:30.
 | Figure 3 Formative variables of dexamethasone-loaded L/NP carriers. |
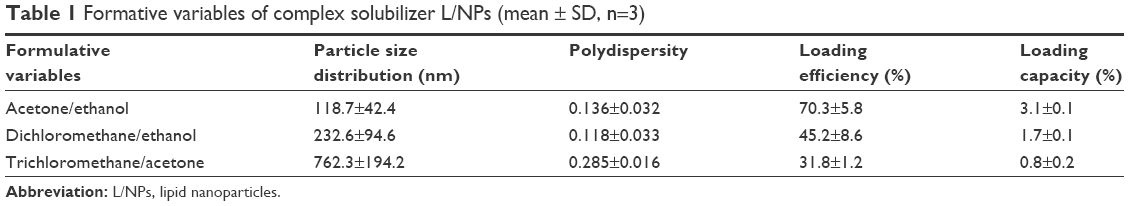
A core structure was formed by dexamethasone and phospholipids in a solution of acetone and ethanol within a soybean oil shell, which spontaneously generated isotropic and thermodynamically stable nano-droplets with an aqueous phase containing Pluronic F68. Unmodified (−) L/NPs were modified with a surface charge by adding the biocompatible cationic material, chitosan. In addition, as a result of the combination of positively charged amino acids in chitosan molecules and the negative charge interaction between hydrophilic hydroxyl and L/NPs, a lipid tear film was formed after eye drop administration (Figure 2).40,41 Consequently, the spreading coefficient changed as a result of contact between the drug and cornea, minimizing the loss of drug in the anterior segment and increasing intraocular bioavailability. Particle size, encapsulation efficiency, and drug loading of L/NPs were affected by solvent composition (Table 1), which could be related to hydrocarbon chain length of volatile organic solvents in the hydration process. In the dichloromethane/acetone system, hydration time was longer, and a large amount of drug leaked from the lipid carrier, resulting in reduced entrapment efficiency.42,43
 | Table 1 Formative variables of complex solubilizer L/NPs (mean ± SD, n=3) |
Permeability is an important feature of L/NPs; particle size and surface charge affect intraocular penetration.44,45 Membrane permeability decreased with increases in particle size, and particles <400 nm showed greater penetration through polarized epithelial cells. The range of particle sizes was 118.2±87.5 to 180.1±74.5 nm.
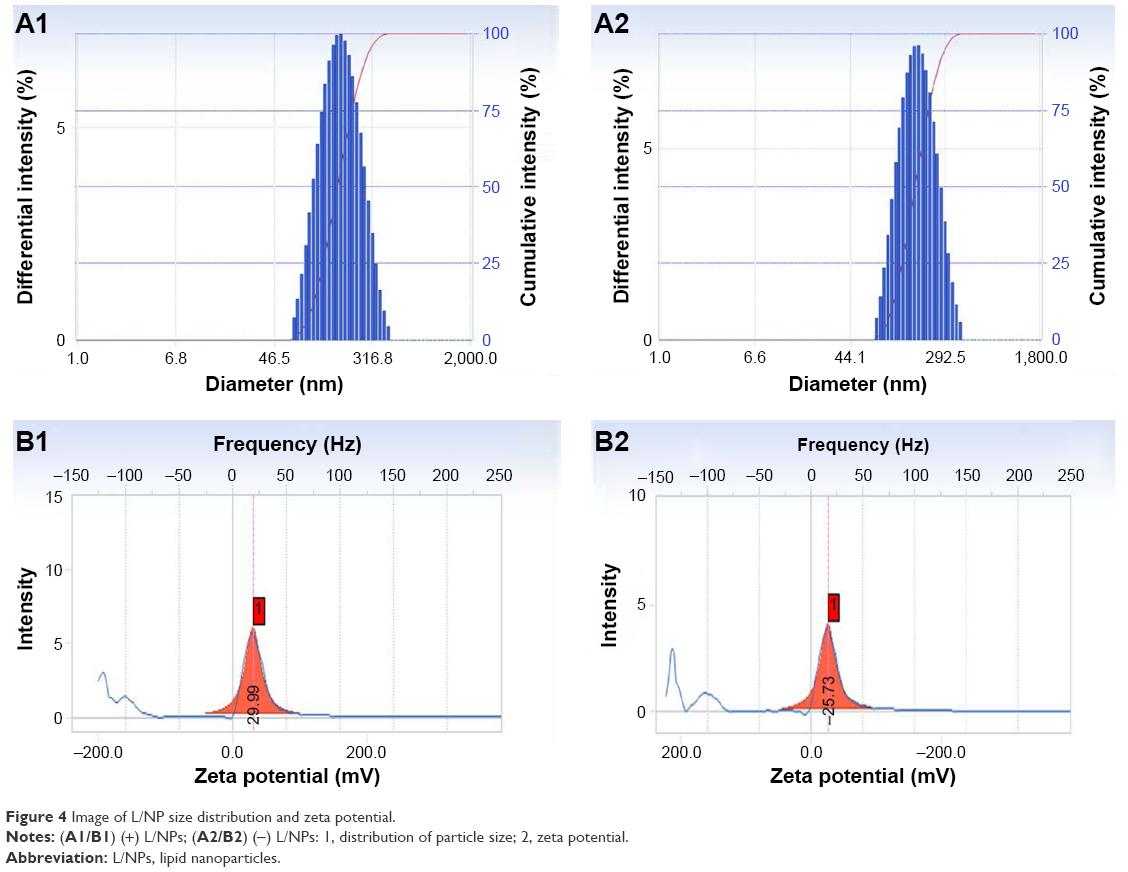
We evaluated the physicochemical properties of (±) L/NPs (Table 2). The average droplet size differed between (−) and (+) L/NPs. The zeta potential was higher for (+) than for (−) L/NPs (Figure 4). L/NPs exhibited cationic characteristics after chitosan modification, which contributed a positive charge to the particle surface, promoting the retention of particles at the corneal surface and stabilizing the system. TEM analysis revealed that the L/NPs had a spherical structure with a smooth surface; the chitosan coating formed a shell around the core but did not alter particle shape (Figure 5).
 | Table 2 Formative variables of L/NPs (mean ± SD, n=3) |
 | Figure 4 Image of L/NP size distribution and zeta potential. |
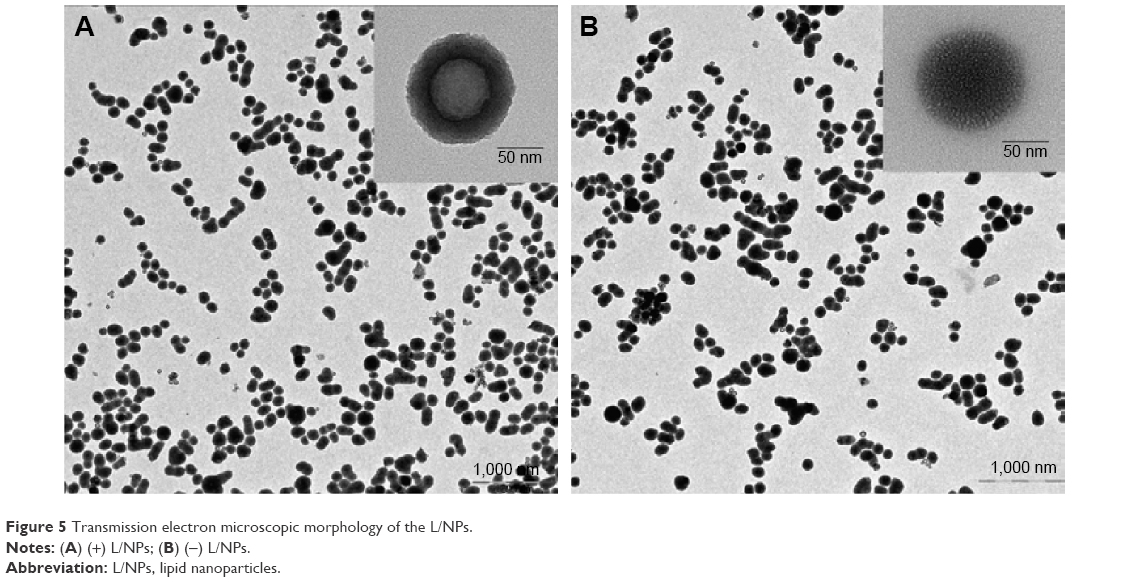 | Figure 5 Transmission electron microscopic morphology of the L/NPs. |
The Jss and Papp obtained for L/NPs and the aqueous solution of dexamethasone are shown in Table 3. Corneal hydration levels in all three samples were within the normal range, 75%–83%; the pH range of the preparations was 6.3–7.4. Since corneal hydration levels were mostly below 80%, the corneal damage appeared to be reversible. These findings demonstrate that L/NPs are safe for use in ocular treatment. Dexamethasone L/NPs and aqueous solution showed good permeation and in vitro bioavailability over a 6 h period. Infiltration of L/NPs modified with chitosan was higher than that of L/NPs without modification, with the respective Papp values being 2.7- and 1.8-fold higher than that of the water solution. This might be attributable to the integration of (+) L/NPs into tear films as a result of electrostatic adsorption between positively and negatively charged L/NPs. Although it improved corneal permeability of the drug, chitosan also prevented the drug from interacting with the intraocular protein, thereby preventing enzyme damage and improving drug bioavailability in the eye.46–48
 | Table 3 Transcorneal permeation parameters of different formulations (mean ± SD, n=3) |
Analysis of in vitro drug release by L/NPs
Methods for investigating in vitro drug release include dialysis, reverse dialysis, and ultrafiltration. There are some caveats for interpreting in vitro drug release kinetics of L/NPs by dialysis. For instance, L/NPs are not fully diluted by the release medium, which is therefore not comparable to intraocular tear composition. The major factor affecting drug release is the concentration gradient between the filter bag and release medium rather than the internal and external phases of the droplet, giving a false appearance of sustained release. In this study, leakage slot conditions were established by reverse osmosis, which can better simulate local application of eye drops such that L/NPs would be diluted and contact a large volume of medium. It also avoided the disadvantage of reduced accuracy of samples that cannot be separated quickly by ultracentrifugation.
To investigate the release characteristics and surface charge of dexamethasone (±) L/NPs, release data were fitted with first-order, Higuchi, Niebergull, Hixson–Crowell, Ritger–Peppas, and Weibull equations. The most suitable equation was determined according to the correlation coefficient R2 and Akaike information criterion. Changes in particle size and area induced by spontaneous formation of tiny droplets during drug release deviated from the first-order kinetic model of drug release behavior in aqueous solution (Table 4). Approximately 50% of L/NPs were released in 36 h, indicating good bioavailability.
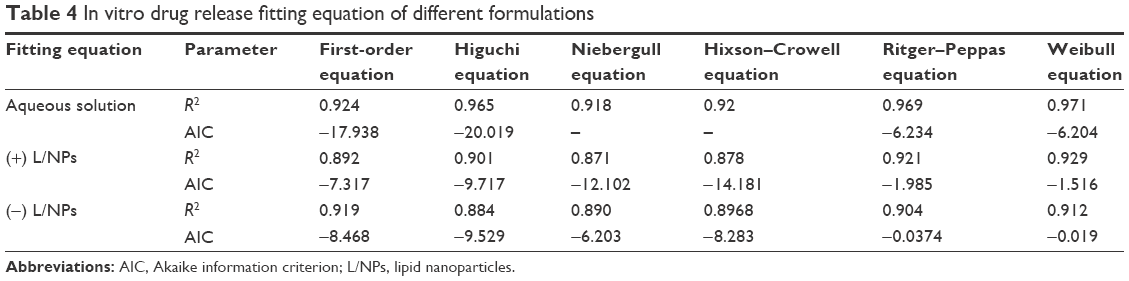 | Table 4 In vitro drug release fitting equation of different formulations |
In vivo assessment of L/NP safety with the eye irritation test
Given that L/NPs are novel drug carriers intended for use as an ocular drug delivery system, it is important to assay their tolerability in living organisms. Histopathological analysis by microscopy was used to assess cell structure and tissue integrity to detect irritation. Representative micrographs of three tissues (cornea, iris, and sclera) treated with different preparations are shown in Figure 6.
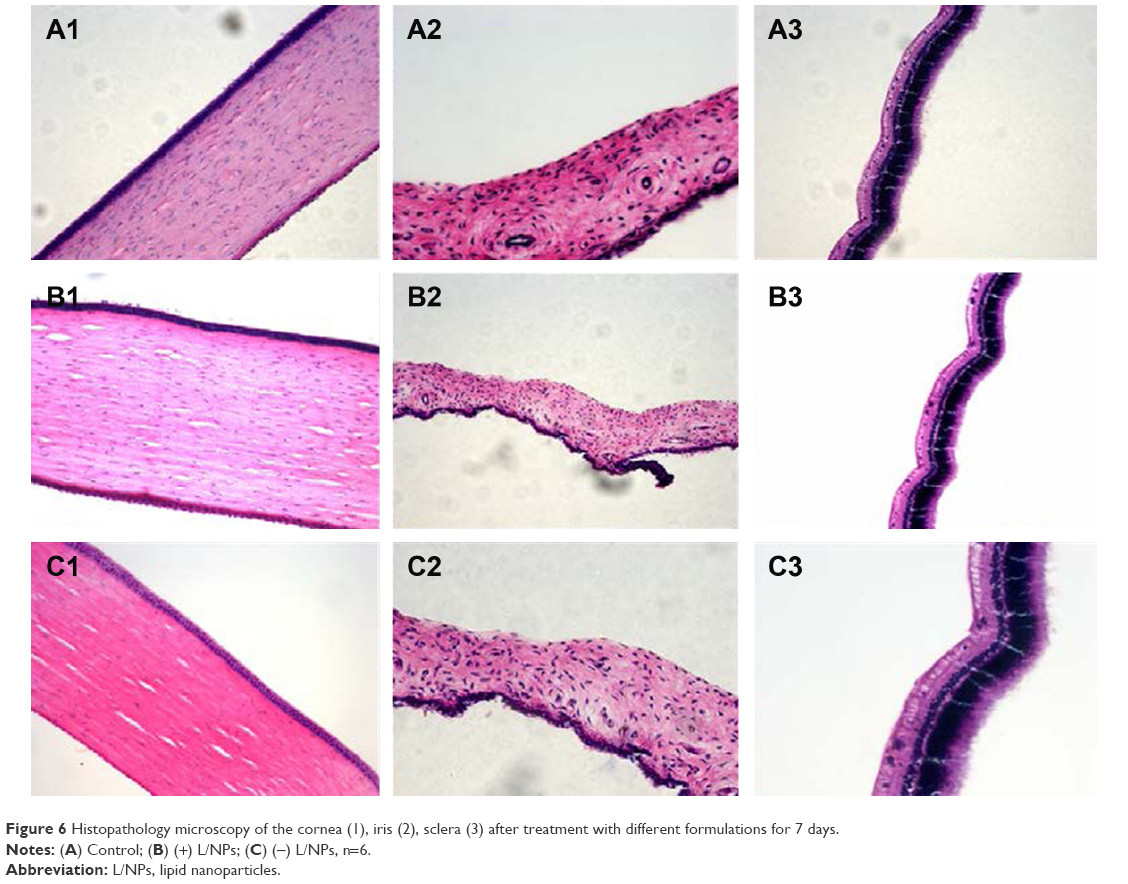 | Figure 6 Histopathology microscopy of the cornea (1), iris (2), sclera (3) after treatment with different formulations for 7 days. |
In control specimens treated with Ringer’s solution (A1–A3), there were no differences in the appearance of the various tissues. Some loose corneal stroma cells, conjunctival epithelial cells with altered appearance, and an increase in the number of inflammatory cells or lymphocytes at the sclera were observed in specimens treated with dexamethasone solution. However, the presence of L/NPs in the (+) L/NPs (B1–B3) and (−) L/NPs (C1–C3) groups had no effect on tissue morphology. These results indicate that L/NPs have good ocular biocompatibility and can in fact reduce the mild tissue damage caused by dexamethasone aqueous solution.
In vivo pharmacokinetics of L/NPs in aqueous humor
L/NPs formulations applied to rabbits’ eyes were well tolerated and showed no evidence of causing ocular inflammation or irritation. Dexamethasone concentrations measured in aqueous humor collected after 0.25, 0.5, 1, 3, 5, 6, 8, 12, and 24 h are shown in Figure 7, and pharmacokinetic parameters calculated by the DAS 3.0 software package (Chinese Pharmacological Society, Shanghai, China) are shown in Table 5. In animals treated with dexamethasone aqueous solution, dexamethasone was detected in the aqueous humor over the 8 h duration of the study period, after which it was no longer detectable, indicating rapid elimination of the drug. In contrast, large quantities of the drug were present in aqueous humor samples for up to 24 h following the topical administration of dexamethasone containing (−) or (+) L/NPs to rabbits’ eyes. Both L/NPs exhibited higher dexamethasone bioavailability (P<0.05) as compared to the aqueous solution (4.69- and 2.12-fold higher, respectively), while the difference between them was negligible. The respective Tmax values of dexamethasone from (+) and (−) L/NPs were 12- and 10-fold higher, whereas the respective Cmax values were 2.37- and 1.17-fold higher, respectively, than the values for the aqueous solution. The calculated Cmax values indicated that there was no difference between the (+) L/NP formulations and the aqueous solution.
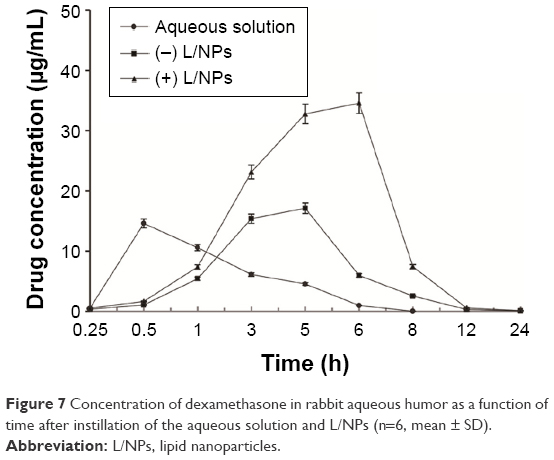 | Figure 7 Concentration of dexamethasone in rabbit aqueous humor as a function of time after instillation of the aqueous solution and L/NPs (n=6, mean ± SD). |
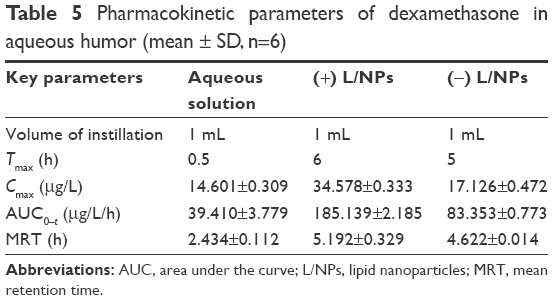 | Table 5 Pharmacokinetic parameters of dexamethasone in aqueous humor (mean ± SD, n=6) |
Conclusion
L/NP-based drug delivery systems can alter the dynamics of intraocular transport. For the delivery of dexamethasone – a drug used in the treatment of ocular fundus diseases such as macular disease – chitosan modification of L/NPs yielded a more sustained and controlled release and showed superior biocompatibility as compared to unmodified L/NPs. Dexamethasone or other drugs in conjunction with a chitosan-modified L/NP carrier can be prepared in the form of eye drops when long-term treatment is required, such as for cataracts and diabetic eye diseases. This system ensures that the drug can achieve therapeutically effective concentrations at a lower dosage in a shorter time, which can in turn improve patient compliance.
Acknowledgments
This work was supported by the R&D Team for Formulation Innovation (grant no: 2015CXQX150); Innovation and Strong School Project of Guangdong Pharmaceutical University (grant no: 2015KQNCX077), and National Natural Science Foundation of China (grant no: 51102052).
Author contributions
All the authors contributed toward data analysis and drafting and revising the article and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Loftssona T, Jarvinen T. Cyclodextrins in ophthalmic drug delivery. Adv Drug Deliv Rev. 1999;36(1):59–79. | ||
Ribeiro AM, Figueiras A, Veiga F. Improvements in topical ocular drug delivery systems: hydrogels and contact lenses. J Pharm Pharm Sci. 2015;18(5):683–695. | ||
Vandamme TF. Microemulsions as ocular drug delivery systems: recent developments and future challenges. Prog Retin Eye Res. 2002;21(1):15–34. | ||
Fialho SL, Da Silva-Cunha A. New vehicle based on a microemulsion for topical ocular administration of dexamethasone. Clin Exp Ophthalmol. 2004;32(6):626–632. | ||
Kaur IP, Smitha R. Penetration enhancers and ocular bioadhesives: two new avenues for ophthalmic drug delivery. Drug Dev Ind Pharm. 2002;28(4):353–369. | ||
Short BG. Safety evaluation of ocular drug delivery formulations: techniques and practical considerations. Toxicol Pathol. 2008;36(1):49–62. | ||
Guanghan D, Junfeng B, Yan Z, et al. Preparation and suitability evaluation of coumarin-6 nanoprobes for ophthalmic use. Chin J Pharm. 2016;47(03):305–309. | ||
Janoria KG, Gunda S, Boddu SH, et al. Novel approaches to retinal drug delivery. Expert Opin Drug Deliv. 2007;4(4):371–388. | ||
Araújo J, Nikolic S, Egea MA, Souto EB, Garcia ML. Nanostructured lipid carriers for triamcinolone acetonide delivery to the posterior segment of the eye. Colloids Surf B Biointerfaces. 2011;88(1):150–157. | ||
Loftsson T, Hreinsdottir D, Stefansson E. Cyclodextrin microparticles for drug delivery to the posterior segment of the eye: aqueous dexamethasone eye drops. J Pharm Pharmacol. 2007;59(5):629–635. | ||
Tanito M, Hara K, Takai Y, et al. Topical dexamethasone-cyclodextrin microparticle eye drops for diabetic macular edema. Invest Ophthalmol Vis Sci. 2011;52(11):7944–7948. | ||
Chan J, Maghraby GM, Craig JP, Alany RG. Phase transition water-in-oil microemulsions as ocular drug delivery systems: in vitro and in vivo evaluation. Int J Pharm. 2007;328(1):65–71. | ||
Baspinar Y, Bertelmann E, Pleyer U, Buech G, Siebenbrodt I, Borchert HH. Corneal permeation studies of everolimus microemulsion. J Ocul Pharmacol Ther. 2008;24(4):399–402. | ||
Raza K, Kumar M, Kumar P, et al. Topical delivery of aceclofenac: challenges and promises of novel drug delivery systems. Biomed Res Int. 2014;2014:406731. | ||
Kumar L, Verma S, Bhardwaj A, Vaidya S, Vaidya B. Eradication of superficial fungal infections by conventional and novel approaches: a comprehensive review. Artif Cells Nanomed Biotechnol. 2014;42(1):32–46. | ||
Saari KM, Nelimarkka L, Ahola V, et al. Comparison of topical 0.7% dexamethasone cyclodextrin with 0.1% dexamethasone sodium phosphate for postcataract inflammation. Graefes Arch Clin Exp Ophthalmol. 2006;244(5):620–626. | ||
Johannesson G, Moya-Ortega MD, Asgrimsdottir GM, et al. Kinetics of gamma-cyclodextrin nanoparticle suspension eye drops in tear fluid. Acta Ophthalmol. 2014;92(6):550–556. | ||
Palma SD, Tartara LI, Quinteros D, Allemandi DA, Longhi MR, Granero GE. An efficient ternary complex of acetazolamide with HP-ss-CD and TEA for topical ocular administration. J Control Release. 2009;138(1):24–31. | ||
Sigurdsson HH, Konráethsdóttir F, Loftsson T, Stefánsson E. Topical and systemic absorption in delivery of dexamethasone to the anterior and posterior segments of the eye. Acta Ophthalmol Scand. 2007;85(6):598–602. | ||
Boddupalli B, Mohammed ZK, Nath R, et al. Mucoadhesive drug delivery system: an overview. J Adv Pharm Technol Res. 2010;1(4):381. | ||
de Campos AM, Diebold Y, Carvalho EL, Sánchez A, Alonso MJ. Chitosan nanoparticles as new ocular drug delivery systems: in vitro stability, in vivo fate, and cellular toxicity. Pharm Res. 2004;21(5):803–810. | ||
Quinteros DA, Ferreira LM, Schaffazick SR, Palma SD, Allemandi DA, Cruz L. Novel polymeric nanoparticles intended for ophthalmic administration of acetazolamide. J Pharm Sci. 2016;105(10):3183–3190. | ||
Urtti A. Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv Drug Deliv Rev. 2006;58(11):1131–1135. | ||
Agrahari V, Agrahari V, Hung WT, Christenson LK, Mitra AK. Composite nanoformulation therapeutics for long-term ocular delivery of macromolecules. Mol Pharm. 2016;13(9):2912–2922. | ||
Mahlumba P, Choonara YE, Kumar P, du Toit LC, Pillay V. Stimuli-responsive polymeric systems for controlled protein and peptide delivery: future implications for ocular delivery. Molecules. 2016;21(8):E1002. | ||
Hirani A, Grover A, Lee YW, Pathak Y, Sutariya V. Triamcinolone acetonide nanoparticles incorporated in thermoreversible gels for age-related macular degeneration. Pharm Dev Technol. 2016;21(1):61–67. | ||
You-yun DU, Jun-Feng B, Yan Z, et al. Preliminary evaluation of in vitro transportation characteristics of triamcinolone acetonide eye drops. Chin Hosp Pharm J. 2016;(12):968–972. | ||
Desai J, Thakkar H. Effect of particle size on oral bioavailability of darunavir loaded solid lipid nanoparticles. J Microencapsul. 2016;33(7):669–678. | ||
Ghanbarzadeh S, Hariri R, Kouhsoltani M, Shokri J, Javadzadeh Y, Hamishehkar H. Enhanced stability and dermal delivery of hydroquinone using solid lipid nanoparticles. Colloids Surf B Biointerfaces. 2015;136:1004–1010. | ||
Wang L, Li H, Wang S, et al. Enhancing the antitumor activity of berberine hydrochloride by solid lipid nanoparticle encapsulation. AAPS Pharm Sci Tech. 2014;15(4):834–844. | ||
Stewart JM, Lee OT, Wong FF, Schultz DS, Lamy R. Cross-linking with ultraviolet-a and riboflavin reduces corneal permeability. Invest Ophthalmol Vis Sci. 2011;52(12):9275–9278. | ||
Li J, Li Z, Zhou T, et al. Positively charged micelles based on a triblock copolymer demonstrate enhanced corneal penetration. Int J Nanomedicine. 2015;10:6027–6037. | ||
Baranowski P, Karolewicz B, Gajda M, et al. Ophthalmic drug dosage forms: characterisation and research methods. Scientific World Journal. 2014;2014:861904. | ||
Gupta H, Aqil M, Khar RK, Ali A, Bhatnagar A, Mittal G. Sparfloxacin-loaded PLGA nanoparticles for sustained ocular drug delivery. Nanomedicine. 2010;6(2):324–333. | ||
Liu X, Zhang Z, Jiang Y, et al. Novel PEG-grafted nanostructured lipid carrier for systematic delivery of a poorly soluble anti-leukemia agent Tamibarotene: characterization and evaluation. Drug Deliv. 2015;22(2):223–229. | ||
Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99(pt A):28–51. | ||
Kalam MA. Development of chitosan nanoparticles coated with hyaluronic acid for topical ocular delivery of dexamethasone. Int J Biol Macromol. 2016;89:127–136. | ||
Andrade LM, Rocha KA, De Sa FA, et al. Voriconazole-loaded nanostructured lipid carriers for ocular drug delivery. Cornea. 2016;35(6):866–871. | ||
Liu R, Wang S, Fang S, et al. Liquid crystalline nanoparticles as an ophthalmic delivery system for tetrandrine: development, characterization, and in vitro and in vivo evaluation. Nanoscale Res Lett. 2016;11(1):254. | ||
Achouri D, Sergent M, Tonetto A, Piccerelle P, Andrieu V, Hornebecq V. Self-assembled liquid crystalline nanoparticles as an ophthalmic drug delivery system. Part II: optimization of formulation variables using experimental design. Drug Dev Ind Pharm. 2015;41(3):493–501. | ||
Zhang W, Wang Y, Lee BT, Liu C, Wei G, Lu W. A novel nanoscale-dispersed eye ointment for the treatment of dry eye disease. Nanotechnology. 2014;25(12):125101. | ||
Amrite AC, Kompella UB. Size-dependent disposition of nanoparticles and microparticles following subconjunctival administration. J Pharm Pharmacol. 2005;57(12):1555–1563. | ||
Barbault-Foucher S, Gref R, Russo P, Guechot J, Bochot A. Design of poly-epsilon-caprolactone nanospheres coated with bioadhesive hyaluronic acid for ocular delivery. J Control Release. 2002;83(3):365–375. | ||
Kaur IP, Kanwar M. Ocular preparations: the formulation approach. Drug Dev Ind Pharm. 2002;28(5):473–493. | ||
Sasaki H, Yamamura K, Mukai T, et al. Enhancement of ocular drug penetration. Crit Rev Ther Drug Carrier Syst. 1999;16(1):85–146. | ||
Motwani SK, Chopra S, Talegaonkar S, Kohli K, Ahmad FJ, Khar RK. Chitosan-sodium alginate nanoparticles as submicroscopic reservoirs for ocular delivery: formulation, optimisation and in vitro characterisation. Eur J Pharm Biopharm. 2008;68(3):513–525. | ||
Ying L, Tahara K, Takeuchi H. Drug delivery to the ocular posterior segment using lipid emulsion via eye drop administration: effect of emulsion formulations and surface modification. Int J Pharm. 2013;453(2):329–335. | ||
Attama AA, Muller-Goymann CC. Investigation of surface-modified solid lipid nanocontainers formulated with a heterolipid-templated homolipid. Int J Pharm. 2007;334(1–2):179–189. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.