")
Back to Archived Journals » Hypoxia » Volume 4
Control of the heart rate of rat embryos during the organogenic period
Authors Ritchie HE, Ragnerstam C, Gustafsson E, Jonsson JM, Webster WS
Received 15 June 2016
Accepted for publication 26 August 2016
Published 8 November 2016 Volume 2016:4 Pages 147—159
DOI https://doi.org/10.2147/HP.S115050
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Helen E Ritchie,1 Carolina Ragnerstam,2 Elin Gustafsson,2 Johanna M Jonsson,2 William S Webster2
1Discipline of Biomedical Science, Sydney Medical School, University of Sydney, Lidcombe, 2Department of Anatomy and Histology, Sydney Medical School, University of Sydney, Sydney, NSW, Australia
Abstract: The aim of this study was to gain insight into whether the first trimester embryo could control its own heart rate (HR) in response to hypoxia. The gestational day 13 rat embryo is a good model for the human embryo at 5–6 weeks gestation, as the heart is comparable in development and, like the human embryo, has no functional autonomic nerve supply at this stage. Utilizing a whole-embryo culture technique, we examined the effects of different pharmacological agents on HR under normoxic (95% oxygen) and hypoxic (20% oxygen) conditions. Oxygen concentrations ≤60% caused a concentration-dependent decrease in HR from normal levels of ~210 bpm. An adenosine agonist, AMP-activated protein kinase (AMPK) activator and KATP channel opener all caused bradycardia in normoxic conditions; however, putative antagonists for these systems failed to prevent or ameliorate hypoxia-induced bradycardia. This suggests that the activation of one or more of these systems is not the primary cause of the observed hypoxia-induced bradycardia. Inhibition of oxidative phosphorylation also decreased HR in normoxic conditions, highlighting the importance of ATP levels. The β-blocker metoprolol caused a concentration-dependent reduction in HR supporting reports that β1-adrenergic receptors are present in the early rat embryonic heart. The cAMP inducer colforsin induced a positive chronotropic effect in both normoxic and hypoxic conditions. Overall, the embryonic HR at this stage of development is responsive to the level of oxygenation, probably as a consequence of its influence on ATP production.
Keywords: embryonic heart rate, embryo, bradycardia, in vitro, ATP, hypoxia
Introduction
The development of mammalian cardiovascular system is dependent on transcription factors that are stabilized only at low oxygen levels. These hypoxia-inducible factors (HIF-1 and HIF-2α) in conjunction with vascular endothelial growth factor (VEGF) are essential for cardiovascular development. Mouse embryos that lack expression of HIF-1 die between gestational days (GD) 9.5 and 10.5, the period when cardiovascular development normally begins.1,2
The stabilization of HIF-1 transcripts starts at about 5% oxygen (37.5 mmHg) and is inversely proportional to oxygen concentrations below 5% with a maximal response at 0.5% (7.5 mmHg).3 These oxygen levels fall within the range reported for the placenta during the first trimester of pregnancy.4 Oxygen tension in the human placenta is <20 mmHg up to 8 weeks gestation increasing to 50 mmHg at 12 weeks.5,6 After the first trimester, embryonic growth alone may be responsible for an appropriate oxygen gradient in the fetus as newly formed cells are pushed away from the developing capillary network. Similarly, in normal mouse embryos, the hypoxia marker pimonidazole has been reported to form adducts, implying oxygen levels in some parts of the embryo are <10 mmHg, <2% oxygen in some tissues.7
Although some degree of tissue hypoxia appears to be important for development, transient severe maternal hypoxia has been observed to rapidly induce bradycardia in mouse embryos8 and can put the embryo at risk. In the human embryo, a heart rate (HR) of 10% or more below normal at 6.3–7.0 weeks gestation was associated with a spontaneous abortion rate of 60.6% compared with 9.1% for those with a normal HR.9 Studies in pregnant rats showed that while many embryos could survive 60–90 minutes of severe hypoxia during the organogenic period, there was an increased frequency of birth defects and growth retardation in the surviving embryos.10–12 The initial aim of the present study was to examine the effects of reduced oxygen concentrations on cardiac activity.
It is not clear as to what extent the first trimester embryo can or needs to control its own oxygen status. The adult heart, in response to increased demand, such as intense physical activity, can increase the HR by approximately threefold from its resting rate, and in conjunction with increased stroke volume can increase cardiac output by approximately eightfold. Adult cardiac activity is modulated by the cardiac parasympathetic and sympathetic innervation and circulating catecholamines. Unlike the adult heart, the embryonic heart during the organogenic period does not have an extrinsic nerve supply13–15 and may not be able to suddenly increase HR and/or cardiac output. This is not necessarily a problem; the first trimester embryo does not undertake much physical activity and its cardiovascular demands are unlikely to vary much, other than a slow increase with development and growth. However, the early embryonic rodent heart does have β-adrenergic16–18 and adenosine19 receptors as well as KATP channels,20 suggesting the potential to respond to changes in oxygen and ATP status.
The second aim of this study was to determine possible mechanisms underlying the decrease in HR induced by hypoxia. There are both direct and indirect ways by which cells detect the changes in oxygen status. As described earlier, HIF-1 transcription factors are stabilized only at lower concentrations of oxygen, setting off a cascade of events resulting in the upregulation of genes that have HIF-responsive elements.21 These include genes involved in glycolysis, angiogenesis and other pathways which increase a cell’s chance of survival.
Another possible mechanism is related to the fall in ATP production, associated with decreased oxygen availability, and the resultant increase in the AMP/ATP ratio22 and accumulation of adenosine.23 Embryonic cardiac cells have several potential systems for detecting such changes. These include 1) activity of the enzyme AMP-activated protein kinase (AMPK), which responds to changes in the AMP/ATP ratio and can switch off ATP-consuming pathways,24 2) adenosine receptors that respond to the increased adenosine levels19 and 3) activation of cardiac KATP channels which are opened only when ATP level decreases.20,25,26 Involvement of one or more of these systems would imply the hypoxia-induced decrease in HR is an ordered process, rather than a passive consequence of ATP depletion.
In order to gain insight into whether the first trimester embryo could control its own HR in response to hypoxia, we used the GD13 whole-rat embryo. The GD13 rat heart is comparable in development to the human heart at 5–6 weeks gestation,27 and like the human embryo, the rat embryo heart at this stage has no functional autonomic nerve supply.15,28 We examined the effects of different pharmacological agonists and antagonists of these various systems on the HR under normoxic and hypoxic conditions.
Materials and methods
The University of Sydney Animal Ethics Committee approved all animal work in this study. All procedures performed in studies involving animals complied with the NSW Animal Research Act (and associated regulations) and the 8th Edition of the Australian code for the care and use of animals for scientific purposes.
Whole-embryo culture
Sprague–Dawley (SD) rats were mated overnight and examined the next morning for a sperm-positive vaginal smear. The rats that had mated were separated and this day was considered GD0. On GD13, the pregnant dams were killed by CO2 inhalation. The uterus was removed and washed in phosphate-buffered saline (Sigma-Aldrich Co., St Louis, MO, USA). The decidua and Reichart’s membrane were removed, leaving the embryo surrounded by an intact yolk sac. A small incision was made in the yolk sac, avoiding damage to major blood vessels, and the embryo was eased through the incision and the amnion was opened. Each embryo was placed in a glass culture bottle containing 2.5 mL DMEM (D1145; Sigma-Aldrich Co.), and then placed in a rotating culture system at 38°C (BTC Engineering, Cambridge, UK). The bottles were rotated at 11 rotations/min and continuously gassed with 95% O2 and 5% CO2. After 1-hour incubation, each embryo, still in its culture bottle, was examined under a dissecting microscope (Leica M 420; Leica Microsystems, Heerbrugg, Switzerland), equipped with a heating stage to maintain the temperature of the culture fluid at 38°C. The embryos were videographed for 20 seconds (Olympus DP70; Olympus Australia Pty Ltd, Melbourne, VIC, Australia) and used in the following experiments. Damaged embryos with HR <160 bpm were rejected at this stage. For each experiment, approximately 10 embryos were included in each treatment group.
Experiment 1: The effect of different concentrations of oxygen on the HR of GD13 embryos
GD13 rat embryos were initially gassed for 1 hour with 95% O2 and 5% CO2. For the next 2–2.5 hours (depending on the experiment), the embryos were continuously gassed with a mixture of O2, N2 and CO2 gases, where the CO2 content was 5% and the O2 content was 95%, 80%, 60%, 40%, 20% or 0% balanced with N2. The embryos were videographed again at the end of the experiment.
Experiment 2: The effect of tested drugs on HR of embryos in 95% or 20% oxygen
GD13 rat embryos were cultured for 1 hour as previously described. After videoing, one of the following test drugs was added to the culture medium and the embryos were incubated for a further 1 or 2 hours while being gassed with either 95% O2, 5% CO2 or 20% O2, 75% N2, 5% CO2. The embryos were videographed again at various time intervals after drug administration depending on the experiment. The 20% O2 level was chosen as the model for hypoxic conditions, as other studies have shown that the GD11 rat embryo cultured in 20% oxygen was markedly hypoxic,29 and in the preliminary studies, this concentration was shown to cause a >10% decrease in HR in the GD13 rat embryo.
Chemicals
Details of chemicals used and concentration ranges tested as well as rationale for use are listed in Table 1.
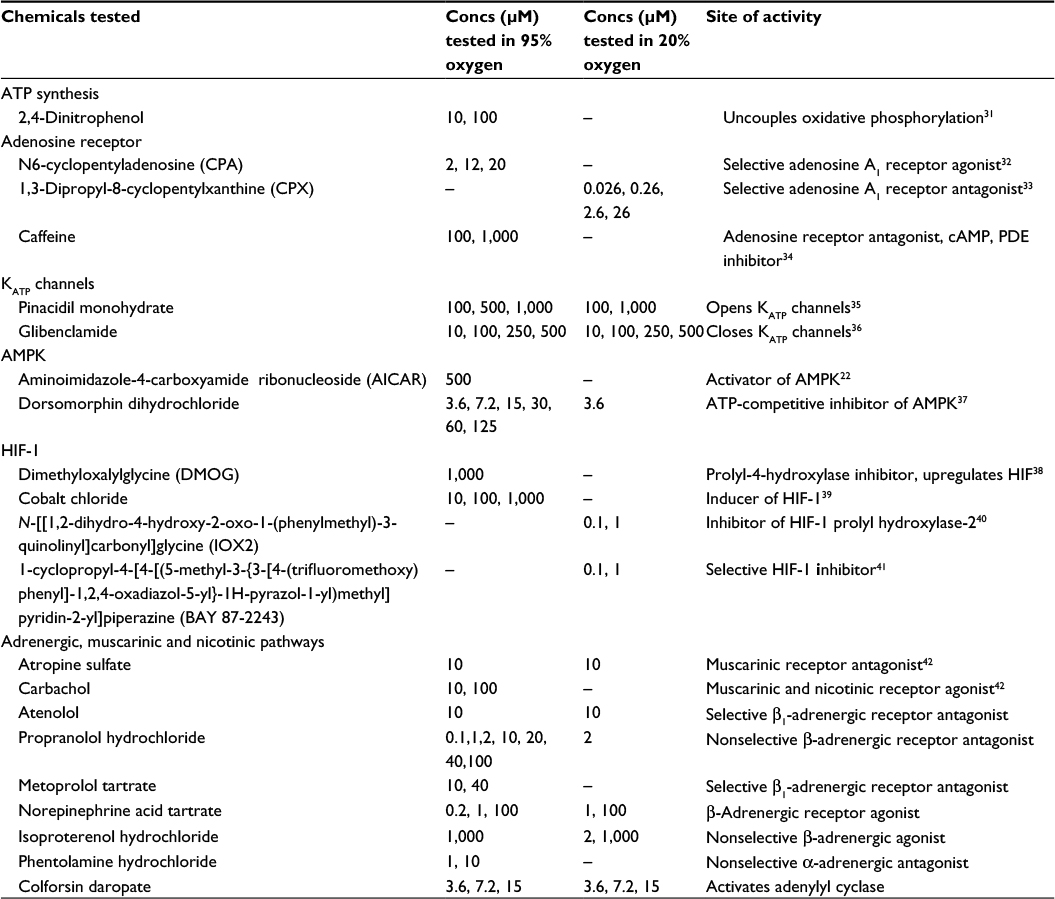 | Table 1 List of chemicals and tested concentration range Abbreviations: Concs, concentrations; AMPK, AMP-activated protein kinase; HIF, hypoxia-inducible factor. |
Aminoimidazole-4-carboxyamide ribonucleoside (AICAR) and colforsin daropate were sourced from Abcam (Cambridge, UK); dorsomorphin dihydrochloride (Chemdea, Ridgewood, NJ, USA); dimethyloxalylglycine (DMOG), N-[[1,2-dihydro-4-hydroxy-2-oxo-1-(phenylmethyl)-3-quinolinyl]carbonyl]glycine (IOX2) and 1-cyclopropyl-4-[4-[(5-methyl-3-{3-[4-(trifluoromethoxy) phenyl]-1,2,4-oxadiazol-5-yl}-1H-pyrazol-1-yl)methyl]pyridin-2-yl]piperazine (BAY 87-2243) from MedChem Express (Princeton, NJ, USA; norepinephrine acid tartrate (Hospira Pty Ltd, Melbourne, VIC, Australia) and epinephrine acid tartrate (Martindale Pharma, High Wycombe, UK). All other reagents were sourced from Sigma-Aldrich Co.
AICAR, N6-cyclopentyladenosine (CPA), 1,3-dipropyl-8-cyclopentylxanthine (CPX), pinacidil monohydrate, glibenclamide, IOX2, BAY 87-2243, atropine and atenolol were dissolved in dimethyl sulfoxide (Sigma-Aldrich Co.), and all other chemicals were dissolved in water. Norepinephrine acid tartrate and epinephrine acid tartrate were purchased as a solution for injection and diluted with water.
For all of these studies, appropriate starting concentrations of drugs were determined by preliminary studies. If solubility permitted, the starting concentration was 1 mM. The tested concentrations were progressively reduced until a no-observable-effect concentration (NOEC) was established. The volume of drug solution or vehicle added to the culture bottle was 25 μL for dimethyl sulphoxide (DMSO) soluble drugs and 50 μL for the water-soluble drugs. Treatment order was randomized.
Image analysis
The video recordings of the embryonic hearts were examined and analyzed using software developed at the University of Uppsala.30 Where possible, the atria and ventricles of the heart were identified on the video image and manually marked. The program measures light–dark intensity changes in the selected area of the heart, and on the basis of these changes can calculate HR and individual inter-beat time over a 15-second period. The video recordings were made at a speed of 30 frames per second.
Data analysis
The great majority of control embryos survive for a period of 2 hours in culture when gassed with 95% oxygen. It is assumed that any embryos that die under these conditions were probably damaged during the explant process. These embryos are not included in HR calculations. These “dead” embryos are recorded separately.
The change in embryonic HR was calculated by the following formula: (HRtime2-HRtime1)/HRtime1×100 (time 1 is the HR immediately before intervention, time 2 is the HR after intervention).
Statistics
In our experience, a sample size of 10 treated embryos is sufficient to detect a difference in the mean change in HR of ~10% with power of 0.8 and a type I error probability of 0.05.
Differences in change in HR before and after addition of drug were compared between control and treated embryos using an independent samples t-test or analysis of variance (ANOVA). Homogeneity of variance was tested by Levene’s test. For post hoc analyses, a Dunnett two-sided t-test was used with comparison to control only. If the variance was not homogenous, a Dunnett T3-test was used. HR (bpm) over time was tested using a repeated measure ANOVA. For all comparisons, differences were considered statistically significant at P<0.05.
Results
Effects of different concentrations of oxygen on the HR of GD13 embryos
GD13 rat embryos kept in 95% oxygen for 1 hour had HR >210 bpm (Figure 1A). The embryos were then maintained for a further 2 hours in 95%, 80%, 60%, 40%, 20% or 0% oxygen. There was no significant difference in the HR of embryos in 95% or 80% oxygen throughout the culture. Embryos maintained HR >200 bpm. In lower oxygen concentrations, the HR had significantly decreased after 30 minutes when compared to HR at start of culture. After 2 hours in 60% oxygen, mean HR had decreased to 193 bpm (compared to 207 bpm in embryos cultured in 95% oxygen). The HR of embryos in 40% oxygen had decreased to 171 bpm, 20% oxygen to 119 bpm and 0% oxygen to 50 bpm. All embryos survived in 95%, 80%, 60% and 40% oxygen. In both 20% and 0% oxygen levels, about one-third of the embryos had no heartbeat after 120 minutes. When these embryos were regassed with 95% oxygen, the heart restarted and for most embryos the HR recovered to normal levels within 30 minutes (Figure 1B).
 | Figure 1 Effects of differing oxygen levels on HR of GD13 rat embryo. Notes: (A) HR (bpm) had significantly decreased compared to starting HR from 30 to 120 minutes when oxygen levels were <80%. (B) HR (bpm) recovered to control levels following return to 95% oxygen. Data are presented as bpm ± SEM. Abbreviation: SEM, standard error of the mean; bpm, beats per minute; HR, heart rate; GD, gestational day. |
Possible causes of hypoxia-induced bradycardia
Role of reduced ATP
Embryos in 95% oxygen were exposed to the oxidative phosphorylation inhibitor 2,4-dinitrophenol (Figure 2A). At 100 mM, the HR was reduced by ~25% and at 10 mM by ~10%.
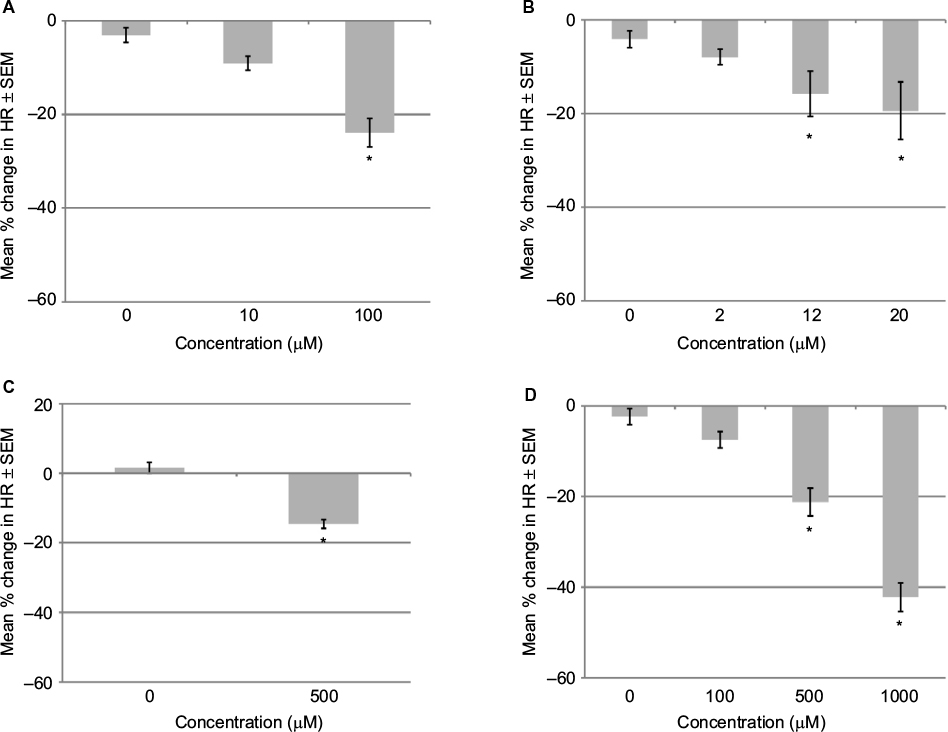 | Figure 2 Effects on HR of GD 13 rat embryo in 95% oxygen after 1 hour. Notes: (A) 2,4-Dinitrophenol induced a concentration-dependent decrease in embryonic HR. (B) N6-cyclopentyladenosine (CPA) induced a concentration-dependent decrease in embryonic HR. (C) Aminoimidazole-4-carboxyamide ribonucleoside (AICAR) induced a decrease in embryonic HR. (D) Pinacidil induced a concentration-dependent decrease in embryonic HR. Changes in HR (expressed as percent change from starting HR). Data are presented as mean ± SEM. *P<0.05 when compared to control. Abbreviations: HR, heart rate; SEM, standard error of the mean; GD, gestational day. |
Role of increased adenosine
Adenosine has an extremely short half-life of only a few seconds in plasma, so a long-lasting A1 adenosine receptor agonist (CPA) was added to GD13 rat embryos that were maintained in 95% oxygen (Figure 2B). CPA at 20 μM decreased the HR by 19.4% after 1 hour. In a 2-hour exposure study, CPA at 12 μM decreased the HR by 17.7% after 30 minutes (P=0.008). The HR gradually recovered with time and was –15.8% after 1 hour and –13.2% after 2 hours. A lower concentration (2 μM) decreased HR by 7.9% after 1 hour. A decrease in HR of 4.3% was seen in the DMSO controls. Preliminary studies with higher concentrations of CPA did not cause more severe HR reduction but was associated with arrhythmias.
To see if blocking the A1 adenosine receptor would alter the response to hypoxia, embryos in 20% oxygen were exposed to a wide range of concentrations of CPX (Table 1). Embryos exposed to both hypoxia and CPX showed a similar pattern of bradycardia as seen with hypoxia alone. The nonspecific adenosine receptor antagonist caffeine was tested at 100 μM and 1 mM, with the same results.
Role of AMPK
AICAR, an AMPK activator, was added to rat embryos in 95% oxygen. High concentrations of AICAR (500 μM) decreased the HR by 15% after 1 hour (Figure 2C). Since this decrease was near the limit of detection, a lower concentration was not tested.
To determine if an inhibitor of AMPK would prevent the hypoxia-induced bradycardia, embryos in 20% oxygen were exposed to dorsomorphin. Preliminary studies showed that dorsomorphin caused severe bradycardia at concentrations of 7.2 μM or higher. Dorsomorphin at 3.6 μM and 1 hour of hypoxia caused a 61.5% decrease in HR compared to 41.8% for hypoxia alone (Figure 3A). Dorsomorphin 3.6 μM was also tested on embryos in 95% oxygen. A decrease in HR of 14% was observed compared with an increase of 3.3% in the controls.
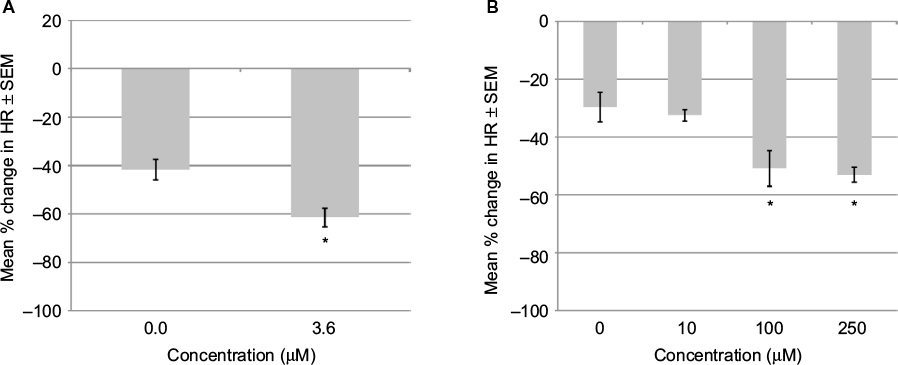 | Figure 3 Effects on HR of GD 13 rat embryo in 20% oxygen after 1 hour. Notes: (A) Dorsomorphin did not prevent the drop in HR induced by 20% oxygen. (B) Glibenclamide did not prevent the drop in HR induced by 20% oxygen. Changes in HR (expressed as percent change from starting HR). Data are presented as mean ± SEM. *P<0.05 when compared to control. Abbreviations: HR, heart rate; SEM, standard error of the mean; GD, gestational day. |
Role of KATP channels
The KATP channel opener, pinacidil, was added to GD13 rat embryos that were maintained in 95% oxygen. A concentration-dependent decrease in HR was observed with a NOEC of 100 μM (Figure 2D). To see whether closing the KATP channels would alter the hypoxia-induced bradycardia, embryos in 20% oxygen were exposed to a range of concentrations of glibenclamide (Table 1). At the highest concentration (250 μM), bradycardia was significantly more severe than that caused by hypoxia alone (Figure 3B). Glibenclamide was also tested on embryos maintained in 95% oxygen with the three highest concentrations (100, 250 and 500 μM) significantly decreasing the HR.
Role of HIF-1
The addition of DMOG at 1 mM to GD13 rat embryos in 95% oxygen showed no effect on HR (Figure 4A). Cobalt chloride, which also stabilizes HIF-1, was also added to embryos in 95% oxygen. At 1 mM concentration, it reduced the HR by 18%. No significant effect was observed at 100 or 10 μM.
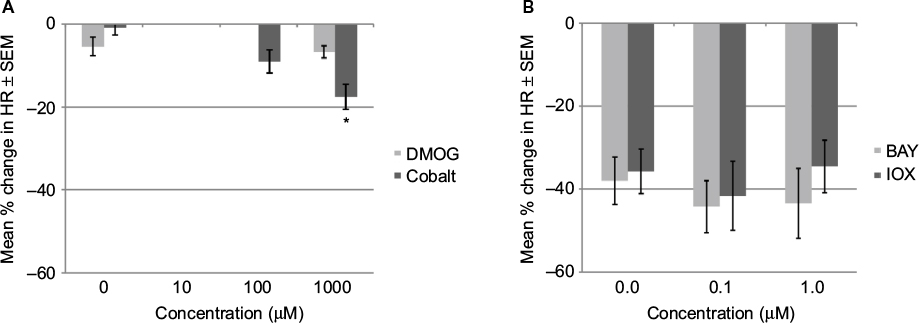 | Figure 4 Effects on HR of GD 13 rat embryo after 1 hour. Notes: (A) DMOG and cobalt chloride induced minimal changes in HR in 95% oxygen. (B) IOX2 or BAY 87-2243 did not prevent drop in heart rate in 20% oxygen. Change in HR (expressed as percent change from starting HR). Data are presented as mean ± SEM. *P<0.05 when compared to control. Abbreviations: HR, heart rate; SEM, standard error of the mean; GD, gestational day; DMOG, dimethyloxalylglycine; IOX2, N-[[1,2-dihydro-4-hydroxy-2-oxo-1-(phenylmethyl)-3-quinolinyl]carbonyl]glycine; BAY 87-2243, 1-cyclopropyl-4-[4-[(5-methyl-3-{3-[4-(trifluoromethoxy) phenyl]-1,2,4-oxadiazol-5-yl}-1H-pyrazol-1-yl)methyl]pyridin-2-yl]piperazine. |
To see if an inhibitor of HIF-1 would alter the hypoxia-induced bradycardia, embryos in 20% oxygen were exposed to the HIF-1 inhibitors IOX2 or BAY 87-2243 (0.1 and 1 mM). At these concentrations, there was no difference in HR compared to that caused by 20% oxygen alone (Figure 4B). Preliminary studies with both drugs at higher concentrations showed more severe reduction in HR.
Activity of autonomic receptors in the embryonic heart
Embryos in 95% oxygen were exposed to the muscarinic receptor agonist carbachol. Carbachol at 10 and 100 μM concentrations did not significantly alter the HR. To test the effect of a muscarinic ACh receptor blocker, atropine 10 μM was added to embryos in 20% or 95% oxygen. There was no significant effect on HR compared to control embryos.
To examine the role of β1-adrenergic receptors, embryos in 95% oxygen were exposed to the receptor antagonists atenolol, propranolol or metoprolol. Atenolol at 10 μM had no effect on HR, propranolol caused a concentration-dependent reduction in HR with an NOEC of 1 μM and metoprolol caused a concentration-dependent reduction in HR with an NOEC <10 μM (Figure 5A).
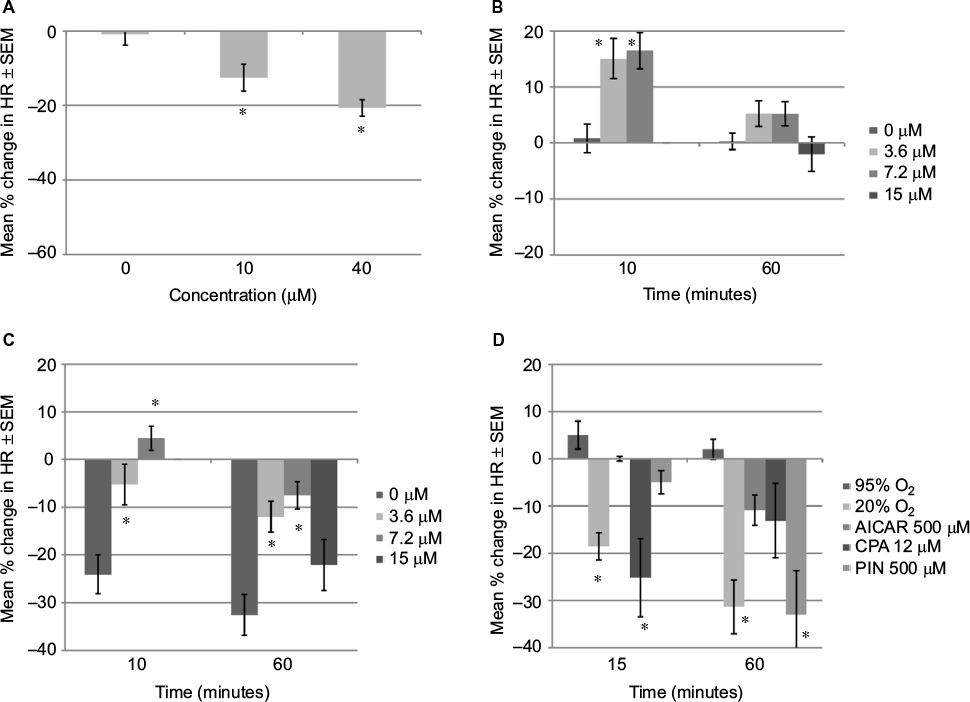 | Figure 5 Effects on HR of GD 13 rat embryo Notes: (A) Metoprolol induced a concentration-dependent decrease in embryonic HR in 95% oxygen after 1 hour. (B) Colforsin induced an increase in embryonic HR in 95% oxygen compared to control after 10 minutes but not 60 minutes. (C) Colforsin (3.6 and 7.2 mM) reduced the drop in embryonic HR in 20% oxygen compared to control after 10 and 60 minutes. (D) Heart rate decreased significantly within the first 15 minutes in 20% oxygen and 12 mM CPA compared to 95% oxygen. Change in HR (expressed as percent change from starting HR). Data are presented as mean ± SEM. *P<0.05 when compared to control. Abbreviations: HR, heart rate; SEM, standard error of the mean; AICAR, aminoimidazole-4-carboxyamide ribonucleoside; CPA, N6-cyclopentyladenosine; PIN, pinacidil. |
Embryos in 20% oxygen were exposed to the β1-adrenergic receptor agonists, norepinephrine or isoproterenol. Rather than increasing the HR, norepinephrine at 1 or 100 μM decreased the HR further over hypoxia alone, although the difference was not significant. Isoproterenol at 2 μM or 1 mM did not alter the HR response to hypoxia. Norepinephrine (0.2, 1 and 100 μM) and isoproterenol (1 mM) were also tested on embryos in 95% oxygen, with no effect on HR.
To investigate whether there is any α-adrenergic activity in the GD13 embryonic heart, embryos maintained in 95% oxygen were exposed to α-adrenergic receptor antagonist phentolamine (1 and 10 μM). The higher concentration caused a severe reduction in HR, whereas 1 μM concentration showed no effect.
An activator of adenylyl cyclase, colforsin, was also tested. This drug increases intracellular cAMP bypassing the β1-adrenergic receptor. Preliminary studies performed to determine the concentrations indicated that 36 μM had a negative effect on the HR of embryos in 95% oxygen but 3.6 and 7.2 μM caused a small increase in HR (+15% and 16%, respectively) within 15 minutes (Figure 5B). This increase had disappeared by the end of culture (60 min). In 20% oxygen, colforsin also showed a positive chronotropic effect (Figure 5C). Within 10 minutes, control HRs had dropped by 24% in 20% oxygen, while embryos cultured with colforsin had only dropped by 5.2% (3.6 μM colforsin) or increased by 4.4% (7.2 μM colforsin). By 1 hour, control embryos in 20% oxygen had an HR reduction of 34.7%, but with colforsin 3.6 or 7.2 μM the reduction was 12.0% or 7.5%, respectively.
Initial embryonic response
To determine how quickly the embryos respond to the changing conditions, the HR of a subset of embryos was recorded at 15 minutes as well as 1 hour after addition of drug or change in gas mixture. Embryos were cultured in either 95% oxygen, 20% oxygen, or 500 μM AICAR, 12 μM CPA or 500 μM pinacidil in 95% oxygen.
Within 15 minutes, HR of embryos in 20% oxygen had dropped by 19% (Figure 5D). Of the three drugs tested that also decreased HR in 95% oxygen, only CPA initiated a rapid response, dropping the HR by 25% within 15 minutes. AICAR and pinacidil showed little or no initial response (0% and 5%, respectively).
Discussion
HR in reduced oxygen concentrations
The GD13 rat embryo used for this study is similar in size and cardiovascular development to a 5–6-week-old human embryo.27 In the present in vitro study, GD13 rat embryos maintained a regular HR of about 210 bpm when the culture medium was in equilibrium with either 95% or 80% oxygen. Although 95% oxygen gives a very high pO2 (~430 mmHg) in the culture media,43 much greater than that expected in vivo, the embryo in vitro does not have a chorioallantoic circulation which is necessary for embryonic respiration from ~GD11 in rats.44 Embryonic pO2 is reported to be 29.5 mmHg in GD12.5 mouse embryos (equivalent to GD14.5 in the rat) when cultured in 95% oxygen.45 Exposure to lower oxygen concentrations (60%, 40%, 20% or 0%) caused a rapid decrease in the HR within 30 minutes. Even when equilibrated with 0% oxygen (95% N2, 5% CO2), most embryos were able to maintain a low level of cardiac activity. When the embryos were regassed with 95% oxygen after remaining for 2 hours without oxygen, the HR rapidly recovered to a normal rate, even in embryos where the heartbeat had stopped. This suggests that the embryos entered a survival mode restricting cellular activity in an ordered manner in response to falling levels of ATP. This is in agreement with studies in pregnant rats that showed many embryos during the organogenic period could survive 60–90 minutes of severe hypoxia caused by obstructing the blood supply to the uterus.10–12
It is likely that the GD13 rat embryonic heart is a major consumer of ATP and reduction in the HR is an expected consequence of hypoxia, since ion transport for the cardiac action potential (AP) is ultimately dependent on ATPase ion pumps.46 The decrease in HR could be a passive consequence of reduced availability of ATP caused by hypoxia or it could be an orchestrated response whereby a fall in ATP is detected at the cellular level and among other responses, HR is reduced. This would suggest processes necessary for cell viability are prioritized.
Limitations of the experimental model
There are limitations in testing the various inhibitors and activators in the embryo culture system. In most cases, compounds were first tested at the highest achievable concentration. If this concentration had no significant effect on the HR, then no further testing was undertaken. If there was a significant reduction in HR, then the compound was further tested at lower concentrations until a no-effect or low-effect concentration was established. However, while a positive result may be due to the primary property of the tested compound, it may also be due to additional chemical properties, eg, cardiac ion channel blockade, that can occur at high concentrations. Information regarding these additional properties is rarely available.
Causes of hypoxia-induced bradycardia in rat embryos
The most likely reason that hypoxia causes bradycardia in rat embryos is that hypoxia causes a reduction in ATP production. In the present study, exposure of rat embryos in 95% oxygen to 100 μM 2,4-Dinitrophenol, an uncoupler of oxidative phosphorylation,31 led to a rapid reduction in HR, equivalent to that seen with exposure to 20% oxygen.47 This shows that reduced ATP alone causes bradycardia and does not need to be accompanied by hypoxia.
The adenosine agonist CPA, the AMPK activator AICAR and the KATP channel opener pinacidil all caused bradycardia in normoxic conditions, suggesting that cardiac adenosine receptors, KATP channels and AMPK are functionally active and capable of influencing embryonic HR. At least in the adults, all the three systems would normally be activated by hypoxia,23–25 resulting in bradycardia. We hypothesized that blocking or deactivating these pathways during hypoxia would prevent this drop in HR.
Adenosine A1 receptor (A1R) mRNA can be detected in the rat embryonic heart from the onset of cardiac contractions.19 Adenosine levels can increase up to 100-fold as a consequence of an hypoxia-induced fall in ATP.23 Using CPA to mimic this response has similarly been reported to inhibit the HR of mouse embryos, where the effect became more pronounced with gestational age.17,48 The activation of adenosine receptors in the adult heart leads to a fall in cAMP and inhibition of the pacemaker current I(f) leading to decreased HR.49 However, our attempts to prevent hypoxia-induced bradycardia using adenosine receptor antagonists, caffeine or CPX have failed. This finding is in contrast to others who reported caffeine prevented hypoxia-induced bradycardia in E9.5 mouse embryo (equivalent to GD11 in the rat) in vitro and reduced the effect in E12.5 isolated hearts.50 In those experiments, control HR was very low compared to well-oxygenated mouse embryos in vitro at this age.45 It is possible that the differences arise due to differing developmental ages or experimental techniques.
AMPK regulates energy metabolism by phosphorylation of key enzymes and is activated by a decrease in the ATP/AMP ratio.51 AMPK activity stimulates glucose uptake for glycolysis and rapidly reduces ATP consumption by suppressing mRNA translation to protein and Na+,K+-ATPase activity.51 Since Na+,K+-ATPase is an essential ion pump for cardiomyocytes, inhibition of this enzyme would be expected to drastically affect AP generation in cardiomyocytes and sinoatrial node (SAN) cells. In our studies, AICAR did induce a drop in embryonic HR in normoxia. However, the AMPK inhibitor, dorsomorphin, failed to prevent the drop in HR in embryos exposed to 20% oxygen. In fact, same concentrations of dorsomorphin induced a similar degree of HR reduction in both 95% oxygen and 20% oxygen levels, making it unlikely that AMPK is involved in the HR reduction caused by hypoxia. Although this was a negative finding, there is concern about the specificity of dorsomorphin as it inhibits other kinases with similar or greater potency.37
KATP channels are numerous in both cardiomyocytes and SAN cells of the adult heart.20 They have also been detected in the rat embryonic heart.52,53 Under normoxic conditions, the channels are closed and play no role in AP generation. However, during hypoxia, the channels open in response to falls in ATP levels and shorten the AP and decrease influx through L-type calcium channels.26,53 Although pinacidil reduced HR in normoxia, the channel closer glibenclamide failed to prevent hypoxia-induced bradycardia. The significance of these results is limited by uncertainty regarding chemical specificity with increasing concentrations. For example, glibenclamide inhibits KATP channels in rat ventricular myocytes with an IC50 of 6 mM,54 and at higher concentrations, it blocks other channels including hERG with an IC50 of 74 mM.55
Although it was demonstrated that agonists/activators for adenosine receptors, KATP channels and AMPK can all induce bradycardia in the GD13 rat embryo, putative antagonists for these systems failed to prevent or ameliorate hypoxia-induced bradycardia. This suggests that activation of one or more of these systems is not the primary cause of hypoxia-induced bradycardia in the GD13 rat embryo. It is unknown whether these systems are activated by hypoxia in the embryo as they are in the adults.
It seems unlikely that HIF-1 is involved in the rapid-onset bradycardia induced by reduced oxygen. The HIF-1 response to hypoxia appears to be a mechanism that prepares cells and tissues for long-term adaption to low oxygen. The system is utilized in embryonic development to coordinate cardiovascular development.7 An attempt was made to increase HIF-1 activity in the presence of adequate oxygenation to determine if HIF-1 stabilization induces bradycardia in the presence of adequate oxygen and ATP. Both cobalt chloride and DMOG have been reported to increase HIF-1 activity in the presence of adequate oxygen.56,57 DMOG at 1 mM had no effect on embryonic HR in adequate oxygen, while cobalt reduced HR by ~18% at 1 mM but had no effect at lower concentrations. Alternatively, if HIF-1 activation has a role in the induction of bradycardia, then destabilization of HIF-1 during hypoxia should limit this effect. However, under hypoxic conditions, the HR of embryos exposed to the HIF-1 inhibitors IOX2 or BAY 87-2243 was not different from that observed in control embryos.
As noted earlier, embryonic HR rapidly responds to a drop in oxygen levels. Of the drugs tested, only the adenosine agonist (CPA) induced a similarly rapid response.
Can the preinnervated heart respond to autonomic stimuli?
Although the midgestation rat embryonic heart does not have a functional autonomic nerve supply, it does have β1-adrenergic receptors.16 It has been suggested that fetal release of norepinephrine is necessary to maintain cardiovascular function during embryonic development.58 In the present study, β-adrenergic receptor agonists, norepinephrine and isoproterenol had either no effect or a negative effect on the embryonic HR during normoxia or hypoxia. The selective β1-adrenergic receptor antagonist metoprolol caused a concentration-dependent decrease in HR (Figure 3A), supporting the idea that there are functional β1-adrenergic receptors in the heart at this stage. The β-adrenergic receptor antagonist bupranolol was also reported to reduce HR by ~37% under high oxygen conditions (95%) using mouse embryos in culture.45 However, other β-adrenergic receptor antagonists (atenolol and propranolol) either showed no effect (atenolol) or slowed the embryonic HR (propranolol) but only at concentrations known to inhibit cardiac sodium and potassium channels.59–61 Unlike propanolol, metoprolol does not appear to inhibit sodium62 or hERG potassium channels63 at the tested concentrations. However, blockade of other important ion channels cannot be excluded as data are unavailable. The absence of an effect by atenolol may be related to its poor lipid permeability and low β1-adrenoceptor affinity compared to metoprolol (6–7 times lower).64
We were unable to detect an effect of β1-adrenergic receptor agonists on the embryonic HR. This suggests that the endogenous catecholamines have already fully activated the β1-adrenergic receptors in the normally oxygenated embryo.
It has been suggested that although the β1-adrenergic receptors are present in the embryo, they may not be fully coupled to their G protein until later in gestation.16,65 In support of this idea, the cAMP inducer colforsin produced a strong signal of activity. It caused a positive chronotropic effect in both normoxia and hypoxia, although the effect in normoxia was transitory. Colforsin directly activates the enzyme adenylyl cyclase and increases intracellular levels of cAMP while bypassing Gs-protein coupled β1-adrenergic receptors.16 These results are interesting as they suggest that in normoxia, the embryonic HR is not purely governed by the availability of ATP, as increasing cAMP by colforsin without increasing oxygen supply was associated with an increase in HR. This implies that embryonic HR is normally dampened to some extent.
Muscarinic receptors appear later in development.17,66 We and others have found no evidence that these receptors, if present, were functional.28,67 Carbachol did not affect the HR of embryos in 95% oxygen, while atropine did not cause a significant increase in HR of embryos in 20% oxygen.
Unlike the adult heart, the embryonic HR at this stage of development shows minimal response to autonomic agents. It is highly responsive to oxygen levels. Others have noted that the relatively high basal levels of adenylyl cyclase as well as protein kinase A and phosphodiesterases in the early embryo.68–70 This results in continuous synthesis and degradation of cAMP. L-type calcium channels are activated by cAMP-stimulated protein kinase A resulting in an influx of calcium ions and cardiomyocyte contraction.69 The early embryo is highly dependent on calcium ions for initiation of the AP.71,72 It is therefore possible that fluctuations in cAMP levels and ATP availability are ultimately responsible for embryonic HR.
Human embryonic HR
The human embryonic HR commences at about day 22 post-conception and the rate gradually increases to ~172 bpm at about 7 weeks gestation, then gradually declines to ~145 bpm at birth.73–75 For most of the organogenic period of development in the human, as in the rat, there is no functional autonomic nerve supply,15,28 and presumably the HR and cardiac output are the result of programmed development at any particular time. It is possible that the heart could respond to increased demand, since the use of colforsin demonstrated the heart has the capacity to beat faster. The human embryo during the organogenic period is small (crown-rump length ~13 mm at 6 weeks, ~30 mm at 8 weeks) and relatively inactive, suggesting cardiovascular demands are reasonably constant compared with postnatal life. However, as seen in this study, the HR is highly dependent on adequate oxygenation of the embryo and production of ATP. There is considerable evidence that oxygenation of the embryo in the first trimester of human development is restricted by placental features that limit placental circulation.5,6 This may be necessary for early development of the embryonic cardiovascular system with its dependence on HIF and VEGF. Insufficient transfer of oxygen, resulting in reduced availability of ATP, is likely to have a negative effect on the embryonic HR. While the embryo has mechanisms that allow it to tolerate such conditions for short periods, more severe bradycardia or bradycardia of prolonged duration are likely to have negative effects on embryonic development.10–12
A number of studies have shown that a slow embryonic HR is associated with a high risk of first trimester spontaneous abortion.9,76–78 In one study, the rate of spontaneous abortion was 60.6% for embryos with a low HR (<110 bpm) at 6.3–7.0 weeks gestation compared with 9.1% for those with a normal HR (120 bpm).9 Low HR could result from inadequate oxygenation as a consequence of poor placental development or as previously demonstrated, many therapeutic drugs including antidepressants, anticonvulsants, antipsychotics and antiarrhythmics have the potential to slow the embryonic heart.79
Conclusion
Hypoxia is an important stimulus in embryonic development, switching on a cascade of genes that are essential for cardiovascular development.80 Indeed, the ability to appropriately respond to physiological hypoxia is critical for embryonic survival, since mouse mutants that lack expression of HIF-1 die between GD9.5 and GD10.5.1,2 However, extreme hypoxia can also lead to embryonic death or malformation.10-12 In the present study, it was shown that embryonic HR is highly dependent on oxygen supply and ATP levels. There was no evidence of muscarinic control of HR. There was some evidence of β1-adrenergic control as the cAMP inducer colforsin induced a markedly positive chronotropic effect in both normoxic and hypoxic conditions. Overall, the embryonic HR at this stage of development is responsive to the level of oxygenation, probably as a consequence of its influence on ATP and cAMP production and subsequently calcium influx.
Disclosure
The authors report no conflicts of interest in this work.
References
Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17(11):3005–3015. | ||
Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–162. | ||
Jiang B-H, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O-2 tension. Am J Physiol. 1996;271(4 Pt 1):C1172–C1180. | ||
Schneider H. Oxygenation of the placental-fetal unit in humans. Respir Physiol Neurobiol. 2011;178(1):51–58. | ||
Jauniaux E, Watson A, Burton G. Evaluation of respiratory gases and acid-base gradients in human fetal fluids and uteroplacental tissue between 7 and 16 weeks’ gestation. Am J Obstet Gynecol. 2001;184(5):998–1003. | ||
Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress – a possible factor in human early pregnancy failure. Am J Pathol. 2000;157(6):2111–2122. | ||
Ream M, Ray AM, Chandra R, Chikaraishi DM. Early fetal hypoxia leads to growth restriction and myocardial thinning. Am J Physiol Regul Integr Comp Physiol. 2008;295(2):R583–R595. | ||
Furukawa S, Tinney JP, Tobita K, et al. Hemodynamic vulnerability to acute hypoxia in day 10.5–16.5 murine embryos. J Obstet Gynaecol Res. 2007;33(2):114–127. | ||
Doubilet PM, Benson CB. Outcome of first-trimester pregnancies with slow embryonic heart rate at 6–7 weeks gestation and normal heart rate by 8 weeks at US. Radiology. 2005;236(2):643–646. | ||
Leist KH, Grauwiler J. Fetal pathology in rats following uterine-vessel clamping on day 14 of gestation. Teratology. 1974;10(1):55–67. | ||
Webster WS, Lipson AH, Brown-Woodman PD. Uterine trauma and limb defects. Teratology. 1987;35(2):253–260. | ||
Franklin JB, Brent RL. Effect of uterine vascular clamping on development of rat embryos three to fourteen days old. J Morphol. 1964;115(2):273–290. | ||
Marvin WJ, Jr, Hermsmeyer K, McDonald RI, Roskoski LM, Roskoski R, Jr. Ontogenesis of cholingergic innervation in the rat heart. Circ Res. 1980;46(5):690–695. | ||
Shigenobu K, Tanaka H, Kasuya Y. Changes in sensitivity of rat heart to norepinephrine and isoproterenol during pre- and postnatal development and its relation to sympathetic innervation. Dev Pharmacol Ther. 1988;11(4):226–236. | ||
Papp JG. Autonomic responses and neurohumoral control in the human early antenatal heart. Basic Res Cardiol. 1988;83(1):2–9. | ||
Slotkin TA, Lau C, Seidler FJ. Beta-adrenergic-receptor overexpression in the fetal rat – distribution, receptor subtypes, and coupling to adenylate-cyclase activity via G-proteins. Toxicol Appl Pharmacol. 1994;129(2):223–234. | ||
Porter JGA, Rivkees SA. Ontogeny of humoral heart rate regulation in the embryonic mouse. Am J Physiol Regul Integr Comp Physiol. 2001;281:R401–R407. | ||
Robkin M, Shepard TH, Baum D. Autonomic drug effects on heart-rate of early rat embryos. Teratology. 1974;9(1):35–44. | ||
Rivkees SA. The ontogeny of cardiac and neural a1 adenosine receptor expression in rats Dev Brain Res. 1995;89(2):202–213. | ||
Toib A, Zhang HX, Broekelmann TJ, et al. Cardiac specific ATP-sensitive K+ channel (K-ATP) overexpression results in embryonic lethality. J Mol Cell Cardiol. 2012;53(3):437–445. | ||
Cummins EP, Taylor CT. Hypoxia-responsive transcription factors. Pflugers Arch. 2005;450(6):363–371. | ||
Hardie DG, Carling D. The AMP-activated protein kinase – fuel gauge of the mammalian cell? Eur J Biochem. 1997;246(2):259–273. | ||
Chu S, Xiong W, Zhang D, et al. Regulation of adenosine levels during cerebral ischemia. Acta Pharmacol Sin. 2013;34(1):60–66. | ||
Viollet B, Athea Y, Mounier R, et al. AMPK: lessons from transgenic and knockout animals. Front Biosci. 2009;14:19–44. | ||
Nichols CG. K-ATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–476. | ||
Koster JC, Knopp A, Flagg TP, et al. Tolerance for ATP-insensitive K-ATP channels in transgenic mice. Circ Res. 2001;89(11):1022–1029. | ||
Sissman NJ. Developmental landmarks in cardiac morphogenesis: comparative chronology. Am J Cardiol. 1970;25(2):141–148. | ||
Robkin MA, Shepard TH, Dyer DC. Autonomic receptors of early rat embryo heart – growth and development. Proc Soc Exp Biol Med. 1976;151(4):799–803. | ||
Chen EY, Fujinaga M, Giaccia AJ, Chen EY, Fujinaga M, Giaccia AJ. Hypoxic microenvironment within an embryo induces apoptosis and is essential for proper morphological development. Teratology. 1999;60(4):215–225. | ||
Khan K, Nilsson M, Danielsson B, Bengtsson E. Fully automatic heart beat rate determination in digital video recordings of rat embryos. In: Perner P, Salivetti O, editors. Advances in Mass Data Analysis of Images and Signals in Medicine, Biotechnology, Chemistry and Food Industry. Berlin, Heidelberg: Springer; 2008:27–37. | ||
Pinchot GB. Mechanism of uncoupling of oxidative phosphorylation by 2’4-dinitrophenol. J Biol Chem. 1967;242(20):4577–4583. | ||
Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-n-6-cyclopentyladenosine – a highly selective agonist at a1 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1988;337(6):687–689. | ||
Noji T, Karasawa A, Kusaka H. Adenosine uptake inhibitors. Eur J Pharmacol. 2004;495(1):1–16. | ||
Conlay LA, Conant JA, deBros F, Wurtman R. Caffeine alters plasma adenosine levels. Nature. 1997;389(6647):136. | ||
Arena JP, Kass RS. Enhancement of potassium-sensitive current in heart cells by pinacidil. Evidence for modulation of the ATP-sensitive potassium channel. Circ Res. 1989;65(2):436–445. | ||
Findlay I. Inhibition of ATP-sensitive K+ channels in cardiac muscle by the sulfony;urea drug glibenclamide J Pharmacol Exp Ther. 1992;261(2):540–545. | ||
Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. | ||
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O-2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | ||
Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278(18):15911–15916. | ||
Chowdhury R, Candela-Lena JI, Chan MC, et al. Selective small molecule probes for the hypoxia inducible factor (HIF) prolyl hydroxylases. ACS Chem Biol. 2013;8(7):1488–1496. | ||
Ellinghaus P, Heisler I, Unterschemmann K, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013;2(5):611–624. | ||
Rang H, Dale M, Flower R, Henderson G. Cholinergic transmission. Rang and Dale’s Pharmacology. 8th ed. London, UK: Elsevier Churchill Livingstone; 2015. | ||
Tarlatzis BC, Sanyal MK, Biggers WJ, Naftolin F. Continuous culture of the postimplantation rat conceptus. Biol Reprod. 1984;31(2):415–426. | ||
New D. Whole-embryo culture and the study of mammalian embryos during organogenesis. Biol. Rev. 1978;53(1):81–122. | ||
Chandra R, Portbury AL, Ray A, Ream M, Groelle M, Chikaraishi DM. B1-adrenergic receptors maintain fetal heart rate and survival. Biol Neonate. 2006;89(3):147–158. | ||
Barth AS, Tomaselli GF. Cardiac metabolism and arrhythmias. Circ Arrhythm Electrophysiol. 2009;2(3):327–335. | ||
Cox SJ, Gunberg DL. Metabolite utilization by isolated embryonic rat hearts in vitro. J Embryol Exp Morphol. 1972;28(2):235–245. | ||
Hofman PL, Hiatt K, Yoder MC, Rivkees SA. A1 adenosine receptors potently regulate heart rate in mammalian embryos. Am J Physiol 1997;273(4 Pt 2):R1374–R1380. | ||
Headrick JP, Peart JN, Reichelt ME, Haseler LJ. Adenosine and its receptors in the heart: regulation, retaliation and adaptation. Biochim Biophys Acta. 2011;1808(5):1413–1428. | ||
Buscariollo DL, Breuer GA, Wendler CC, Rivkees SA. Caffeine acts via A1 adenosine receptors to disrupt embryonic cardiac function. PLoS One. 2011;6(12):e28296. | ||
Wheaton WW, Chandel NS. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am J Physiol Cell Physiol. 2011;300(3):C385–C393. | ||
Xie L-H, Takano M, Noma A. Development of inwardly rectifying K+ channel family in rat ventricular myocytes. Am J Physiol. 1997;272(4 Pt 2):H1741–H1750. | ||
Nie L, Tang M, Zeng Y, et al. Properties and functions of KATP during mouse perinatal development. Biochem Biophys Res Commun. 2012;418(1):74–80. | ||
Ripoll C, Lederer WJ, Nichols CG. On the mechanism of inhibition of Katp channels by glibenclamide in rat ventricular myocytes. J Cardiovasc Electrophysiol. 1993;4(1):38–47. | ||
Rosati B, Rocchetti R, Zaza A, Wanke E. Sulfonylureas blockade of neural and cardiac HERG channels. FEBS Lett. 1998;440(1–2):125–130. | ||
Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene-expression and hypoxia-inducible factor-1 DNA-binding activity – implications for models of hypoxia signal-transduction. Blood. 1993;82(12):3610–3615. | ||
Barrett TD, Palomino HL, Brondstetter TI, et al. Pharmacological characterization of 1-(5-chloro-6-(trifluoromethoxy)-1H-benzoimidazol-2-yl)-1H-pyrazole-4-ca rboxylic acid (JNJ-42041935), a potent and selective hypoxia-inducible factor prolyl hydroxylase inhibitor. Mol Pharmacol. 2011;79(6):910–920. | ||
Portbury AL, Chandra R, Groelle M, et al. Catecholamines act via a b-adrenergic receptor to maintain fetal heart rate and survival. Am J Physiol Heart Circ Physiol. 2003;284(6):H2069–H2077. | ||
Kawakami K, Nagatomo T, Abe H, et al. Comparison of HERG channel blocking effects of various beta-blockers – implication for clinical strategy. Br J Pharmacol. 2006;147(6):642–652. | ||
Wang DW, Mistry AM, Kahlig KM, Kearney JA, Xiang J, George AL, Jr. Propranolol blocks cardiac and neuronal voltage-gated sodium channels. Front Pharmacol. 2010;1:144. | ||
Xie LH, Takano M, Noma A. The inhibitory effect of propranolol on ATP-sensitive potassium channels in neonatal rat heart. Br J Pharmacol. 1998;123(4):599–604. | ||
Besana A, Wang DW, George AL, Jr, Schwartz PJ. Nadolol block of Nav1.5 does not explain its efficacy in the long QT syndrome. J Cardiovasc Pharmacol. 2012;59(3):249–253. | ||
Dupuis DS, Klaerke DA, Olesen SP. Effect of beta-adrenoceptor blockers on Human Ether-a-go-go-Related gene (HERG) potassium channels. Basic Clin Pharmacol Toxicol. 2005;96(2):123–130. | ||
Abrahamsson T, Ek B, Nerme V. The beta 1- and beta 2-adrenoceptor affinity of atenolol and metoprolol. A receptor-binding study performed with different radioligands in tissues from the rat, the guinea pig and man. Biochem Pharmacol. 1988;37(2):203–208. | ||
Yasui K, Liu W, Opthof T, et al. I(f) current and spontaneous activity in mouse embryonic ventricular myocytes. Circ Res. 2001;88(5):536–542. | ||
Franco D, Moorman AF, Lamers WH. Expression of the cholinergic signal-transduction pathway components during embryonic rat heart development. Anat Rec. 1997;248(1):110–120. | ||
Chen FH, Klitzner TS, Weiss JN. Autonomic regulation of calcium cycling in developing embryonic mouse hearts. Cell Calcium. 2006;39(5):375–385. | ||
Song GL, Tang M, Liu CJ, et al. Developmental changes in functional expression and β-adrenergic regulation of I(ƒ) in the heart of mouse embryo. Cell Res. 2002;12(5–6):385–394. | ||
Yan X, Gao S, Tang M, et al. Adenylyl cyclase/cAMP-PKA-mediated phosphorylation of basal L-type Ca2+ channels in mouse embryonic ventricular myocytes. Cell Calcium. 2011;50(5):433–443. | ||
Maltsev VA, Ji GJ, Wobus AM, Fleischmann BK, Hescheler J. Establishment of beta-adrenergic modulation of L-type Ca2+ current in the early stages of cardiomyocyte development. Circ Res. 1999;84(2):136–145. | ||
Creazzo T. Functional developmental biology of the myocardium. In: Kirby M, editor. Cardiac Development. Oxford: Oxford University Press; 2007:53–68. | ||
Nilsson MF, Ritchie H, Webster WS. The effect on rat embryonic heart rate of Na+, K+, and Ca2+ channel blockers, and the human teratogen phenytoin, changes with gestational age. Birth Defects Res B Dev Reprod Toxicol. 2013;98(5):416–427. | ||
Britten S, Soenksen DM, Bustillo M, Coulam CB. Very early (24–56 days from last menstrual period) embryonic heart rate in normal pregnancies Hum Reprod. 1994;9(12):2424–2426. | ||
Makikallio K, Jouppila P, Rasanen J. Human fetal cardiac function during the first trimester of pregnancy. Heart. 2005;91(3):334–338. | ||
Papaioannou GI, Syngelaki A, Poon LCY, Ross JA, Nicolaides KH. Normal ranges of embryonic length, embryonic heart rate, gestational sac diameter and yolk sac diameter at 6–10 weeks. Fetal Diagn Ther. 2010;28(4):207–219. | ||
Park I, Shin J, Kim C. Prognosis of threatened abortion by embryonic/fetal heart beat rate. Ultrasound Obstet Gynecol. 2006;28(4):482. | ||
Chittacharoen A, Herabutya Y. Slow fetal heart rate may predict pregnancy outcome in first-trimester threatened abortion. Fertil Steril. 2004;82(1):227–229. | ||
Arleo EK, Troiano RN. Outcome of early first-trimester pregnancies (<6.1 weeks) with slow embryonic heart rate. Am J Roentgenol. 2011;197(1):252–255. | ||
Webster WS, Nilsson M, Ritchie H. Therapeutic drugs that slow the heart rate of early rat embryos. Is there a risk for the human? Curr Pharm Des. 2014;20(34):5364–5376. | ||
Patterson AJ, Zhang L. Hypoxia and fetal heart development. Curr Mol Med. 2010;10(7):653–666. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.