")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Concomitant Use of rFVIIa and Emicizumab in People with Hemophilia A with Inhibitors: Current Perspectives and Emerging Clinical Evidence
Authors Linari S, Castaman G
Received 18 March 2020
Accepted for publication 23 April 2020
Published 22 May 2020 Volume 2020:16 Pages 461—469
DOI https://doi.org/10.2147/TCRM.S205310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Silvia Linari, Giancarlo Castaman
Department of Oncology, Center for Bleeding Disorders and Coagulation, Careggi University Hospital, Florence, Italy
Correspondence: Giancarlo Castaman
Department of Oncology, Center for Bleeding Disorders and Coagulation, Careggi University Hospital, Largo G. Brambilla 3, Florence 50134, Italy
Tel +39 055 7947587
Fax +39 055 7947794
Email [email protected]
Abstract: Emicizumab, a humanized, bi-specific, monoclonal antibody subcutaneously administered, mimicking the function of FVIIIa, represents a milestone in treatment of patients affected by hemophilia A complicated with inhibitors. The HAVEN 1 and 2 studies have clearly established its superiority compared to bypassing agents for routine prophylaxis in preventing or reducing bleeding episodes in adult and pediatric patients with inhibitors. However, its protection against bleeding is only partial, and concomitant use of a bypassing agent may be required with potential prothrombotic risk. The emicizumab Phase III trials (HAVEN 1, 2 and 4) have shown that the traditional bypassing agents, activated prothrombin complex concentrates or recombinant activated factor VII (rFVIIa), may be necessary for the treatment of breakthrough bleeds or surgery management. A post hoc analysis in particular has shown that the concomitant use of emicizumab and rFVIIa is safe and no thrombotic events have been described. The review describes the state of the art of the concomitant use of emicizumab and rFVIIa for treating acute bleeding and surgeries, its efficacy and safety and the lack of thrombotic events associated with this treatment modality. Data still derive mainly from HAVEN trials; however, the availability of emicizumab in clinical practice is progressively increasing the number of patients treated and no adverse events directly attributed to this agent have occurred . The availability of guidelines for the use and dosing of rFVIIa during emicizumab prophylaxis is useful in clinical practice for managing suspected or ongoing bleeding, emergency situations and elective invasive procedures. In the next years, careful prospective post-licensure surveillance to monitor safety of rFVIIa use during prophylaxis with emicizumab is highly recommended.
Keywords: hemophilia A, FVIII inhibitors, emicizumab, bypassing agents, recombinant FVIIa, safety
Introduction
The occurrence of neutralizing alloantibodies (inhibitors) following exposure to therapeutically infused factor VIII (FVIII) represents the most important complication of treatment of hemophilia A. The cumulative incidence of inhibitor may range from 20% to 40% in severe hemophilia A, usually within the first 10–15 days of exposure, and approximately 5–10% in moderate or mild disease.1–3 The inhibitor risk is significantly lower when patients are exposed to FVIII for more than 50–150 days. The pathophysiology of inhibitor development is a complex and multi-causal process, including the interaction of genetic and environmental determinants.4,5 As a result of the neutralizing alloantibodies onset, replacement therapy with FVIII concentrates becomes ineffective, and usual long-term prophylaxis is not feasible. Patients are hence at an increased risk of mortality, morbidity, and disability with a significantly worse quality of life because also bleeding episodes are more difficult to control.6,7 When inhibitors occur, patients with a low-responding inhibitor (<5 Bethesda Units) may still be treated with specific factor replacement at much higher doses to neutralize the antibody and to allow FVIII to increase to stop bleeding. On the other hand, patients with high-responding inhibitors (>5 Bethesda Units) present a high risk of anamnestic response upon treatment and must be treated with bypassing agents (BPAs), which represented the standard of care for many years. Two BPAs are available such as activated prothrombin complex concentrates (aPCC)8 or recombinant activated factor VII (rFVIIa).9,10 The efficacy of BPAs, however, is not 100% guaranteed and these patients often require frequent intravenous administrations, even on the same day, and the lack of suitable laboratory tests to monitor their efficacy makes clinical outcome more unpredictable. Therefore, immune tolerance induction (ITI) to eradicate inhibitors has represented the primary aim in patients with a high-responding inhibitor, to restore the use of FVIII replacement treatment.11 This approach requires daily, long-term administration of FVIII ultimately resulting in a down-regulation of the production of neutralizing antibodies in 60% to 80% of patients.12–14 However, ITI represents a very demanding treatment, both for the need of an easy and safe venous access and its considerable cost.15
The development of agents targeting different key proteins in the coagulation process to restore thrombin generation in patients with hemophilia has been the focus of recent studies. These new agents aim at maintaining the coagulation to generate thrombin (Emicizumab) or at inhibiting natural anticoagulant pathways at different levels (Concizumab, Fitusiran and molecules targeting activated protein C or protein S).16,17
The subcutaneous route of administration and the long half-life are additional novel potential advantages of these agents, resulting in an improved compliance and protection. Emicizumab (Hemlibra`) has been recently approved as the first non-factor-based therapy for routine prophylaxis in patients affected by hemophilia A with inhibitors, thus representing a milestone in their treatment. However, the traditional BPAs may be still required for the treatment of breakthrough bleeds or to manage surgery, with a potential thrombotic risk.
This review analyzes the state of the art of concomitant use of emicizumab and BPAs, in particular rFVIIa.
Agents for Treating Patients with Hemophilia A and Inhibitors
Today, in clinical practice, the available drugs for the treatment of patients affected by hemophilia A with inhibitors are emicizumab, aPCC and rFVIIa (Table 1).
 |
Table 1 Drugs for Treatment of Patients Affected by Hemophilia A with Inhibitors |
Emicizumab
Emicizumab (Hemlibra, Roche Genentech, South San Francisco, CA, USA) is a recombinant, humanized, bi-specific monoclonal antibody (Mab) which mimics the procoagulant activity of activated FVIII (FVIIIa).18 Bi-specificity has been established by constructing an antigen-binding fragment (Fab) recognizing FIXa while the other Fab has FX as its substrate. The simultaneous binding of FX and FIXa by emicizumab facilitates the proteolytic activation of FX by FIXa without the cofactor activity of FVIIIa,19,20 thus increasing deficient thrombin generation with peak thrombin generation showing a bell-shaped curve and a close dose depending on emicizumab concentrations between 10 and 100 µg/mL.20 However, emicizumab and FVIII have some differences including the affinity for the antigen, topology, FIXa enhancing activity and regulation.21 While emicizumab binds to FIXa only at a single epidermal growth factor-1 like (EGF-1) domain site, FVIII binds to several heavy and light chain domains of FIXa.21 As a consequence, it has a lower affinity for FIXa and FX (10-fold and 6-fold, respectively) compared to FVIII. The antibody replaces partially FVIIIa cofactor activity and its activity is directly dependent on the amount of FIXa generated. Emicizumab-induced coagulation has no regulation at variance with the physiological FVIIIa-induced coagulation, characterized by exceeding concentrations of FIXa and FX. This imbalance disrupts the physiological on-and-off switch mechanism.21 Emicizumab has been approved in the United States in November 2017 for routine prophylaxis in patients with hemophilia A and inhibitors of all ages22 and in European Union in February 2018.23 The same dosing schedule consisting of 4 weekly loading doses of 3 mg/kg followed by weekly dosing of 1.5 mg/kg by subcutaneous injection is used in all the patients. Alternative treatment schemes are 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks after the initial 4-week loading schedule. Emicizumab half-life elimination is 24–30 days, but the complete disappearance of any drug-related effect can theoretically require as long as 150 days (5 half-lives).24,25 Apart from body weight, no dosage adjustment for age, renal or hepatic function is required and emicizumab concentration remains acceptably stable once the plateau has been achieved.25 Very few cases of immune response against emicizumab have been described.26 Most of these antibodies bind to emicizumab, without neutralizing its activity and treatment is usually continued. However, the onset of bleeding episodes previously prevented by emicizumab prophylaxis should suggest checking for emicizumab concentration or, if available, for the presence of anti-drug antibody (ADA).26 In patients on emicizumab prophylaxis, routine assays are not useful to monitor treatment. The drug strongly influences activated partial thromboplastin time (aPTT), which normalises at concentrations well below the expected therapeutic range. Therefore, a normal aPTT in a patient on emicizumab does not reflect an in-vivo coagulation ability. In order to accurately measure FVIII and inhibitor titer in these patients, chromogenic assays using either human reagents (to assess emicizumab’s factor tenase activity) or bovine reagents (to assess tenase activity of residual FVIII) are required.
aPCC
aPCC (FEIBA, Factor Eight Inhibitor Bypassing Activity, Takeda Pharmaceutical Company Limited, Lexington, MA, USA) is a plasma-derived BPA comprising activated coagulation factors, and in particularly high concentrations of FXa and FII.8 Typically, the recommended dose for treating bleeding episodes is 50–100 IU/kg every 8–12 h. A capped daily dose of ≤200 IU/kg is recommended to minimize the potential of thrombotic complications. Up to 30% of patients receiving aPCC may show a variable anamnestic increase of inhibitor levels caused by the presence in the product of small amounts of FVIII.27 However, inhibitor titer gradually decreases in more than 50% of patients on regular aPCC treatment and the anamnestic response is not associated with a reduction of clinical efficacy.27
rFVIIa
rFVIIa (Novoseven, eptacog alfa, NovoNordisk, Bagvaerd, Denmark) is a single-chain glycoprotein produced in baby hamster kidney (BHK) cell line, genetically modified. During purification, rFVII is converted in its activated form. rFVIIa is able to directly activate FX and increases thrombin generation on the surface of activated platelets in the absence of FVIII or FIX. The platelet-specific generation of FIIa by rFVIIa is thought to localize the hemostatic process to the sites of active bleeding and tissue injury.28,29 The pharmacokinetics of rFVIIa shows a half-life of 2–3 h in adults and 1.5–3 h in children, with significant individual variations,9 and thus it must be infused every 2–6 h. Recently, an automated pump for the administration of rFVIIa has been developed to simplify the administration of rFVIIa in the future.30 rFVIIa has been initially approved at a dose of 90 μg/kg every 2–3 h until clinical evidence of good hemostasis is achieved; then, a single bolus 270 μg/kg has been also used in clinical practice to meet the requirement of less infusions, especially in pediatric populations, faster response rates, thus allowing longer intervals between infusions.31,32 Prospective clinical studies on rFVIIa efficacy have demonstrated that a single 270 μg/kg dose is comparable to three 90 μg/kg doses administered every 3 h.10,31,32 Moreover, the higher dose is safe, with a low risk of thrombosis risk in patients with inhibitors.33
With regard to hemostatic efficacy, the FENOC study showed similar overall results with resolution in 70–80% of bleeding events in a controlled head-to-head comparison of aPCC and rFVIIa.34 An improved response to one BPA versus the other, changing over time also in the same patient, has been frequently observed.35 Moreover, sequential regimens of both aPCC and rFVIIa may be required to control 10–20% of bleeds not effectively managed with a single BPAs.36
Both BPAs are associated with a potential risk of thrombotic complications, including venous thromboembolism, disseminated intravascular coagulation and myocardial infarction. These events are very rare in hemophilia patients, but they could be theoretically increased in patients with preexisting conditions (atherosclerotic disease, liver disease, prolonged immobilization).37,38
aPCC and rFVIIa are used in prophylactic regimens in patients with inhibitors, even if their benefits are not as evident as with the usual FVIII prophylaxis for patients without inhibitor. Prophylaxis with BPAs may be considered before starting ITI, during ITI or in those patients who failed ITI. Two prospective, randomized trials, the Pro–FEIBA and PROOF studies demonstrated a 60–72% reduction of bleeding episodes compared to on-demand treatment with aPCC.39,40 Only a single prospective, randomized trial has evaluated the efficacy of rFVIIa, showing up to 60% reduction of bleeding episodes compared to the pre-prophylactic period.41 However, the short half-life of the BPAs, their variable efficacy and the relevant costs are important drawbacks limiting their use in the real-world practice.42
Furthermore, notwithstanding many years of experience with BPAs, there is no laboratory test easily available for monitoring their hemostatic efficacy, and dose and duration of treatment is mainly decided by clinical assessment.
Emicizumab in HAVEN Trials
Data on the efficacy of emicizumab derives by an extensive clinical program of phase III trials (HAVEN 1, HAVEN 2 and HAVEN 4) in patients with hemophilia A with inhibitors (Table 2).43–45
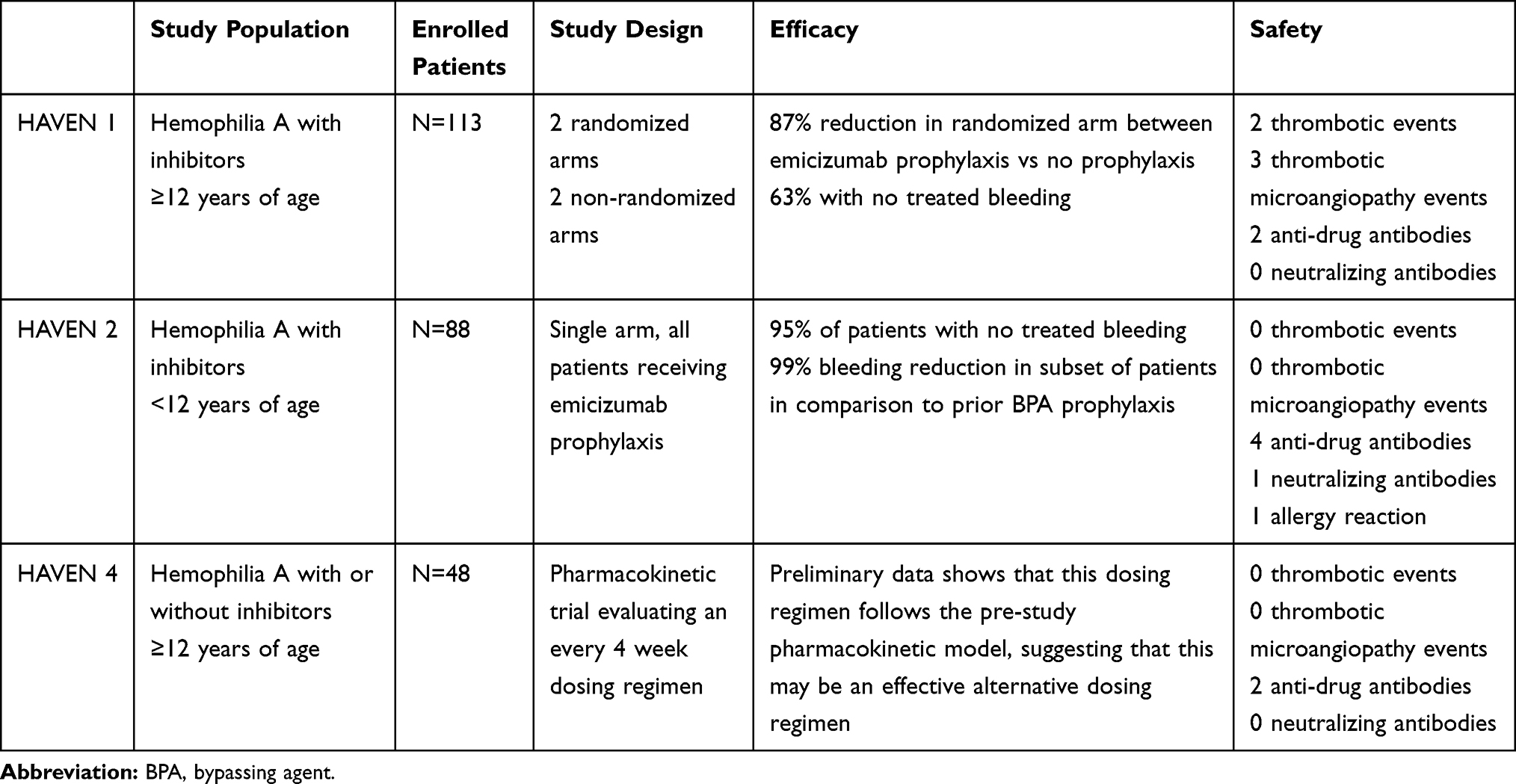 |
Table 2 Emicizumab Clinical Program of Phase III Trials in Hemophilia A with Inhibitors |
In HAVEN 1, an inter-individual comparison of the number of treated bleeds during 24 weeks between patients with hemophilia A and high titer inhibitors aged ≥12 years receiving episodic BPA treatment compared to emicizumab prophylaxis was the primary end point.43 The median annualized bleeding rate (ABR) was 2.9 (95% confidence interval [CI]:1.7–5.0) in patients on emicizumab prophylaxis versus 23.3 (95% CI: 12.3–43.9) among those treated on on-demand with BPAs, with a 87% difference favouring emicizumab (p<0.001).43 Secondary bleeding-related end points, including all bleeding, spontaneous bleeding, joint bleeding and target joint bleeding also were significantly lower in patients on emicizumab prophylaxis and 63% of them did not experience any bleeding episodes requiring treatment with BPAs compared to only 6% of those on episodic BPAs treatment. In the intra-individual analysis, emicizumab prophylaxis showed a 79% reduction of bleeds requiring treatment (p<0.001) compared to previous prophylaxis with BPAs. Furthermore, there was a significant improvement in overall health-related quality of life and the physical health subscores.43
HAVEN 2 was a single-arm trial evaluating the efficacy, safety and pharmacokinetic profile of emicizumab prophylaxis in pediatric hemophilia A patients with high titer inhibitors.44 Eighty-eight children, previously receiving episodic or mostly prophylactic BPAs, were treated with emicizumab 1.5 mg/kg weekly (n=68), 3 mg/kg every 2 weeks (n=10) or 6 mg/kg every 4 weeks (n=10).44 With once-weekly emicizumab prophylaxis, the ABR was 0.3 (95% CI, 0.17–0.50) and 77% of patients experienced no treated bleeds. A 99% reduction of the number of treated bleeds in the intra-patient analysis of those on BPAs prophylaxis before enrollment was observed, with a drop of ABR from 21.1 (95% CI:15.99–27.82) to 0.3.44 The ABRs in participants receiving emicizumab every 2 weeks or every 4 weeks were 0.2 (95% CI:0.03–1.72) and 2.2 (95% CI:0.69–6.81), respectively, with 90% and 60% of patients having reporting zero treated bleeds.44
HAVEN 4 trial evaluated prophylaxis with emicizumab 6 mg/kg every 4 weeks in hemophilia A patients aged ≥12 years, with or without inhibitors.45 Data on 41 patients have shown a consistent high efficacy and acceptable safety profile of such schedule dosing with a median ABR for treated bleeds of 0.0 with 56.1% (95% CI: 39.7–71.5) of patients without treated bleeds and 90.2% (95% CI: 76.9–97.3) having ≤3 treated bleeds.45
Data on emicizumab efficacy are undoubtedly significant to recommend its use for prophylaxis in patients with inhibitors. However, it should be kept in mind that protection against bleeding is not absolute and the risk of bleeding could be roughly estimated to be similar to that of patients with mild hemophilia.21,46 Breakthrough bleeds may still occur (trauma, emergency surgery, etc.) and co-administration of a BPAs required, with potential prothrombotic risk. In HAVEN 1 four serious thrombotic events (2 venous thrombosis and 2 thrombotic microangiopathy (TMA)), occurred in subjects who received aPCC at high doses (>100 IU/kg/day) for ≥24 hours for the treatment of breakthrough bleeds.43 A fifth patient developed TMA and died of rectal hemorrhage after the refusal of blood transfusions. However, TMA was improving before he died, and the death was judged unrelated to emicizumab.43 No venous thromboembolic or TMA episodes were reported in HAVEN 2 and HAVEN 4.44,45
The synergistic interaction between emicizumab and aPCC and the inherent thrombotic risk are well explained by the presence in aPCC of FIX and FX and their activated forms, which are the substrates for the bi-specific antibody, thus exceedingly increasing thrombin generation. In fact, the enzymatic activity of FIXa in aPCC can be increased in an unregulated manner up to 20,000-fold by emicizumab.,21 Thus, some guidelines have been provided about the dosing of BPAs during emicizumab prophylaxis and the avoidance of aPCC or choosing the lowest dose approved BPAs.47–50
No thrombotic complications have been reported when using emicizumab alone or in combination with rFVIIa. The lack of events when co-administrating the two drugs may be explained by the short half-life of rFVIIa, the fact that emicizumab does not bind to rFVIIa and the physiological antithrombin-mediated inactivation process able to prevent excessive rFVIIa-mediated thrombin generation.51
Co-Administration of Emicizumab and rFVIIa
Management of Bleeding Episodes
Recently, an extensive analysis assessing the safety of emicizumab and rFVIIa co-administration in HAVEN 1, 2 and 4 clinical trials was jointly performed by Roche and Novo Nordisk.52
Overall, 61 enrolled patients received rFVIIa for one or more events to manage or as prophylaxis of bleeding episodes. A total of 210 bleeding episodes for which rFVIIa treatment was used were analysed.52
The large majority of bleeds were observed in HAVEN 1, in which 46/113 patients had at least one rFVIIa treatment.43 A total of 193 bleeds (84% in joints or muscles) in 37 patients were managed with rFVIIa and. A 100 ± 20 μg/kg initial dose of rFVIIa was given in the majority of cases. Dosing interval and cumulative dosing were in keeping with prescribing information and current practice. The median duration of treatment per bleed was 1 day for more than half (61.7%) of events.43
In HAVEN 2, 11 patients received rFVIIa to treat a bleed (10 bleeding trauma-related) and 4 as a prophylaxis before activity.44 The cumulative median dose per bleed was 164.21 μg/kg and the median number of infusions was one. The dosing interval was at least 2 h.44
In HAVEN 4, a single patient out of five with inhibitors was treated with rFVIIa for three bleeds, two of which occurring at the start of treatment, when emicizumab concentration was still not at the steady state yet.45 The rFVIIa cumulative dose per bleed was 276.94 μg/kg for the first bleeding and 449.15 μg/kg for the second bleeding.45
Overall, it appeared that the co-administration of rFVIIa for treating breakthrough bleeds in patients on emicizumab was safe and control of bleeding excellent.52
Management of Surgery
In HAVEN trials, patients with surgeries already planned were not enrolled, with the exception of minor procedures. However, unplanned emergency surgeries have been required in some enrolled patients receiving emicizumab.43–45 Perioperative management was left at the investigator’s discretion on clinical judgement and specific guidance (per protocol) on surgical management with regard to BPAs use was provided.
Only preliminary information on the management of surgeries and procedures are described so far and final data are going to be presented.53–55
In HAVEN 1, 13 patients underwent surgical procedures using perioperatively rFVIIa.43 Two surgeries were major (a total hip replacement and a knee arthroscopy with synovectomy, chondroplasty, and debridement). The others were minor surgeries or procedures as dental surgery, radiosynovectomy, endoscopy and central venous access devices (CVADs) placement or removal. In HAVEN 2, 21 patients underwent CVADs removal with rFVIIa prophylaxis only in 4 of them.44
A total knee replacement in a patient enrolled in HAVEN 1 and perioperative management with rFVIIa have been described.56 A preoperative rFVIIa bolus of 200 μg/kg was infused, followed by doses of 100 μg/kg every 2 h during surgery and for the first postoperative day. A bolus dose of 100 μg/kg was administered every 3 h on day 2 and then the infusion intervals were prolonged up to every 4 and 6 h, respectively, on days 3 and 4. The patient remained on every 6-h dosing until postoperative day 11, with a subsequent tapered treatment every 8 h until day 14. Hemostatic efficacy was excellent, without clinical evidence of thrombosis or TMA.56 Another case report describes the perioperative management of a total hip arthroplasty with a preoperative rFVIIa bolus of 180 μg/kg followed by a bolus of 90 μg/kg every 3 h until day 3, when the frequency of administration was tapered to every 6 h on postoperative days 4–7.57 Since day 8 to 12, rFVIIa was infused every 8 h, then every 12 h until day 14. No additional rFVIIa was given. Co-administration of Emicizumab and rFVIIa was effective without adverse events.57
Conclusions
Emicizumab represents a milestone in hemophilia A treatment, offering the opportunity to improve the prophylactic approach even in patients with inhibitors. HAVEN trials have shown that the concomitant use of traditional BPAs may be required for the treatment of breakthrough bleeds or for surgery with a potential thrombotic risk.43–45 The same studies have also shown that the concomitant use of emicizumab and rFVIIa is safe and effective and no thrombotic events have been described.52 However, the modulation of usual therapeutic plans to manage suspected or ongoing bleeding or scheduled invasive procedures has been required. This also for the issues related to monitoring emicizumab therapy, as routine assays cannot be used. Emicizumab strongly shortens the aPTT, even very low concentrations, far from the theoretical therapeutic range. This spurious effect may be misinterpreted as a normalization of the coagulation potential in the patients. The concomitant use of rFVIIa makes it currently impossible to perform a laboratory monitoring that reflects the in vivo activation of hemostasis.
Therefore, due to thrombotic episodes observed in HAVEN 1 in patients receiving aPCC at doses ≥100 IU/kg/day for 24 h or longer to treat breakthrough bleeds 43 and according to the guidance released by the manufacturer for the use and dosing of BPAs in patients on emicizumab prophylaxis, the indication to avoid aPCC and to prescribe the lowest doses of approved BPA is commonly accepted.47–50
rFVIIa represents the first-line option for bleeds requiring hemostatic replacement therapy in patients with inhibitors on emicizumab. An initial dose of 90–120 µg/Kg is suggested, to be repeated 2–4 h apart according to the clinical severity and response.48,49 rFVIIa megadose (270 µg/kg) should be avoided, even as a single infusion. Even in surgery with major bleeding risk, is rFVIIa represents the first-line treatment, at a dose of 90–120 µg/Kg starting 15 min preoperatively and then post-surgery every 2–3 h with a progressive interval between injections.48,49
The availability of emicizumab in clinical practice is progressively increasing the number of patients treated and the knowledge of the drug by clinicians. No new thrombosis or TMA events have been reported when concomitant use of BPAs, according to available practical guidance. Post-licensure carefully designed prospective together with registry data will be of utmost importance to continue monitoring the safety of concomitant use of rFVIIa and Emicizumab in a real-world scenario as well in guiding the best clinical practice.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
GC acted as a speaker at company satellite symposia during scientific meetings for Roche, Sobi, Novo Nordisk, Werfen, and Kedrion; is a member of the steering committee of Uniqure; and funding research was directly provided to his Institution from CSL Behring, Pfizer and SOBI. He participated in advisory boards of Ablynx, Alexion, Bayer, Baxalta/Shire, CSL Behring, Novo Nordisk, Pfizer, Roche, SOBI and Uniqure. He also reports grants and personal fees from CSL Behring and Sobi, personal fees from Roche, Novo Nordisk, Bayer, Kedrion, Shire, Sanofi, Werfen, and Uniqure, and grants from Pfizer, outside the submitted work.
SL participated in advisory boards of CSL Behring, Roche, Sobi, and Novo Nordisk.
The authors report no other possible conflicts of interest in this work.
References
1. Wight J, Paisly S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9(4):418–435. doi:10.1046/j.1365-2516.2003.00780.x
2. Astermark J, Altisent C, Batorova A, et al. European haemophilia therapy standardisation board non-genetic risk factors and the development of inhibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010;16(5):747–766. doi:10.1111/j.1365-2516.2010.02231.x
3. Gouw S, van der Berg H, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046–4055. doi:10.1182/blood-2012-09-457036
4. Astermark J. Inhibitor development: patient-determined risk factors. Haemophilia. 2009;16(102):66–70. doi:10.1111/j.1365-2516.2008.01923.x
5. Santagostino E, Mancuso ME, Rocino A, et al. Environmental risk factors for inhibitor development in children with haemophilia A: a case-control study. Br J Haematol. 2005;130(3):422–427. doi:10.1111/j.1365-2141.2005.05605.x
6. Morfini M, Haya S, Tagariello G, et al. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia. 2007;13(5):606–612. doi:10.1111/j.1365-2516.2007.01518.x
7. Gringeri A, Mantovani LG, Scalone L, Mannucci PM. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: the COCIS study group. Blood. 2003;102(7):2358–2363. doi:10.1182/blood-2003-03-0941
8. Negrier C, Gomperts ED, Oldenburg J. The history of FEIBA: a lifetime of success in the treatment of haemophilia complicated by an inhibitor. Haemophilia. 2006;12:4–13.
9. Lindley CM, Sawyer WT, Macik G, et al. Pharmacokinetics and pharmacodynamics of recombinant factor VIIa. Clin Pharmacol Ther. 1994;55:638–648.
10. Young G, Shafer FE, Rojas P, Seremetis S. Single 270 micro kg(−1)-dose rFVIIa vs. Standard 90 micro kg(−1)-dose rFVIIa and aPCC for home treatment of joint bleeds in haemophilia patients with inhibitors: a randomized comparison. Haemophilia. 2008;14(2):287–294. doi:10.1111/j.1365-2516.2007.01601.x
11. Brackmann HH, White GC, Berntorp E, Andersen T, Escuriola‐Ettingshausen C. Immune tolerance induction: what have we learned over time? Haemophilia. 2018;24 Suppl 3(Suppl3):3–14. doi:10.1111/hae.13445
12. DiMichele DM. The North American Immune Tolerance Registry: contributions to the thirty-year experience with immune tolerance therapy. Haemophilia. 2009;15(1):320–328. doi:10.1111/j.1365-2516.2008.01880.x
13. Hay CRM, DiMichele DM. The principal results of the international immune tolerance study: a randomized dose comparison. Blood. 2012;119(6):1335–1344. doi:10.1182/blood-2011-08-369132
14. Valentino LA, Kempton CL, Krause-Jarres R, Mathew P, Meeks SL, Reiss UM, International Immune Tolerance Induction Study Investigators. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia. 2015;21(5):559–567. doi:10.1111/hae.12730
15. Rocino A, Cortesi PA, Scalone L, et al. Immune tolerance induction in patients with haemophilia A and inhibitors: effectiveness and cost analysis in an European Cohort (The ITER Study). Haemophilia. 2016;22(1):96–102. doi:10.1111/hae.12780
16. Peyvandi F, Garagiola I, Biguzzi E. Advances in the treatment of bleeding disorders. J Thromb Haemost. 2016;14(11):2095–2106. doi:10.1111/jth.13491
17. Castaman G, Linari S. Current and emerging biologics for the treatment of hemophilia. Expert Opin Biol Ther. 2019;19(8):801–810. doi:10.1080/14712598.2019.1614163
18. Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18(10):1570–1574. doi:10.1038/nm.2942
19. Shima M, Hanabusa H, Taki M, et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044–2053. doi:10.1056/NEJMoa1511769
20. Kitazawa T, Esaki K, Tachibana T, et al. Factor VIIIa-mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost. 2017;117(7):1348–1357. doi:10.1160/TH17-01-0030
21. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;130(23):2463–2468. doi:10.1182/blood-2017-08-801662
22. U.S. Department of Health and Human Services. Food and Drug Administration. FDA approves emicizumab-kxwh for prevention and reduction of bleeding in patients with hemophilia A with factor VIII inhibitors. Available from. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm585650.htm.
23. Roche. European Commission approve Roche’s Hemlibra for people with hemophilia A with factor VIII inhibitors. Press release. Basel, Switzerland, 2018. Available from: https://www.roche.com/media/store/releases/med-cor-2018-02-27.htm.
24. Uchida N, Sambe T, Yoneyama K, et al. A first-in-human phase 1 study of ACE910, a novel factor VIII-mimetic bispecific antibody, in healthy subjects. Blood. 2016;127(13):1633–1641. doi:10.1182/blood-2015-06-650226
25. Yoneyama K, Schmitt C, Kotani N, et al. A pharmacometric approach to substitute for a conventional dose-finding study in rare diseases: example of phase III dose selection for emicizumab in hemophilia A. Clin Pharmacokinet. 2018;57(9):1123–1134. doi:10.1007/s40262-017-0616-3
26. Paz-Priel I, Chang T, Asikanius E, et al. Immunogenicity of emicizumab in people with hemophilia A: results from the HAVEN 1-4 studies. Blood. 2018;132:633.
27. Negrier C, Goudemand J, Sultan Y, Bertrand M, Rothschild C, Lauroua P; The members of the French FEIBA Study Group. Multicenter retrospective study on the utilization of FEIBA in France in patients with factor VIII and factor IX inhibitors. Thromb Haemost. 1997;77(6):1113–1119.
28. Monroe DM, Hoffman M, Oliver JA, Roberts HR. Platelet activity of high dose factor VIIa is independent of tissue factor. Br J Haematol. 1997;99(3):542–547. doi:10.1046/j.1365-2141.1997.4463256.x
29. Hoffman M, Monroe DM, Roberts HR. Activated factor VII activates FIX and FX on the surface of activated platelets: thoughts on the mechanism of action of high dose activated factor VII. Blood Coagul Fibrinolysis. 1998;9 Suppl 1:S61–S65.
30. Negrier C, Chamouard V, Lienhart A, Nougier C, Fleury R. A novel protocol for accurate and reliable postoperative bolus administration of recombinant factor VIIa using an automated mini-pump system. Haemophilia. 2019;25(6):1020–1027. doi:10.1111/hae.13863
31. Kavakli K, Makris M, Zulfikar B, et al. Novoseven trial (F7HAEM-1510) investigators. Home treatment of haemarthroses using a single dose regimen or recombinant activated factor VII in patients with haemophilia and inhibitors. A multi-centre, randomized, double-blind, cross over trial. Thromb Haemost. 2006;95(4):600–605.
32. Santagostino E, Mancuso ME, Rocino A, et al. A prospective randomized trial of high and standard dosages of recombinant factor VIIa for treatment of haemarthroses in haemophiliacs with inhibitors. J Thromb Haemost. 2006;4:367–371.
33. Neufeld EJ, Negrier C, Arkhammar P, et al. Safety update on the use of recombinant activated factor VII in approved indications. Blood Rev. 2015;29(Suppl1):S34–41. doi:10.1016/S0268-960X(15)30006-0
34. Astermark J, Donfield SM, DiMichele DM, et al. FENOC Study Group. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA novoseven comparative (FENOC) study. Blood. 2007;109(2):546–551. doi:10.1182/blood-2006-04-017988
35. Hayashi T, Tanaka I, Shima M, et al. Unresponsiveness to factor VIII inhibitor bypassing agents during haemostatic treatment for life-threatening massive bleeding in a patient with haemophilia A and a high responding inhibitor. Haemophilia. 2004;10(4):397–400. doi:10.1111/j.1365-2516.2004.00924.x
36. Gringeri A, Fischer K, Karafoulidou A, et al. Sequential combined bypassing therapy is safe and effective in the treatment of unresponsive bleeding in adults and children with haemophilia and inhibitors. Haemophilia. 2011;17(4):630–635. doi:10.1111/j.1365-2516.2010.02467.x
37. Roberts HR. Clinical experience with activated factor VII: focus on safety aspects. Blood Coagul Fibrinolysis. 1998;9:S115–S118.
38. Ehrlich HJ, Henzl MJ, Gomperts ED. Safety of factor VIII inhibitor bypass activity (FEIBA): 10-year compilation of thrombotic adverse events. Haemophilia. 2002;8(2):83–90. doi:10.1046/j.1365-2516.2002.00532.x
39. Leissinger C, Gringeri A, Antmen B, et al. Anti-inhibitor coagulant complex prophylaxis in hemophilia with inhibitors. N Engl J Med. 2011;365(18):1684–1692. doi:10.1056/NEJMoa1104435
40. Antunes SV, Tangada S, Stasyshyn O, et al. Randomized comparison of prophylaxis and on-demand regimens with FEIBA NF in the treatment of haemophilia A and B with inhibitors. Haemophilia. 2014;20(1):65–72. doi:10.1111/hae.12246
41. Konkle BA, Ebbesen LS, Erhardtsen E, et al. Randomized, prospective clinical trial of recombinant factor VIIa for secondary prophylaxis in hemophilia patients with inhibitors. J Thromb Haemost. 2007;5(9):1904–1913. doi:10.1111/j.1538-7836.2007.02663.x
42. Ljung R. Aspects of prophylactic treatment of hemophilia. Thromb J. 2016;14:S60–63.
43. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia with inhibitors. N Engl J Med. 2017;377(9):809–818. doi:10.1056/NEJMoa1703068
44. Young G, Liesner R, Chang T, et al. A multicenter, open-label phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134(24):2127–2138. doi:10.1182/blood.2019001869
45. Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomized phase 3 study. Lancet Haematol. 2019;6(6):e295–e305. doi:10.1016/S2352-3026(19)30054-7
46. Kotani N, Yoneyama K, Katakami N, et al. Relative and absolute bioavailability study of emicizumab to bridge drug products and subcutaneous injection sites in healthy volunteers. Clin Pharmacol Drug Dev. 2018;8(6):702–12. Epub ahead of print.
47. Collins PW, Liesner R, Makris M, et al. Treatment of bleeding episodes in haemophilia A complicated by a factor VIII inhibitor in patients receiving Emicizumab. Interim guidance from UKHCDO inhibitor working party and executive committee. Haemophilia. 2018;24(3):344–347. doi:10.1111/hae.13495
48. Castaman G, Santoro C, Coppola A, et al. Emergency management in patients with haemophilia A and inhibitors on prophylaxis with emicizumab: AICE practical guidance in collaboration with SIBioC, SIMEU, SIMEUP, SIPMeL and SISET. Blood Transfus. 2019. doi:10.2450/2019/.0186-19
49. Susen S, Gruel Y, Godier A, et al. Management of bleeding and invasive procedures in haemophilia A patients with inhibitor treated with emicizumab (Hemlibra): proposal from the French network on inherited bleeding disorders (MHEMO), the french reference centre on haemophilia, in collaboration with the french working group on perioperative haemostasis. Haemophilia. 2019;25(5):731–737. doi:10.1111/hae.13817
50. MASAC Document #255, approved on November 21, 2018
51. Agerso H, Brophy DF, Pelzer H, et al. Recombinant human factor VIIa (rFVIIa) cleared principally by antithrombin following intravenous administration in hemophilia patients. J Thromb Haemost. 2011;9(2):333–338. doi:10.1111/j.1538-7836.2010.04152.x
52. Levy GG, Asikanius E, Kuebler P, et al. Safety analysis of rFVIIa with emicizumab dosing in congenital hemophilia A with inhibitors: experience from the Haven clinical program. J Thromb Haemost. 2019;17(1):1–8. doi:10.1111/jth.14351
53. Kruse-Jarres R, Callaghan MU, Croteau SE, et al. Surgical experience in two multicenter, open label phase 3 studies of Emicizumab in person with hemophilia A with inhibitors (HAVEN 1 and HAVEN 2). Blood. 2017;130:89.
54. Santagostino E, Mancuso ME, Novembrino C, Solimeno LP, Tripodi A, Peyvandi F. Rescue factor VIII replacement to secure hemostasis in a patient with hemophilia A and inhibitors on emicizumab prophylaxis undergoing hip replacement. Haematologica. 2019;104(8):e380–e382. doi:10.3324/haematol.2018.215129
55. Santagostino E, Oldenburg J, Chang T, et al. Surgical experience from four phase III studies (HAVEN 1-4) of emicizumab in persons with haemophilia A (PwHA) with or without FVIII inhibitors. Res Pract Haemost. 2019;3(S1):115.
56. Kizilocak H, Yukhtman CL, Marquez-Casas E, et al. Management of perioperative hemostasis in a severe hemophilia A patient with inhibitors on emicizumab using global hemostasis assays. Ther Adv Hematol. 2019;10:1–9.
57. Seaman CD, Ragni MV. Emicizumab use in major orthopaedic surgery. Blood Adv. 2019;3(11):1722–1724. doi:10.1182/bloodadvances.2019000228
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.