")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Copanlisib for treatment of B-cell malignancies: the development of a PI3K inhibitor with considerable differences to idelalisib
Authors Krause G , Hassenrück F , Hallek M
Received 27 March 2018
Accepted for publication 21 May 2018
Published 21 August 2018 Volume 2018:12 Pages 2577—2590
DOI https://doi.org/10.2147/DDDT.S142406
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Video abstract presented by Günter Krause.
Views: 486
Günter Krause, Floyd Hassenrück, Michael Hallek
Department I of Internal Medicine, University of Cologne, Center of Integrated Oncology Köln Bonn, Cologne Cluster of Excellence on Cellular Stress Responses in Aging-Associated Diseases (CECAD), Cologne, Germany
Abstract: On the occasion of its recent approval for relapsed follicular lymphoma, we review the design and development of the pan-class I PI3K inhibitor copanlisib as a drug for the treatment of B-cell malignancies in comparison with other kinase inhibitors targeting B-cell-receptor signaling, in particular with strictly isoform-δ-selective idelalisib. In agreement with previously defined PI3K-inhibitor chemotypes, the 2,3-dihydroimidazo[1,2-c]quinazoline scaffold of copanlisib adopts a flat conformation in the adenine-binding pocket of the catalytic p110 subunit and further extends into a deeper-affinity pocket in contrast to idelalisib, the quinazoline moiety of which is accommodated in a newly created selectivity pocket. Copanlisib shows higher potency than other clinically developed PI3K inhibitors against all four class I isoforms, with approximately tenfold preference for p110α and p110δ. Owing to its potency and isoform profile, copanlisib exhibits cell-type-specific cytotoxicity against primary chronic lymphocytic leukemia cells and diffuse large B-cell lymphoma (DLBCL) cell lines at nanomolar concentrations. Moreover, copanlisib differs from idelalisib in regard to intravenous versus oral administration and weekly versus twice-daily dosing. In regard to adverse effects, intermittent intravenous treatment with copanlisib leads to fewer gastrointestinal toxicities compared with continuous oral dosing of idelalisib. In relapsed follicular lymphoma, copanlisib appears more effective and especially better tolerated than other targeted therapies. Copanlisib extends existing treatment options for this subtype of indolent non-Hodgkin lymphoma and also shows promising response rates in DLBCL, especially of the activated B-cell type.
Keywords: targeted therapy, B-cell receptor signaling, p110 isoforms, non-Hodgkin lymphoma, leukemia
Introduction
Typically for B cell malignancies as a whole, pharmacotherapy for chronic lymphocytic leukemia (CLL) started from alkylating agents and purine analogs, to evolve into the continuing era of the incorporation of monoclonal antibodies into immunochemotherapeutic regimens, until small-molecule-targeted therapies emerged in the last decade.1 These drugs are designed specifically to inhibit signaling pathways that are crucial for the survival and interaction of neoplastic cells with the microenvironment, eg, by targeting kinases involved in B-cell receptor (BCR) signaling, such as PI3K, BTK, or antiapoptotic mitochondrial Bcl2 proteins. These molecules are targeted by idelalisib, ibrutinib, or venetoclax, which already have been approved for the treatment of CLL in North America and Europe.
PI3Ks produce the second messenger PIP3 and determine pivotal cell functions like proliferation, survival, and metabolism, but also oncogenesis.2 Expression of the PI3K p110δ isoform is restricted to cells of hematopoietic origin, where it is intricately involved in BCR signaling regulating cell proliferation and survival. Therefore, in malignant B cells, the PI3K pathway is constitutively activated and dependent on PI3Kδ.3 Consequently the PI3Kδ inhibitor idelalisib selectively induces apoptosis of CLL cells while sparing normal T or natural killer cells.4 Idelalisib inhibits the migration of CLL cells to CXCL12 and the secretion of chemokines by activated CLL cells.5 At the beginning of idelalisib treatment, transient redistribution lymphocytosis is frequently observed, as with other inhibitors of BCR signaling.6,7 This can be interpreted as egress of malignant B lymphocytes from protective compartments to the peripheral blood in line with these drugs exerting modest direct cytotoxicity against CLL cells while displaying predominant effects on the microenvironmental dialogue.
Here, the design and development of the pan-class I PI3K inhibitor copanlisib as a drug for the treatment of B cell malignancies are reviewed in comparison with other kinase inhibitors targeting BCR signaling, in particular idelalisib. While preclinical data are available for B-cell acute lymphoblastic leukemia (B-ALL), mantle-cell lymphoma (MCL), CLL, diffuse large B-cell lymphoma (DLBCL), and multiple myeloma (MM), copanlisib has gained approval for relapsed follicular lymphoma (FL) with ongoing clinical trials for several subtypes of indolent and aggressive B-cell non-Hodgkin lymphoma (B-NHL).
Molecular pharmacology
Design and structure–activity relationship
In an effort to develop PI3K inhibitors with potential as antiasthmatic agents, Bayer identified by high-throughput screening the 2,3-dihydroimidazo[1,2-c]quinazoline scaffold as a lead that was further developed finally to yield copanlisib.8 This scaffold is also the basis of PIK90, which represents one of eleven chemotypes in a pharmacological map of PI3K inhibitors docked to p110γ.9 Lead optimization and docking to the p110γ structure established important features of the structure–activity relationship (Figure 1). For instance, the imidazoline N1 was postulated to bind to the hinge region of the p110γ-binding pocket, since its substitution or oxidation diminishes inhibitor potency. Moreover, comparison of various substituents at C5 and C7 indicates that the C5 amide fits into the affinity pocket and C7 points to the sugar pocket. Similarly, C8 was found to face toward the solvent at the entrance of the binding pocket, which offered the opportunity of tuning physical, chemical, and pharmacokinetic properties of the molecule.
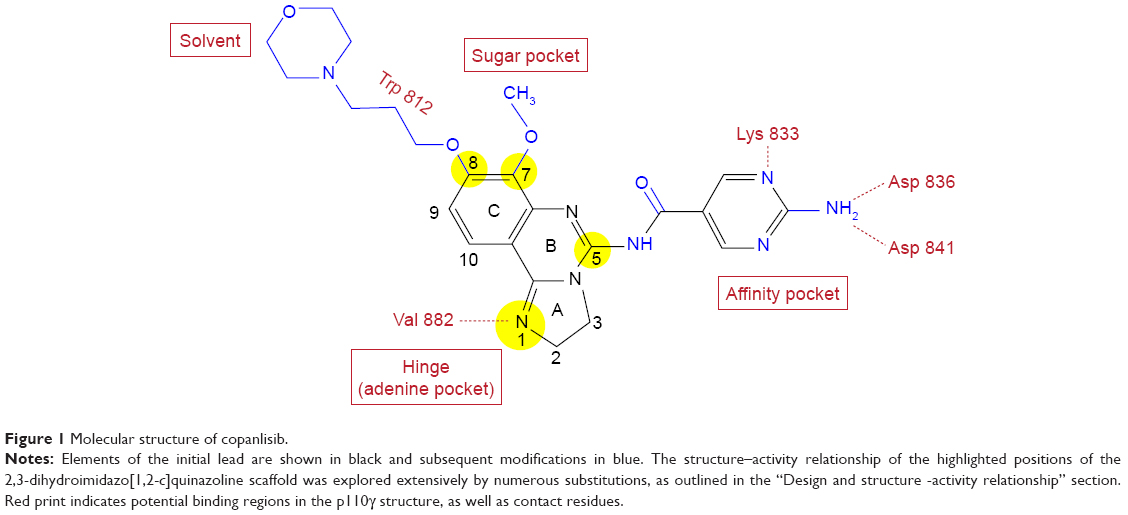 | Figure 1 Molecular structure of copanlisib. |
Inhibitor binding to p110, the catalytic subunit of PI3K
In docking studies on the basis of PI3K inhibitors and the X-ray structure of PI3Kγ available at the time, clearly distinct binding modes were found for PI3K-inhibitor chemotypes classified as multitargeted versus isoform-selective.9 Representatives of these classes are PIK90 and PIK39, which were later developed into the drugs copanlisib and idelalisib, respectively. While multitargeted PI3K inhibitors adopt a flat conformation in the adenine-binding pocket of the catalytic p110 subunit and extend further into a deeper-affinity pocket, isoform-selective PI3K inhibitors extend in different dimensions, eg, as the propeller-shaped molecules PIK39 and idelalisib. For their binding to the catalytic PI3K subunit, these inhibitors exploit a conformationally mobile region including Met752, which leads to an induced fit of one of the three rotor blades of the inhibitor molecules, namely the quinazoline moiety, into a newly created selectivity pocket. Consequently, mutation of this methionine to isoleucine (M752I) affects binding of isoform-selective but not multitargeted PI3K inhibitors to p110γ and leads to resistance to PIK39, while sensitivity to PIK90 is maintained.9
These proposed binding modes were confirmed by the X-ray structures of cocrystals of p110γ and p110δ with copanlisib and idelalisib, respectively.8,10 The corresponding structural information was deposited as entries 5G2N and 4XE0 in the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). The structures are available for interactive viewing on the RCSB PDB website (www.rcsb.org) by means of the integrated NGL viewer.11,12 In the view of the binding pocket of p110γ accommodating copanlisib (Figure 2A), the inhibitor molecule is shown in a similar orientation as in Figure 1 to facilitate comparison with the structure–activity relationship of copanlisib. Moreover, the views of copanlisib and idelalisib bound to the binding pockets of p110γ and p110δ, respectively, are aligned according to the methionine residues M804 or M752 and tryptophan residues W812 or W760 of p110γ or p110δ, respectively, to show the induced fit of idelalisib (Figure 2).
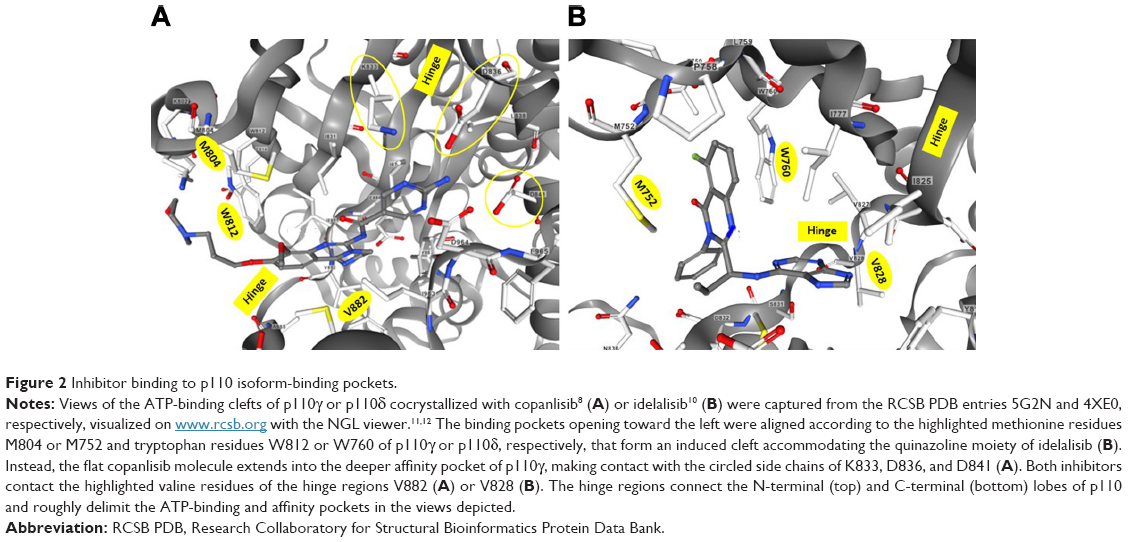 | Figure 2 Inhibitor binding to p110 isoform-binding pockets. |
Isotype selectivity of some PI3K inhibitors in clinical development
The isotype selectivity of PI3K inhibitors was assessed by determination of the inhibitor concentrations that lead to half-maximal inhibition of PI3K activity. For this purpose, activity assays were performed on purified recombinant PI3K isoforms in the presence of different inhibitor concentrations, mainly with detection of the reaction product PIP3. This affords the comparison of clinically used and developed PI3K inhibitors by virtue of IC50 values for individual p110 isoforms (Figure 3).13–20 Further PI3K inhibitors in clinical development for treatment of B-NHL, namely umbralisib (TGR1202), ACP319 (previously named AMG319), and acalisib are p110δ-selective, but show approximately tenfold-lower potency against p110δ activity than idelalisib.21–23
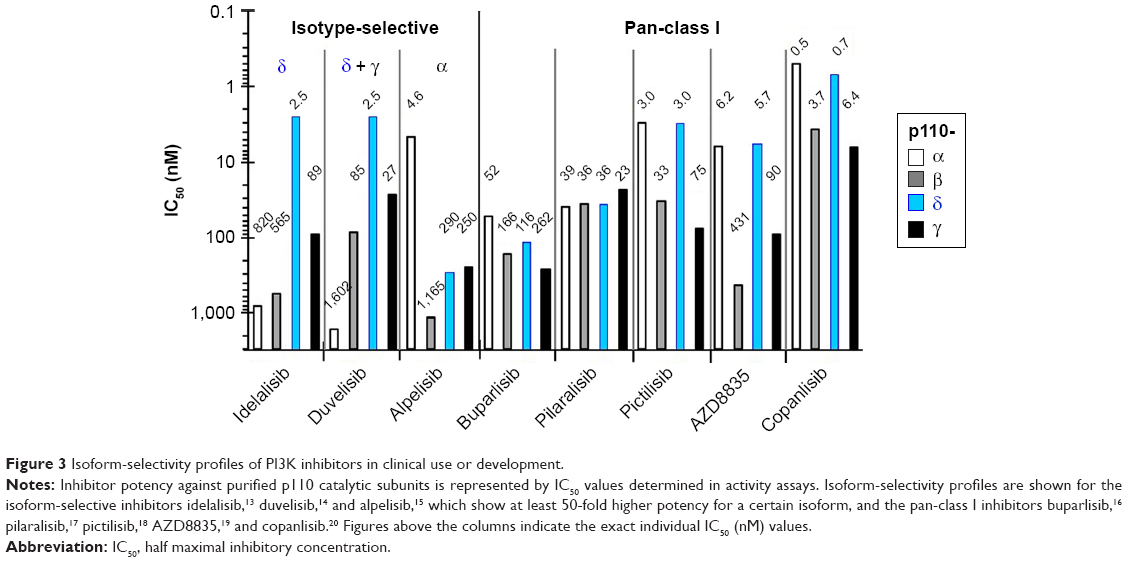 | Figure 3 Isoform-selectivity profiles of PI3K inhibitors in clinical use or development. |
Differences in the binding modes of PI3K-inhibitor chemotypes were correlated with their isoform selectivity.9 Copanlisib and idelalisib had been developed from PIK90 and PIK39 scaffolds respectively, which represent multitargeted and isotype-selective chemotypes of PI3K inhibitors. To qualify as isoform-selective in activity profiling, PI3K inhibitors must exhibit an approximately 30-fold difference in potency between isoforms. This threshold is reached by the isoform-selective PI3K inhibitors idelalisib, duvelisib and alpelisib, but not by the multitargeted PI3K inhibitors buparlisib, pictilisib, AZD8835, or copanlisib (Figure 3). Duvelisib has a very similar molecular structure to idelalisib, but compared to idelalisib, potency against p110γ or p110α is shifted slightly up or down, respectively, putting emphasis on the selectivity against p110γ. The same high level of selectivity that idelalisib has for p110δ recently has also been achieved for the p110α isoform with the design of alpelisib.15 Of note, the high potency of idelalisib and duvelisib against p110δ is surpassed only by copanlisib in the present selection and particularly among multitargeted inhibitors.
Copanlisib belongs to the group of multitargeted pan-class I PI3K inhibitors without relevant potency against other classes of PI3K or mTOR, for which it shows an IC50 of 45 nM,20 and shows higher potency than other clinically developed PI3K inhibitors against all four class I isoforms, with approximately tenfold preference for p110α and p110δ. The selectivity of AZD8835 for p110α and p110δ compared to the other class I isoforms is approximately 15-fold, and thus exceeds that of other multitargeted PI3K inhibitors that predominantly target p110α and p110δ, namely pictilisib and copanlisib. In spite of its predominant selectivity for p110α and p110δ, copanlisib is the most potent inhibitor of p110γ in the present selection and in particular a more potent p110γ inhibitor than duvelisib. The potency of copanlisib against p110γ also surpasses that of voxtalisib, which is a similar molecule to pilaralisib, with enhanced activity against p110γ and especially mTOR.24 According to their biochemical pharmacology, copanlisib and idelalisib can be regarded as PI3K inhibitors with flat versus propeller-shaped structure, different binding modes to the active site of the catalytic p110 subunit, and comparatively higher potency versus more stringent isoform selectivity.
Impact of copanlisib on cell functions
At the next level of investigation, one needs to ask how the biochemical pharmacology of PI3K inhibitors influences the action of these substances on malignant B cells and their microenvironment. In this regard, the consequences of pan-class I inhibition with preferential targeting of p110α and p110δ will be compared to that of purely p110δ selective idelalisib. Its pan-class I selectivity profile led to copanlisib being considered also for treatment of solid tumors.20,25–28 Apart from cytotoxicity against tumor cells, preclinical investigation of copanlisib must also comprise that against cells of the tumor microenvironment and of the immune system. Of note, even other completely unrelated mechanisms may be involved in the cytotoxicity of PI3K inhibitors, as recently shown for buparlisib, which apart from action as an ATP-competitive PI3K inhibitor, also interferes with microtubule polymerization.29 Before we specifically address the relationship of the biochemical pharmacology of copanlisib, especially its PI3K isoform-selectivity profile, with preferentially targeted B-NHL types, we discuss two issues on a more general note, namely the role of lipid kinases in the signal transduction occurring in malignant B cells and the biology of B-cell malignancies.
PI3K in BCR signaling
Class I phosphatidylinositide-3-kinases catalyze the formation of the lipid second messenger PIP3, which is capable of binding to the pleckstrin-homology domain of important signaling mediators, eg, Akt and BTK.2 The predominantly expressed and functionally relevant catalytic PI3K subunit, namely p110δ,30 cooperates with other kinases of BCR signaling (Figure 4). Activation of PI3Kδ in mature B cells occurs chiefly via binding of the Src homology 2 (SH2) domain of the regulatory p85 subunit to a phosphotyrosine motive of the receptor CD19. Since CD19 lacks intrinsic or associated kinase activity, the required tyrosine phosphorylation is carried out by the Src family kinase Lyn, which is associated with the BCR. In addition, rather than exclusively by receptor tyrosine kinases, PI3Kδ in B cells is commonly activated by chemokine receptors that belong to the group of G-protein-coupled receptors, which in other cell types usually activate the PI3Kγ isoform. While both Akt and BTK may be recruited to the cytoplasmic membrane via their pleckstrin-homology domains and PIP3, this may also alternatively occur by additional routes, eg, in the case of Akt via the lipid second messenger PI(4,5)P2, which results from dephosphorylation of PIP3 by SHIP or via the SH2 domain of BTK. PI3K inhibition, eg, by copanlisib, in contrast to inhibition of AKT alone, also blocks the ensuing RAF–MEK–ERK cascade,25 which is also affected by inhibitors of BTK. A major effect of Akt activity is the phosphorylation and inactivation of the transcription factor FOXO. Other consequences of Akt activity include phosphorylation of the inhibitory IKK complex, which leads to activation of NFκB, and of the proapoptotic Bcl2 protein BAD, which triggers its own degradation.
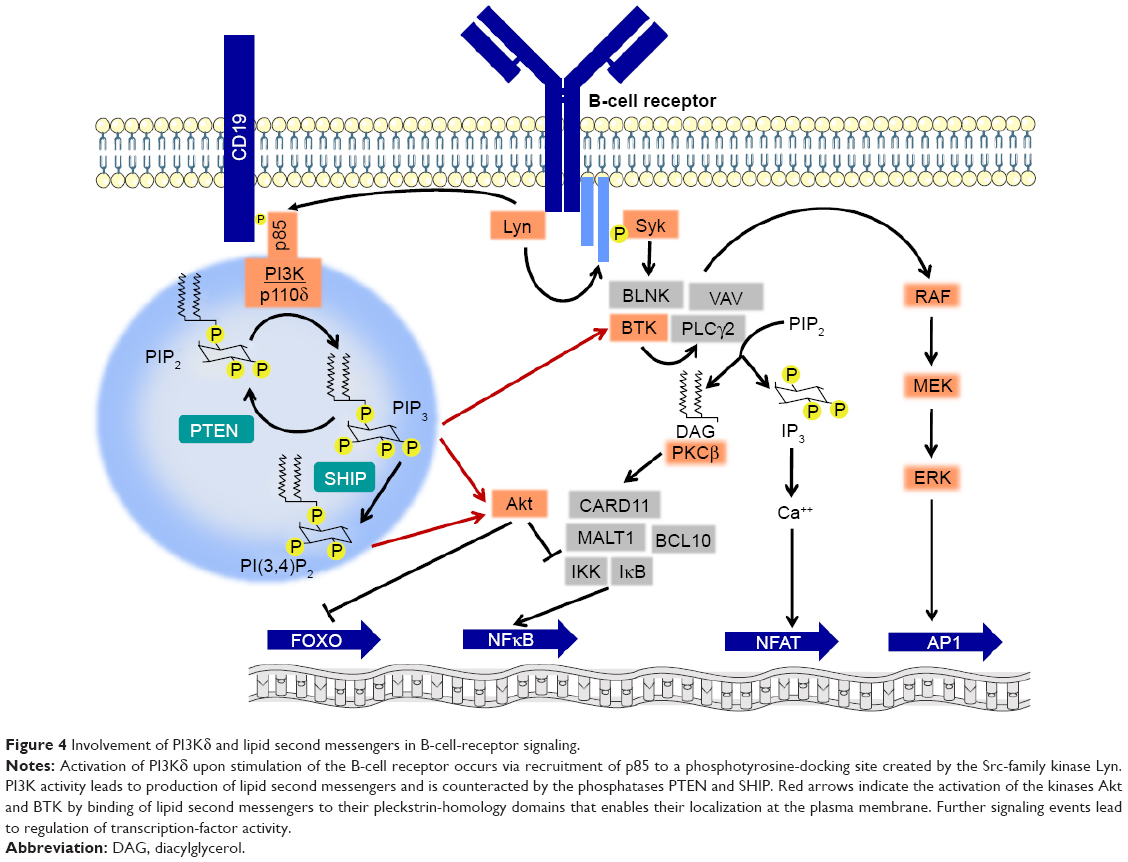 | Figure 4 Involvement of PI3Kδ and lipid second messengers in B-cell-receptor signaling. |
BCR signaling is now accepted as a common driving force of B-cell malignancies, and inhibitors have become available for kinases at several interception points in these pathways, with those targeting p110δ or the tyrosine kinases BTK and Syk already in or close to clinical use.31 While p110δ appears indispensable for the BCR pathway stimulated by antigen, p110α may play a role in tonic BCR signaling and inhibitor resistance. Similarly to other inhibitors of the BCR pathway, idelalisib exhibits only modest direct cytotoxicity, so that disturbance of interactions with the microenvironment, specifically inhibition of chemotaxis to CXCL12 and secretion of the chemokines CCL3 and CCL4, may offer an explanation for its clinical activity.5 Exactly these important functions of neoplastic B cells also depend on two other kinases involved in BCR signaling, namely BTK and Lyn, since these cell functions are rescued from inhibition by the tyrosine-kinase inhibitors ibrutinib or dasatinib, owing to expression of inhibitor-resistant kinase mutants in cell-line models.32 Interrogation of the role of the same kinases in the pathogenesis of CLL using the Eμ-TCL1 mouse model revealed that loss of Lyn in leukemic cells reduces BCR signaling, including BTK phosphorylation, but does not affect leukemic cell expansion, in contrast to transplantation of syngeneic CLL cells into Lyn- or BTK-deficient recipients, which results in strongly delayed leukemic progression and prolonged survival.33 Therefore, the microenvironment plays a predominant role in the pathogenesis of CLL compared to neoplastic B cells, which implies that clinically effective inhibitors, such as ibrutinib and idelalisib, do not chiefly target BCR signaling in the literal sense, but different (as yet incompletely defined) signaling pathways and cell functions in accessory cells. Small-molecule inhibitors targeting the BCR pathway, especially during prolonged treatment, have the potential to promote the development of secondary lymphoma, since disruption of BCR signaling in normal and neoplastic B cells, especially by inhibition of PI3Kδ, enhances the expression of AID and increases translocation frequency and genomic instability.34 Whether a different balance of PI3K-isoform inhibition, as could be achieved for instance with copanlisib, could change this effect is as yet unknown.
Different types of B-cell malignancies
The definition of B-NHL by the World Health Organization comprises B-cell malignancies irrespective of their appearance as solid tumors or leukemias. Accordingly, these diseases can be arranged according to the development stage of their presumed cells of origin (Table 1).35 In the course of B-cell development, pre-B cells from the bone marrow migrate to the lymph nodes and specifically germinal centers, where they experience antigen contact, V(D)J recombination and finally isotype switch before long-lived plasma cells return to the bone marrow. As reference for the preclinical and clinical data reported in this review, molecular features of B-NHL36 and their incidence37 are indicated. Although the malignancies listed38–47 are often heterogeneous within the subgroups, one can distinguish more indolent, eg, CLL, marginal-zone lymphoma, FL, and more aggressive malignancies, eg, B-ALL, MCL, Burkitt lymphoma, and DLBCL. CLL patients can be divided into prognostic subgroups with unmutated versus mutated IGHV genes with more aggressive and indolent course.48 Similarly, gene-expression profiling distinguishes DLBCL of the germinal center or activated B-cell (ABC) subtypes.49
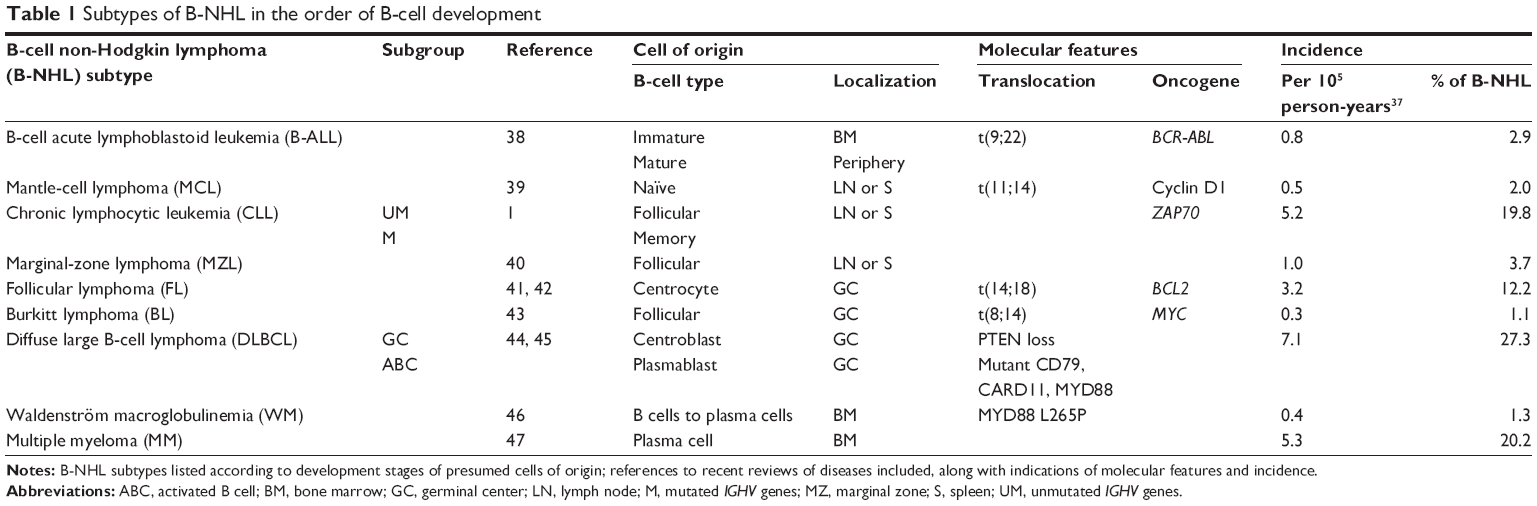 | Table 1 Subtypes of B-NHL in the order of B-cell development |
Cytotoxicity of copanlisib against B-NHL subtypes
Given the different PI3K-isoform profiles of copanlisib and idelalisib, the sensitivity of specific B-NHL subtypes to these inhibitors could depend on the expression and functional involvement of p110α in these entities. Genome analysis of patient samples indicates amplification of PI3KCA in MCL50 and CLL.51 In MCL, p110δ inhibition by idelalisib blocks BCR-stimulated, but not constitutive PI3K activation, and pictilisib, which predominantly targets p110α and p110δ, shows significantly more cytotoxicity against MCL cell lines and primary samples than idelalisib.52 Accordingly, a panel of MCL cell lines exhibits high sensitivity to copanlisib.53
Relatively easy access to B lymphocytes from the peripheral blood of CLL patients enables the assessment of PI3K inhibitors with different isoform selectivity on such samples. Multitargeted PI3K inhibitors, including PIK90, from the scaffold of which copanlisib was derived, show stronger inhibition of the phosphorylation of Akt and apoptosis induction in CLL cells than p110δ-selective inhibitors.54 From analysis of the survival of CLL cells treated with different isoform-selective PI3K inhibitors, one can conclude that in contrast to normal B cells, where p110δ is mostly involved in signaling through the IL4 receptor and the BCR,55 the most important PI3K isoform in the survival of CLL cells is p110α, followed by p110δ, whereas the viability of CLL cells is hardly affected by inhibition of p110γ.56 Copanlisib exhibits cytotoxicity selectively against CLL cells compared to normal B and T cells and the concentrations leading to half-maximal reduction of CLL-cell survival are more than tenfold lower for copanlisib than for idelalisib or duvelisib.57 In addition, treatment with copanlisib also overcomes the survival advantage that CLL cells gain from stroma-cell support.
Immunohistochemistry shows higher PI3Kα expression in DLBCL than in FL samples.58 ABC-DLBCL cell lines are sensitive to 0.5 μM idelalisib only if p110α is knocked down by shRNA, and their proliferation and survival are inhibited by AZD8835 that predominantly targets p110δ and p110α.59 Similarly, simultaneous inhibition of p110δ and p110α by copanlisib or a combination of idelalisib and alpelisib leads to significantly enhanced inhibition of the growth of ibrutinib-sensitive and -resistant ABC-DLBCL cell lines.58 Since activating CARD11 mutations seem to cause resistance against PI3K inhibition, a thorough molecular characterization is necessary for effective use of targeted therapeutics in DLBCL.59
In agreement with isoform-expression profiles of MM samples, which show higher levels of p110α and p110β than normal samples, with further increases during disease progression, the survival of primary MM cells and MM cell lines is inhibited by p110α-selective alpelisib more strongly than that of normal peripheral blood mononuclear cells.60 Therefore, potency against p110α may contribute to the inhibition of constitutive and IGF1-induced Akt phosphorylation in MM cell lines by copanlisib concentrations of 100 nM or lower.61 Copanlisib inhibits cell-cycle progression and survival of primary MM cells and cell lines and the growth of subcutaneous xenografts in nude mice.
Interactions with the microenvironment
The clinical efficacy of idelalisib cannot be explained by its direct cytotoxicity against CLL cells, but by the disturbance of CLL-microenvironment interactions, such as redistribution of neoplastic B cells and production of cytokines.5 In the survival of CLL cells, p110 isoforms appear to be involved to different degrees, namely p110α more than p110δ and especially p110γ,56 and pharmacological inhibition of the chemotaxis of CLL cells to CXCL12, eg, by PIK90, can also be connected with targeting p110α.54 In a head-to-head comparison with 1 μM each of the clinically developed inhibitors copanlisib and duvelisib, which target p110γ in addition to p110δ, inhibited the chemotaxis of CLL cells to CXCL12 significantly more efficiently than idelalisib, which implies involvement of p110γ in this process.57 This role of p110γ in the chemotaxis of CLL cells was substantiated by up- and downregulation of p110γ expression and use of an exclusively p110γ-selective inhibitor.62 In addition to showing increased effects on chemotaxis compared to idelalisib, the p110δ and γ-selective PI3K inhibitor duvelisib also impacts on the BCR-mediated secretion of the chemokines CCL3 and CCL4.63 According to its isoform profile and high potency, copanlisib may also be expected to affect this microenvironment interaction to a high extent. Apart from their impact on neoplastic B cells, it will be essential to understand how PI3K inhibitors influence the function of myeloid cells, T cells, and natural killer cells. For instance, selective inactivation of macrophage PI3Kγ stimulates and prolongs NFκB activation and inhibits C/EBPβ activation, thus promoting an immunostimulatory transcriptional program that restores CD8+ T-cell activation and cytotoxicity.64
Copanlisib in drug combinations against B-NHL
BCR–ABL-positive leukemias can be efficiently controlled by inhibitors of the tyrosine kinase Abl, unless acquired resistance arises, eg, via the T315I mutation in ABL. In cell lines expressing wild-type and mutant BCR–ABL, the cytotoxic effects of Abl inhibitors are additively enhanced by copanlisib. Remarkably, treatment with a combination of ponatinib and copanlisib abolishes tumor growth in mice after irradiation and subcutaneous or intravenous transplantation with allografts consisting of BaF3 cells expressing BCR–ABL carrying the T315I-resistance mutation.65
The occurrence of the t(9;22) translocation with concomitant deregulation of cyclin-dependent kinases in >90% of MCL cases prompted the assessment of the CDK4 inhibitor palbociclib in combination with PI3K inhibitors in the context of acquired ibrutinib resistance.66 The prolonged early G1 arrest induced by palbociclib sensitizes DT40 chicken-lymphoid cells to cell killing by ibrutinib if they express wild-type BTK, and to PI3K inhibitors, even in the presence of the BTKC481S mutant. Owing to the role of p110α in MCL cells,52 it is reasonable to assume that copanlisib can be more cytotoxic for them than idelalisib, particularly in combinations with CDK inhibitors and for overcoming ibrutinib resistance.
The irreversible BTK inhibitor ibrutinib is clinically used for treatment of CLL and MCL and acquired resistance owing to the BTKC481S mutation is observed in both entities. By targeting a different interception point in BCR signaling, namely PI3K, duvelisib is capable of overcoming this ibrutinib resistance.67 As a PI3K inhibitor exhibiting greater cytotoxicity against CLL cells than duvelisib,57 copanlisib can be expected to function in a similar manner. Also in ABC-DLBCL cell lines, combinations of PI3K and BTK inhibitors lead to synergistic enhancement of cytotoxic effects and overcome ibrutinib resistance.68
Owing to independent mechanisms of action, monoclonal antibodies are considered promising combination partners of kinase inhibitors; however, with the risk that kinase inhibition interferes with killing mechanisms of antibodies, eg, with antibody-dependent cell-mediated cytotoxicity (ADCC). As such, the irreversible BTK inhibitor ibrutinib antagonizes the ADCC of rituximab,69 while in the presence of the PI3Kδ inhibitor idelalisib, the ADCC of alemtuzumab is maintained.4 Survival inhibition of CLL cells by copanlisib and idelalisib is enhanced by the therapeutic anti-CD20 antibodies rituximab and obinutuzumab, while ADCC by alemtuzumab and peripheral blood mononuclear cells is not substantially impaired by idelalisib or copanlisib.57 In a more sensitive and refined analysis of ADCC using genetically modified NK92 cells expressing CD16-176V as effector cells, copanlisib led to a considerably stronger impairment of the ADCC of anti-CD20 antibodies than idelalisib or duvelisib at the same clinically unachievable concentration of 10 μM, but significant ADCC of obinutuzumab against Raji cells was maintained in the presence of 1 μM copanlisib.70 In CLL cells, PI3K inhibitors enhance the direct cytotoxicity of anti-CD37 antibodies, eg, otlertuzumab, owing to opposing signaling by N- and C-terminal tyrosine residues of CD37.71 On primary CLL cells, the combination of the anti-CD37 antibody BI836826 with copanlisib is particularly efficacious, which can be attributed to high single agent anti-apoptotic activity of both drugs.72
Emerging clinical evidence
Preclinical investigation of copanlisib in vitro was followed by in vivo studies in rat- and mouse-xenograft models.20,25,61 Despite an extremely short half-life in mice of only 0.7 hours, pulsatile administration of copanlisib led to much better efficacy against subcutaneous xenografts of the breast cancer cell line BT-474 than continuous treatment.25 Based on preclinical evidence including xenograft models,20 copanlisib was investigated in a first-in-human dose-expansion trial with patients suffering from solid and hematological tumors.73 In this trial, an intermittent dosing schedule of 60 mg copanlisib on days 1, 8, and 15 of a 4-week cycle was established. In contrast, 150 mg idelalisib was administered twice daily,6 yielding a ≥35-fold higher drug load than with copanlisib.
Adverse effects
The most common toxicities of idelalisib include elevation of liver transaminases, diarrhea, and neutropenia. These can be divided into inflammatory toxicities of presumed autoimmune origin, as well as bacterial and opportunistic infections that are possibly caused by impaired T-cell function. Concerns have emerged about the risks associated with oral PI3Kδ-targeting agents, such as infections, pneumonitis, and colitis.74,75 Unexpected aggravation of such adverse effects in combined applications of idelalisib with rituximab and lenalidomide or with the Syk inhibitor entospletinib led to premature termination of the trials of these combinations.76,77 In addition safety concerns in prematurely discontinued Phase III trials of combinations containing idelalisib and rituximab with or without bendamustine were attributed to opportunistic infections, notably with Pneumocystis jirovecii and Cytomegalovirus.78 This raises the possibility that serious adverse effects of isoform-δ-selective PI3K inhibition may be avoided by inhibitors with broader isoform selectivity or different dosing. Since data from Phase II trials with similar numbers of participating patients suffering from relapsed indolent B-NHL were available, the adverse effects of copanlisib79 or idelalisib80 reported in these trials were compared (Figure 5). In this comparison, hyperglycemia, an expected on-target effect of PI3Kα inhibition, and hypertension were observed exclusively with copanlisib, while idelalisib led to higher incidence of gastrointestinal symptoms and hematological toxicities. Transient hyperglycemia is a recognized on-target effect of PI3Kα inhibition that is related to insulin-receptor signaling9,81 and occurred also in trials of orally administered multitargeted pan class I PI3K inhibitors, namely buparlisib,82 pictilisib,83 and pilaralisib.84
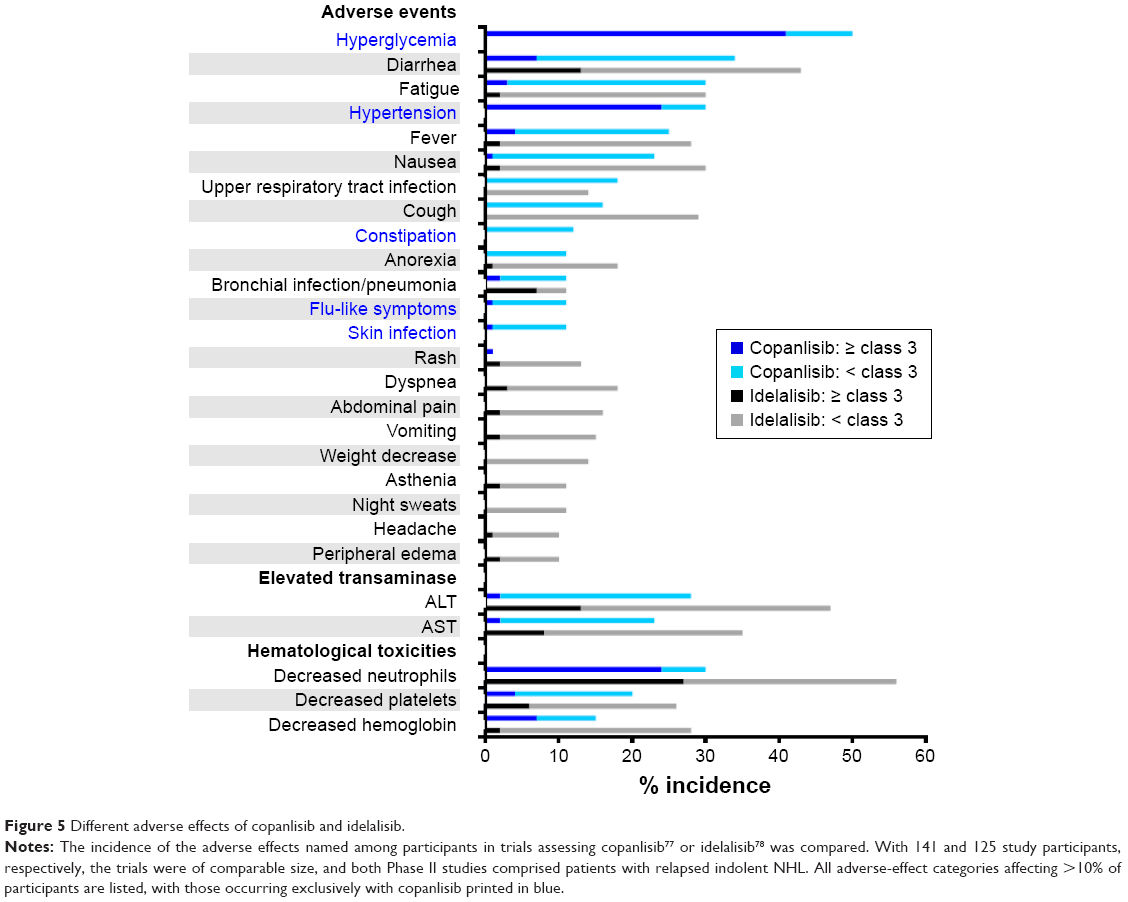 | Figure 5 Different adverse effects of copanlisib and idelalisib. |
Efficacy against different B-NHL subtypes
Although p110α is expressed to a lesser extent than p110δ in most subtypes of B-NHL, copanlisib might offer greater efficacy than idelalisib in situations where alternative isoforms are upregulated in response to selective PI3Kδ inhibition as a resistance mechanism. The selection of B-NHL entities (Table 1) for potential clinical use of copanlisib may be guided by the molecular properties of the inhibitor and by preclinical studies, but is mainly influenced by insights with regard to adverse effects and clinical efficacy gained from pilot trials, which sometimes comprise several entities. Also for the assessment of copanlisib, a trial was conducted with participants suffering from different subtypes of relapsed/refractory indolent or aggressive lymphomas,85 which was continued as the CHRONOS-1 trial focusing on indolent B-NHL,79 where a prespecified threshold of 40% overall response was exceeded, leading to accelerated approval of copanlisib for FL. Accelerated approvals allow drugs for serious conditions that fill an unmet medical need to be cleared for marketing based on a surrogate rather than measurable clinical benefit. Three placebo-controlled Phase III trials of copanlisib have been started in patients with indolent B-NHL, namely on rituximab-refractory patients (CHRONOS-2) and of copanlisib in combination with rituximab (CHRONOS-3) or chemoimmunotherapy (CHRONOS-4).86
As measures for the efficacy of copanlisib, response rates were determined in single-armed Phase I and II trials with patients suffering from relapsed/refractory indolent or aggressive B-NHL, including FL, DLBCL, and MCL. Participating patients were heavily pretreated with chemoimmunotherapy, usually containing rituximab. Comparisons of response rates from such single-arm trials have to be done with caution, because of differences between trials, but are the only way to estimate the clinical efficacy of copanlisib and competitor drugs before results from randomized placebo-controlled trials become available. Accordingly, overall and complete response rates obtained in a trial comprising indolent and aggressive B-NHL subtypes85 were compared with those from trials assessing different PI3K inhibitors.87 A similar comparison of recently published results of trials of copanlisib in relapsed indolent B-NHL, including FL, as well as more aggressive DLBCL and MCL, is presented in Table 2. In trials of indolent B-NHL and FL, the response rates observed with copanlisib77 were superior to those with idelalisib,78 duvelisib,86 and buparlisib87 to different degrees, particularly in regard to the frequency of complete responses (Table 2), but in addition to its efficacy, the more favorable side-effect profile of copanlisib compared to p110δ-selective agents contributes to its advantages.
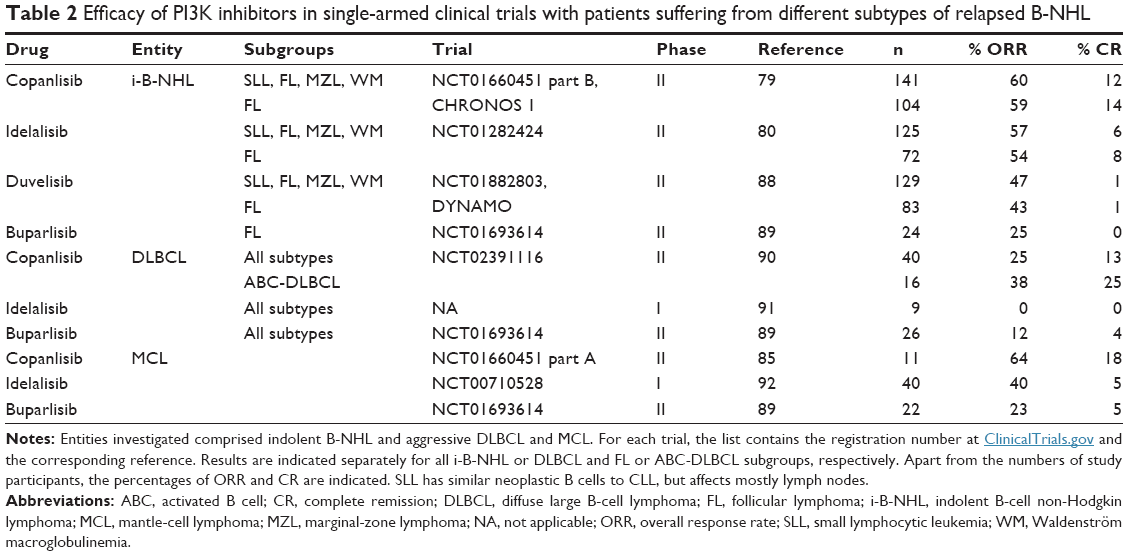 | Table 2 Efficacy of PI3K inhibitors in single-armed clinical trials with patients suffering from different subtypes of relapsed B-NHL |
With chemoimmunotherapy, ABC-DLBCL has a cure rate of 40%, which is lower than that for germinal-center DLBCL. BCR-pathway inhibitors are considered for the treatment of relapsed patients, particularly of the ABC subtype. The efficacy of PI3K inhibitors for treating DLBCL observed in Phase I and II trials is compiled in Table 2. The clinical usefulness of PI3K inhibition for the treatment of DLBCL has been called into question, when in an early trial this drug was not effective in the few studied DLBCL patients, in contrast to other B-NHL subtypes.92 However, copanlisib treatment of relapsed or refractory DLBCL patients resulted in encouraging responses, especially in the ABC subtype,90 which may be explained by the requirement for simultaneous inhibition of both p110δ and p110α.59 Since treatment with single-agent ibrutinib, or copanlisib, with ~40% response has only limited efficacy against ABC-DLBCLs,93 combined inhibition of BTK and p110δ and p110α is suggested on the basis of xenograft models.59
The percentages of ORR and CR achieved with copanlisib against MCL, similarly as against ABC-DLBCL, surpass those obtained with idelalisib to a higher extent than observed in the treatment of FL (Table 2). However, the data available so far are from single-arm Phase I trials and must be interpreted with caution, even more so for ongoing or preliminary studies with low numbers of evaluated patients.
Summarizing the properties of copanlisib and idelalisib as representatives of multitargeted versus p110δ-selective PI3K inhibitors (Table 3) does not immediately reveal why copanlisib was first approved for the treatment of FL, rather than for an entity, where targeting p110α in addition to p110δ would offer an advantage, such as ABC-DLBCL or MCL. While copanlisib exhibits at least 50-fold higher cytotoxicity against malignant B cells compared to only approximately five times lower clinically achievable concentrations than idelalisib, the differences between these inhibitors in regard to disturbance of the microenvironmental dialogue are still incompletely explored. Therefore, future clinical trials may reveal a favorable pharmacological profile of copanlisib against additional types of NHL.
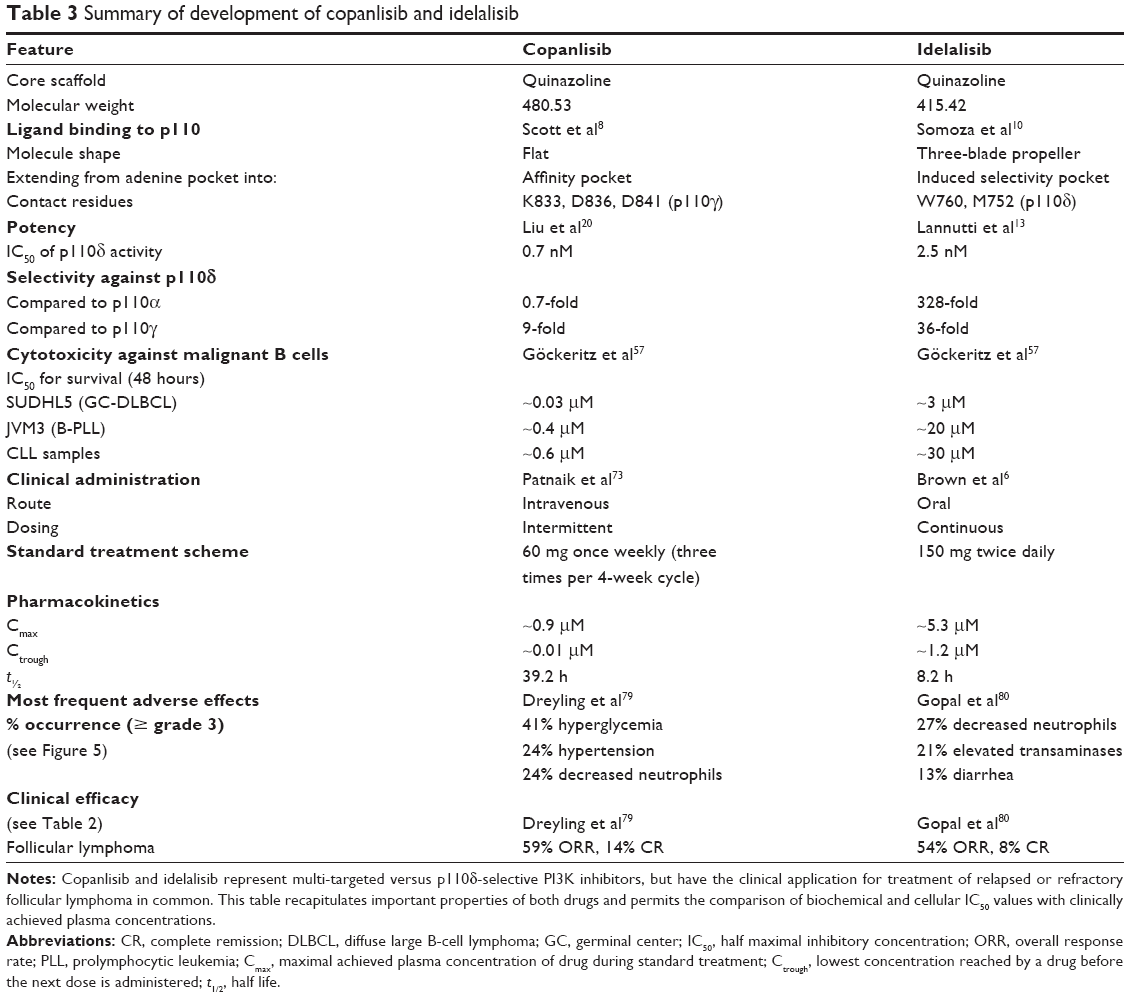 | Table 3 Summary of development of copanlisib and idelalisib |
Conclusion
Copanlisib and idelalisib are PI3K inhibitors with flat versus propeller-shaped structures and belong to multitargeted versus isoform-selective PI3K inhibitors. Copanlisib shows higher potency than other clinically developed PI3K inhibitors against all four class I isoforms, with approximately tenfold preference for p110α and p110δ. Of note, the high potency of idelalisib and duvelisib against p110δ is surpassed only by copanlisib among currently available multitargeted inhibitors. Simultaneous targeting of p110δ and p110α was most convincingly shown to be of advantage in MCL and ABC-DLBCL cells, but targeting p110γ may be of paramount importance in interruption of the microenvironmental dialogue. Apart from targeting all four class I isoforms versus only one, copanlisib differs from idelalisib also in regard to intravenous versus oral administration and weekly versus twice-daily dosing. These properties may contribute to fewer and less severe gastrointestinal toxicities with copanlisib compared to idelalisib. Owing to efficacy and an adverse-effect profile distinct from idelalisib, copanlisib has already expanded the drug armamentarium against relapsed FL, with promising prospects for future applications, eg, against ABC-DLBCL and potentially further B-cell malignancies.
Disclosure
GK has received research funding from Bayer, Roche, and Boehringer Ingelheim. MH has received consultancy fees and honoraria from AbbVie, Mundipharma, GlaxoSmithKline, Gilead, and Celgene; consultancy, honoraria, and speakers bureau fees from Janssen; consultancy and speakers bureau fees from Pharmacyclics; consultancy, research funding, and speakers bureau fees from Roche; and funding from Gilead. The authors report no other conflicts of interest in this work.
References
Hallek M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2017;92(9):946–965. | ||
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. | ||
Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3(4):317–330. | ||
Herman SE, Gordon AL, Wagner AJ, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078–2088. | ||
Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603–3612. | ||
Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390–3397. | ||
Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997–1007. | ||
Scott WJ, Hentemann MF, Rowley RB, et al. Discovery and SAR of novel 2,3-dihydroimidazo[1,2-c]quinazoline PI3K inhibitors: identification of copanlisib (BAY 80-6946). ChemMedChem. 2016;11(14):1517–1530. | ||
Knight ZA, Gonzalez B, Feldman ME, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125(4):733–747. | ||
Somoza JR, Koditek D, Villasenor AG, et al. Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase δ. J Biol Chem. 2015;290(13):8439–8446. | ||
Rose AS, Hildebrand PW. NGL Viewer: a web application for molecular visualization. Nucleic Acids Res. 2015;43:W576–W579. | ||
Rose AS, Bradley AR, Valasatava Y, et al. Web-based molecular graphics for large complexes. ACM Proceedings of the 21st International Conference on Web3D Technology (Web3D '16). 2016;185–186. | ||
Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. | ||
Winkler DG, Faia KL, DiNitto JP, et al. PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol. 2013;20(11):1364–1374. | ||
Fritsch C, Huang A, Chatenay-Rivauday C, et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–1129. | ||
Maira SM, Pecchi S, Huang A, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–328. | ||
Foster P, Yamaguchi K, Hsu PP, et al. The selective PI3K inhibitor XL147 (SAR245408) inhibits tumor growth and survival and potentiates the activity of chemotherapeutic agents in preclinical tumor models. Mol Cancer Ther. 2015;14(4):931–940. | ||
Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18):5522–5532. | ||
Barlaam B, Cosulich S, Delouvrie B, et al. Discovery of 1-(4-(5-(5-amino-6-(5-tert-butyl-1,3,4-oxadiazol-2-yl)pyrazin-2-yl)-1-ethyl-1,2,4-triazol-3-yl)piperidin-1-yl)-3-hydroxy-propan-1-one (AZD8835): a potent and selective inhibitor of PI3Kα and PI3Kdelta for the treatment of cancers. Bioorg Med Chem Lett. 2015;25(22):5155–5162. | ||
Liu N, Rowley BR, Bull CO, et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12(11):2319–2330. | ||
Shugg RP, Thomson A, Tanabe N, et al. Effects of isoform-selective phosphatidylinositol 3-kinase inhibitors on osteoclasts: actions on cytoskeletal organization, survival, and resorption. J Biol Chem. 2013;288(49):35346–35357. | ||
Cushing TD, Hao X, Shin Y, et al. Discovery and in vivo evaluation of (S)-N-(1-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (AMG319) and related PI3Kδ inhibitors for inflammation and autoimmune disease. J Med Chem. 2015;58(1):480–511. | ||
Burris HA, Patel MR, Lanasa MC, et al. Activity of TGR-1202, a novel once-daily PI3Kδ inhibitor, in patients with relapsed or refractory hematologic malignancies. J Clin Oncol. 2014;32(15 Suppl):2513. | ||
Yu P, Laird AD, Du X, et al. Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol Cancer Ther. 2014;13(5):1078–1091. | ||
Will M, Qin AC, Toy W, et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov. 2014;4(3):334–347. | ||
Schneider P, Schön M, Pletz N, et al. The novel PI3 kinase inhibitor, BAY 80-6946, impairs melanoma growth in vivo and in vitro. Exp Dermatol. 2014;23(8):579–584. | ||
Elster N, Cremona M, Morgan C, et al. A preclinical evaluation of the PI3K alpha/delta dominant inhibitor BAY 80-6946 in HER2-positive breast cancer models with acquired resistance to the HER2-targeted therapies trastuzumab and lapatinib. Breast Cancer Res Treat. 2015;149(2):373–383. | ||
Kim RD, Alberts SR, Peña C, et al. Phase I dose-escalation study of copanlisib in combination with gemcitabine or cisplatin plus gemcitabine in patients with advanced cancer. Br J Cancer. 2018;118(4):462–470. | ||
Bohnacker T, Prota AE, Beaufils F, et al. Deconvolution of buparlisib’s mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention. Nat Commun. 2017;8:14683. | ||
Okkenhaug K, Burger JA. PI3K signaling in normal B cells and chronic lymphocytic leukemia (CLL). Curr Top Microbiol Immunol. 2016;393:123–142. | ||
Jerkeman M, Hallek M, Dreyling M, Thieblemont C, Kimby E, Staudt L. Targeting of B-cell receptor signalling in B-cell malignancies. J Intern Med. 2017;282(5):415–428. | ||
Göckeritz E, Vondey V, Guastafierro A, et al. Establishing a chemical genetic link between Bruton tyrosine kinase activity in malignant B cells and cell functions involved in the micro-environmental dialogue. Br J Haematol. 2017;178(6):949–953. | ||
Nguyen PH, Fedorchenko O, Rosen N, et al. Lyn kinase in the tumor microenvironment is essential for the progression of chronic lymphocytic leukemia. Cancer Cell. 2016;30(4):610–622. | ||
Compagno M, Wang Q, Pighi C, et al. Phosphatidylinositol 3-kinase δ blockade increases genomic instability in B cells. Nature. 2017;542(7642):489–493. | ||
Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18(3):148–167. | ||
Shaffer AL 3rd, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol. 2012;30:565–610. | ||
Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood. 2006;107(1):265–276. | ||
Zhou Y, You MJ, Young KH, et al. Advances in the molecular pathobiology of B-lymphoblastic leukemia. Hum Pathol. 2012;43(9):1347–1362. | ||
Perez-Galan P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26–38. | ||
Spina V, Rossi D. Molecular pathogenesis of splenic and nodal marginal zone lymphoma. Best Pract Res Clin Haematol. 2017;30(1–2):5–12. | ||
Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood. 2016;127(17):2055–2063. | ||
Dreyling M, Ghielmini M, Rule S, Salles G, Vitolo U, Ladetto M. Newly diagnosed and relapsed follicular lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27 Suppl 5:v83–v90. | ||
Jacobson C, LaCasce A. How I treat Burkitt lymphoma in adults. Blood. 2014;124(19):2913–2920. | ||
Nogai H, Dörken B, Lenz G. Pathogenesis of non-Hodgkin’s lymphoma. J Clin Oncol. 2011;29(14):1803–1811. | ||
Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125(1):22–32. | ||
Hunter ZR, Yang G, Xu L, Liu X, Castillo JJ, Treon SP. Genomics, signaling, and treatment of Waldenström macroglobulinemia. J Clin Oncol. 2017;35(9):994–1001. | ||
Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–1060. | ||
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig VH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854. | ||
Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. | ||
Psyrri A, Papageorgiou S, Liakata E, et al. Phosphatidylinositol 3′-kinase catalytic subunit α gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res. 2009;15(18):5724–5732. | ||
Brown JR, Hanna M, Tesar B, et al. Integrative genomic analysis implicates gain of PIK3CA at 3q26 and MYC at 8q24 in chronic lymphocytic leukemia. Clin Cancer Res. 2012;18(14):3791–3802. | ||
Iyengar S, Clear A, Bödör C, et al. P110α-mediated constitutive PI3K signaling limits the efficacy of p110δ-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood. 2013;121(12):2274–2284. | ||
Gaudio E, Kwee I, Spriano F, et al. The pan class-I PI3K inhibitor copanlisib has preclinical activity in mantle cell lymphoma, marginal zone lymphoma and chronic lymphocytic leukemia as single agent and in combination with other targeted and conventional agents. Blood. 2016;128:4185. | ||
Niedermeier M, Hennessy BT, Knight ZA, et al. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood. 2009;113(22):5549–5557. | ||
Bilancio A, Okkenhaug K, Camps M, et al. Key role of the p110δ isoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110δ function in B cells. Blood. 2006;107(2):642–650. | ||
de Frias M, Iglesias-Serret D, Cosialls AM, et al. Isoform-selective phosphoinositide 3-kinase inhibitors induce apoptosis in chronic lymphocytic leukaemia cells. Br J Haematol. 2010;150(1):108–111. | ||
Göckeritz E, Kerwien S, Baumann M, et al. Efficacy of phosphatidylinositol-3 kinase inhibitors with diverse isoform selectivity profiles for inhibiting the survival of chronic lymphocytic leukemia cells. Int J Cancer. 2015;137(9):2234–2242. | ||
Paul J, Soujon M, Wengner AM, et al. Simultaneous inhibition of PI3Kδ and PI3Kα induces ABC-DLBCL regression by blocking BCR-dependent and -independent activation of NF-κB and Akt. Cancer Cell. 2017;31(1):64–78. | ||
Erdmann T, Klener P, Lynch JT, et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood. 2017;130(3):310–322. | ||
Azab F, Vali S, Abraham J, et al. PI3KCA plays a major role in multiple myeloma and its inhibition with BYL719 decreases proliferation, synergizes with other therapies and overcomes stroma-induced resistance. Br J Haematol. 2014;165(1):89–101. | ||
Glauer J, Pletz N, Schön M, et al. A novel selective small-molecule PI3K inhibitor is effective against human multiple myeloma in vitro and in vivo. Blood Cancer J. 2013;3:e141. | ||
Ali AY, Wu X, Eissa N, et al. Distinct roles for phosphoinositide 3-kinases γ and δ in malignant B cell migration. Leukemia. Epub 2018 Jan 31. | ||
Balakrishnan K, Peluso M, Fu M, et al. The phosphoinositide-3-kinase (PI3K)-δ and γ inhibitor, IPI-145 (duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia. 2015;29(9):1811–1822. | ||
Kaneda MM, Messer KS, Ralainirina N, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–442. | ||
Okabe S, Tauchi T, Tanaka Y, Sakuta J, Ohyashiki K. Combination therapy with copanlisib and ABL tyrosine kinase inhibitors against Philadelphia chromosome-positive resistant cells. Oncotarget. 2016;7(33):53116–53126. | ||
Chiron D, Di Liberto M, Martin P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–1035. | ||
Dong S, Guinn D, Dubovsky JA, et al. IPI-145 antagonizes intrinsic and extrinsic survival signals in chronic lymphocytic leukemia cells. Blood. 2014;124(24):3583–3586. | ||
Yahiaoui A, Meadows SA, Sorensen RA, et al. PI3Kδ inhibitor idelalisib in combination with BTK inhibitor ONO/GS-4059 in diffuse large B cell lymphoma with acquired resistance to PI3Kδ and BTK inhibitors. PLoS One. 2017;12(2):e0171221. | ||
Kohrt HE, Sagiv-Barfi I, Rafiq S, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123(12):1957–1960. | ||
Hassenrück F, Knödgen E, Göckeritz E, et al. Sensitive detection of the natural killer cell-mediated cytotoxicity of anti-CD20 antibodies and its impairment by B cell receptor pathway inhibitors. Biomed Res Int. 2018;2018:1023490. | ||
Lapalombella R, Yeh YY, Wang L, et al. Tetraspanin CD37 directly mediates transduction of survival and apoptotic signals. Cancer Cell. 2012;21(5):694–708. | ||
Krause G, Baki I, Kerwien S, et al. Cytotoxicity of the CD37 antibody BI 836826 against chronic lymphocytic leukaemia cells in combination with chemotherapeutic agents or PI3K inhibitors. Br J Haematol. 2016;173(5):791–794. | ||
Patnaik A, Appleman LJ, Tolcher AW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol. 2016;27(10):1928–1940. | ||
Pongas G, Cheson BD. PI3K signaling pathway in normal B cells and indolent B-cell malignancies. Semin Oncol. 2016;43(6):647–654. | ||
Greenwell IB, Flowers CR, Blum KA, et al. Clinical use of PI3K inhibitors in B-cell lymphoid malignancies: today and tomorrow. Expert Rev Anticancer Ther. 2017;17(3):271–279. | ||
Cheah CY, Nastoupil LJ, Neelapu SS, et al. Lenalidomide, idelalisib, and rituximab are unacceptably toxic in patients with relapsed/refractory indolent lymphoma. Blood. 2015;125(21):3357–3359. | ||
Barr PM, Saylors GB, Spurgeon SE, et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood. 2016;127(20):2411–2415. | ||
Cheah CY, Fowler NH. Idelalisib in the management of lymphoma. Blood. 2016;128(3):331–336. | ||
Dreyling M, Santoro A, Mollica L, et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol. 2017;35(35):3898–3905. | ||
Gopal AK, Kahl BS, de Vos S, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008–1018. | ||
Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J. 1998;333(Pt 3):471–490. | ||
Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–290. | ||
Sarker D, Ang JE, Baird R, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(1):77–86. | ||
Brown JR, Davids MS, Rodon J, et al. Phase I trial of the pan-PI3K Inhibitor pilaralisib (SAR245408/XL147) in patients with chronic lymphocytic leukemia (CLL) or relapsed/refractory lymphoma. Clin Cancer Res. 2015;21(14):3160–3169. | ||
Dreyling M, Morschhauser F, Bouabdallah K, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017;28(9):2169–2178. | ||
Markham A. Copanlisib: first global approval. Drugs. 2017;77(18):2057–2062. | ||
Batlevi CL, Younes A. Revival of PI3K inhibitors in non-Hodgkin’s lymphoma. Ann Oncol. 2017;28(9):2047–2049. | ||
Zinzani P, Wagner-Johnston N, Miller C, et al. DYNAMO: a phase 2 study demonstrating the clinical activity of duvelisib in patients with double-refractory indolent non-Hodgkin lymphoma. Hematol Oncol. 2017;35 Suppl 2:69–70. | ||
Younes A, Salles G, Martinelli G, et al. Pan-phosphatidylinositol 3-kinase inhibition with buparlisib in patients with relapsed or refractory non-Hodgkin lymphoma. Haematologica. 2017;102(12):2104–2112. | ||
Lenz G, Hawkes E, Verhoef G, et al. Phase II study of single-agent copanlisib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). J Clin Oncol. 2017;35(15 Suppl):7536. | ||
Kahl B, Byrd JC, Flinn IW, et al. Clinical safety and activity in a phase 1 study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase p110δ, in patients with relapsed or refractory non-Hodgkin lymphoma. Blood. 2010;116(21):1777. | ||
Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398–3405. | ||
Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21(8):922–926. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.