")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 12
Computational evaluation of natural compounds as potential inhibitors of human PEPCK-M: an alternative for lung cancer therapy
Authors Baptista LPR , Sinatti VVC , Da Silva JHM, Dardenne LE , Guimarães AC
Received 5 December 2018
Accepted for publication 20 March 2019
Published 7 August 2019 Volume 2019:12 Pages 15—32
DOI https://doi.org/10.2147/AABC.S197119
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Luiz Phillippe R Baptista,1 Vanessa VC Sinatti,1 Joao HM Da Silva,2 Laurent Emmanuel Dardenne,3 Ana Carolina Guimarães1
1Laboratory for Functional Genomics and Bioinformatics, Fiocruz, Oswaldo Cruz Institute, Rio de Janeiro, RJ, Brazil; 2Group for Computational Modelling, Fiocruz, Oswaldo Cruz Foundation, Eusébio, CE, Brazil; 3Group for Molecular Modelling of Biologic Systems, National Laboratory of Scientific Computing, Petrópolis, RJ, Brazil
Background: Lung cancer is the leading cause of cancer-related death worldwide. Among its subtypes, non-small cell lung cancer (NSCLC) is the most common. Recently, the mitochondrial isoform of the enzyme phosphoenolpyruvate carboxykinase (HsPEPCK-M) was identified as responsible for the metabolic adaptation in the NSCLC allowing tumor growth even under conditions of glucose deficiency. This adaptation is possible due to the role of HsPEPCK-M in gluconeogenesis, converting the oxaloacetate to phosphoenolpyruvate in the presence of GTP, which plays an important role in the energetic support of these tumors. In this context, it was shown that the inhibition or knockdown of this enzyme was able to induce apoptosis in NSCLC under low glucose conditions.
Purpose: In this study, novel putative inhibitors were proposed for the human PEPCK-M (HsPEPCK-M) based on a computer-aided approach.
Methods: Comparative modeling was used to generate 3D models for HsPEPCK-M. Subsequently, the set of natural compounds of the ZINC database was screened against HsPEPCK-M models using structure-based pharmacophore modeling and molecular docking approaches. The selected compounds were evaluated according to its chemical diversity and clustered based on chemical similarity.
Results: The pharmacophore hypotheses, generated based on known PEPCK inhibitors, were able to select 7,124 candidate compounds. These compounds were submitted to molecular docking studies using three conformations of HsPEPCK-M generated by comparative modeling. The aim was to select compounds with high predicted binding affinity for at least one of the conformations of HsPEPCK-M. After molecular docking, 612 molecules were selected as potential inhibitors of HsPEPCK-M. These compounds were clustered according to their structural similarity. Chemical profiling and binding mode analyses of these compounds allowed the proposal of four promising compounds: ZINC01656421, ZINC895296, ZINC00895535 and ZINC02571340.
Conclusion: These compounds may be considered as potential candidates for HsPEPCK-M inhibitors and may also be used as lead compounds for the development of novel HsPEPCK-M inhibitors.
Keywords: lung cancer, PEPCK-M, virtual screening, pharmacophore modeling, molecular docking, natural products
Introduction
Lung cancer is the most common of all malignant tumors and the primary cause of cancer-related mortality worldwide with more than 1.7 million deaths per year.1 The non-small cell lung cancer (NSCLC) is the main type of lung cancer corresponding to approximately 85% of all cases. Despite the available treatments, a low survival rate is observed in most of the world’s populations, averaging 10–15%. This fact along with the high prevalence of this type of lung cancer highlight the need for the development of effective treatment strategies for NSCLC.2,3
In general, although angiogenesis occurs in growing tumors, the process is highly disorganized resulting in substantial nutrient and oxygen deprivation.4 In these cases, the glucose concentration may be 3 to 10-fold lower in tumors than in normal tissues.5 To circumvent this problem, it is described that lung cancer cells, in particular, NSCLC, use some steps of gluconeogenesis from sources such as glutamine and lactate to compensate the lack of glucose and overcome the metabolic stress.6,7
Recent studies have shown that the gluconeogenic enzyme phosphoenolpyruvate carboxykinase (PEPCK; EC 4.1.1.32) plays an important role in the bioenergetic support of tumors.8,9 The enzyme PEPCK catalyzes the reversible conversion of oxaloacetate (OAA) + GTP to phosphoenolpyruvate (PEP) + GDP + CO2 – performing a major role in the de novo synthesis of glucose. This enzyme occurs in two isoforms in the human genome: PEPCK-C, the cytoplasmic isoform whose gene is located on chromosome 20; and PEPCK-M, the mitochondrial isoform whose gene is located on chromosome 14.10 Although there are studies exploring the expression of the enzyme PEPCK-C, the PEPCK-M isoform has not been extensively studied.11 In the literature, the expression of PEPCK-C is associated with differentiated liver, small intestine, kidney cortex, and adipose tissue.12 PEPCK-M, in contrast, appears to be expressed in a variety of cell types, including T- and B-cells, pancreatic β-cells, and neurons.12
In previous studies, it was observed a high PEPCK-M expression under low-glucose conditions in tumor tissues from NSCLC patients and very low expression of PEPCK-C.6,7 In this context, PEPCK-M was indicated as responsible for mediate metabolic adaptation, enabling biosynthesis of important metabolites and glucose-independent tumor growth.6,7 This adaptation process is possible through a re-wiring of the tricarboxylic acid (TCA) cycle and early steps of gluconeogenesis by PEPCK-M. The substrate OAA – supplied by glutamine or other anapleurotic substrates – is metabolized in PEP, maintaining TCA cycle function and production of glycolytic intermediates for biosynthetic pathways required in tumor cell proliferation.6 The importance of PEPCK-M in the metabolism of NSCLC is corroborated by a study showing that inhibition by a known HsPEPCK-M inhibitor (3-mercaptopicolinic acid, MPA) or knockdown assays of this enzyme, under low glucose conditions, enhanced apoptosis and cell death in lung cancer cells (A549 and H23 cells).7 This same treatment, under the condition of high glucose concentration, did not have the effect of inducing apoptosis on A549 and H23 cells.7 From this perspective, PEPCK-M is a promisor therapeutic drug target since its inhibition may interrupt the metabolic adaptation under glucose deprivation observed on NSCLC.
To date, there are no experimentally determined structures for the human mitochondrial isoform of PEPCK (HsPEPCK-M). However, structural studies were performed with other related PEPCKs, particularly human PEPCK-C (HsPEPCK-C) and Rattus norvegicus PEPCK-C (RnPEPCK-C). From these studies, it is known that the enzyme needs two divalent metal ions (usually manganese) to function properly – these cations together with positively charged residues help to stabilize the reaction’s intermediate enolate.13 Three regions show flexibility in PEPCK-C structures that are important to its function: (i) the 10-residue Ω-loop, (ii) P-loop, (iii) R-loop.13 The Ω-loop is a highly flexible region on the surface of the protein that functions as a cover for the active site. The P-loop, also called the kinase-1a domain, helps in positioning the nucleotide in the catalytic cleft.14 The R-loop is near to the OAA/PEP binding site and is important for stabilizing the closed Ω-loop lid.15
Besides the 3-mercaptopicolinic acid (MPA), which as previously mentioned can inhibit the enzyme PEPCK-M, there are no other inhibitors identified for the mitochondrial isoform.16 Nevertheless, experiments were performed to identify new inhibitors for PEPCK-C from different organisms though their effectiveness in inhibiting the PEPCK-M was not tested.17,18
Natural products have inherent characteristics that have been exploited classically in infectious diseases and cancers with success.19 Especially, natural compounds present complex frameworks that have evolved to exhibit protein binding motifs.20 This wide variety is not found in synthetic libraries: approximately 50% of the natural products in the Dictionary of Natural Products database do not have a synthetic equivalent.21
The process of discovering new drugs is notoriously expensive.22 Computer-aided drug design (CADD) approaches can reduce the time and costs related to the development process for new medicines.23 Therefore, in the present work, CADD strategies were used to find new potential HsPEPCK-M inhibitors that could be used as alternatives for NSCLC treatment. The drug discovery approach is represented in Figure 1. Considering the promising anti-cancer therapeutic features of natural products, as high efficacy and low adverse effect compared to synthetic drugs,24 we employed structure-based virtual screening approaches to screening the natural compounds subset from the ZINC database. To this end, we applied a pharmacophore modeling method using the available crystal PEPCK structures to recover, from the natural compounds’ subset, a library enriched with potential PEPCK inhibitors with new scaffolds. Next, we used comparative modeling to access the three conformations adopted by PEPCK-M active site during the catalysis. These modeled conformations and the enriched library were used in molecular docking assays. As the outcome, binding mode analysis and chemical diversity evaluation enabled the proposition of four novel potential PEPCK-M inhibitors.
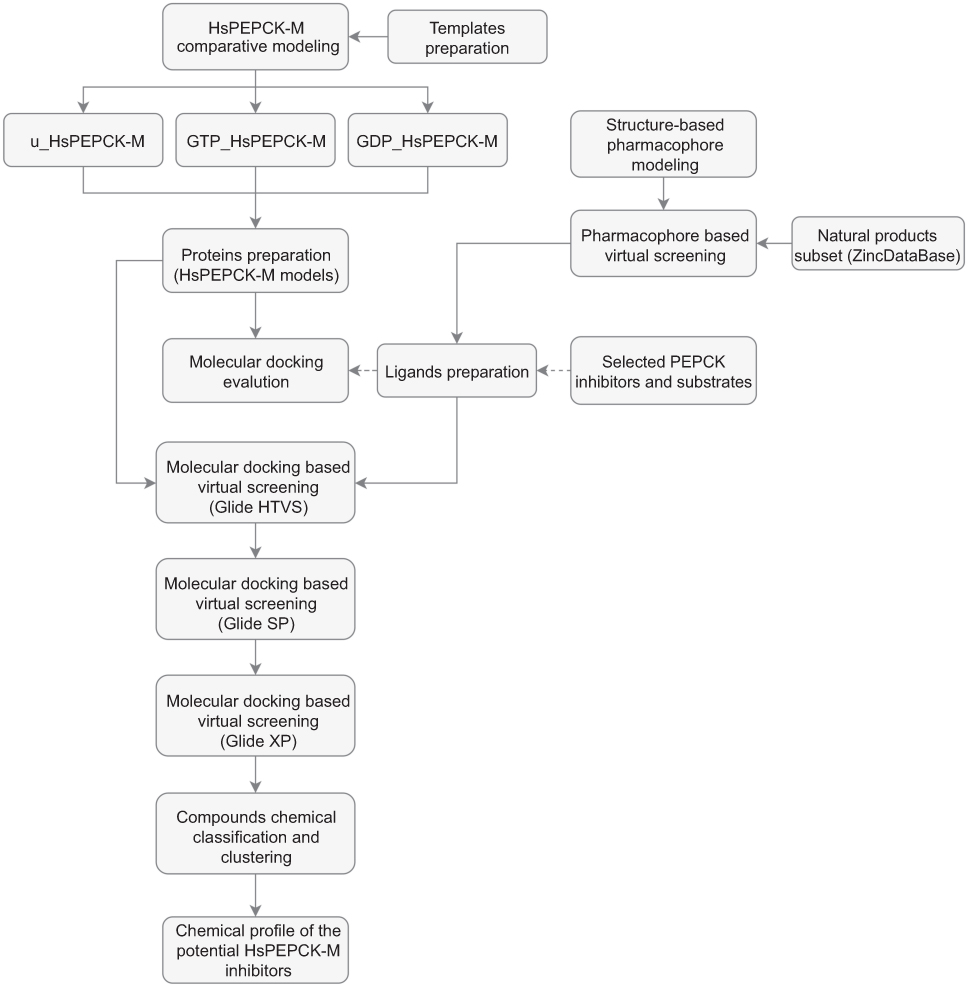 |
Figure 1 Schematic overview of the computer-aided drug design approach used in the identification of HsPEPCK-M potential inhibitors. Abbreviations: u_HsPEPCK-M, modeled protein without ligands; GTP_HsPEPCK-M, modeled protein complexed with GTP and OAA; GDP_HsPEPCK-M, modeled protein complexed with GDP and PEP. |
Materials and methods
Template preparation and comparative modeling
Since there is no three-dimensional (3D) experimentally determined structure for the human PEPCK-M protein (HsPEPCK-M), models were constructed using comparative modeling techniques. The amino-acid sequence of HsPEPCK-M (UniProt code Q16822) was used to retrieve suitable template structures from the protein data bank (https://www.rcsb.org/). The selected structures were: (i) PDB ID 1KHG (resolution 2.34 Å), represented HsPEPCK-C enzyme without substrates; (ii) PDB ID 1KHE (resolution 2.4 Å) to represent HsPEPCK-C complexed with a GTP analogue; (iii) PDB ID 1KHF (resolution 2.02 Å) to characterize HsPEPCK-C containing PEP substrate; and (iv) PDB ID 2FAH (resolution 2.09 Å) to represent PEPCK-M enzyme of Gallus gallus complexed with GDP (GgPEPCK-M).25,26
The final objective was to construct models considering the three conformations that the enzyme adopts with its substrates: (i) the unbound conformation (HsPEPCK-M), (ii) the enzyme bound to GTP + OAA (GTP_HsPEPCK-M), (iii) and the enzyme bound to GDP + PEP (GDP_HsPEPCK-M). It is important to note that 1KHF structure was determined complexed with PEP substrate but did not have the GDP substrate. To circumvent this problem, GDP molecule was added by superimposing the active site of 1KHF with the rat PEPCK-C complexed with PEP + GDP (PDB 4GMW). Then, similarly to what was done for 1KHF, we modified 1KHE (complexed with GTP) structure to add oxaloacetate (OAA) present in the rat PEPCK-C 2QF2 structure. The third structure, 1KHG, was maintained without substrates as determined experimentally.
The detailed analysis of the structures 1KHG, 1KHE and 1KHF revealed that these PDBs had incomplete regions – with no 3D structure defined for the canonical amino acid sequence. The regions comprised: (i) nine N-terminal residues, (ii) T465-G472 portion and (iii) K547, A548. To solve this problem, we used the MODELLER 9.17 loop optimization with the loopmodel class followed by refinement with “refine.slow” option – this was performed to complete the structures and optimize sidechains.27 Five structures were generated for each PDB, and the best result was selected using the DOPE score value.28 These prepared structures were used as templates for the HsPEPCK-M models.
The model for HsPEPCK-M unbound conformation (u_HsPEPCK-M) was built using prepared HsPEPCK-C – PDB ID 1KHG (sequence identity of 71% and 84% of similarity) and the GgPEPCK-M – PDB ID 2FAH (sequence identity of 70% and 79% of similarity). The models generated to represent the conformations of HsPEPCK-M bound to GTP (GTP_HsPEPCK-M) and GDP (GDP_HsPEPCK-M) were built based on the HsPEPCK-C – PDB ID 1KHE and HsPEPCK-C – PDB ID 1KHF, respectively; and also based on GgPEPCK-M structure (PDB ID 2FAH) – this structure was used since it presents all the loops ordered, without missing residues.
A hundred models were generated for each configuration (u_HsPEPCK-M, GTP_HsPEPCK-M, and GDP_HsPEPCK-M) using the automodel class of MODELLER 9.17. The three final monomeric models were selected based on the DOPE scoring function and then evaluated for their stereochemical quality using ERRAT and VERIFY 3D scores from SAVES server (http://services.mbi.ucla.edu/SAVES/) and MOLPROBITY (http://molprobity.biochem.duke.edu/).29–31
Structure-based pharmacophore modeling and ZINC database virtual screening
Structure-based pharmacophore hypotheses were generated with Pharmer software,32 using the crystallographic structure of HsPEPCK-C complexed to PEP (PDB ID 1KHF). No structure of human PEPCK-C complexed with OAA was available. All interactions between the ligand and the crystallographic protein were mapped. The final selection of key pharmacophoric features used to generate the hypotheses was based on the analysis of HsPEPCK-C active site and common interaction patterns observed in RnPEPCK-C structures complexed with known inhibitors – oxalic acid, phosphonoformic acid, 2-phosphoglycolic acid, 3-phosphonopropanoic acid and sulfoacetic acid (PDB IDs 2RK7, 2RK8, 2RKA, 2RKD and 2RKE, respectively).18
These structure-based pharmacophore hypotheses were used as queries in ZINCPharmer web server aiming at screening compounds from ZINC database (natural products subset) that fit into the pharmacophore hypotheses.33,34 These compounds, therefore, have the potential to act as HsPEPCK-C inhibitors and were used to build the compounds library for virtual screening in this study.
Protein preparation for molecular docking
The monomeric structures for each protein conformation (u_HsPEPCK-M, GTP_HsPEPCK-M, and GDP_HsPEPCK-M) were prepared with Protein Preparation Wizard, Maestro (Schrödinger, LLC, New York, NY, 2014). This step aims at adding hydrogens, assigning the rotation of Chi (Chi flip) angles of asparagine, glutamine and histidine residues and correcting errors in the structures. Based on the proposed structural mechanism of catalysis for rat PEPCK-C13 and the pKa data predicted by PROPKA35 the protonation states of the ionizable side chains at pH 7 were defined. Residues Arg104, Lys261, Lys262 and Arg423, were considered protonated. The divalent Manganese ions, essential to the reaction, were kept in the active site.
In addition, an analysis of conserved water in the active site of PEPCK enzymes was carried out using the Maestro program (Schrödinger, LLC, New York, NY, 2014). The RnPEPCK-C (PDB ID 2QF1, 2QEY, 2QF2, and 4GMW) and HsPEPCK-C (PDB ID 1KHG, 1KHE and 1KHF) structures with a set of ligands (oxaloacetate, 1,2-ethanediol, oxaloacetate ion/pyruvic acid, phosphoenolpyruvate, no ligand, phosphomethylphosphonic acid guanylate ester, phosphoenolpyruvate, respectively) were selected for the analysis of structural water.14,26,36 The water molecules present in 60% or more of the structures within the catalytic site were maintained in the HsPEPCK-M modeled structures. The list of conserved water molecules considered in each modeled structure is in the Table S1.
Ligand preparation for molecular docking
The PEPCK substrates phosphoenolpyruvate (1, CID 1005), oxaloacetate (2, CID 164,550) and seven analogue compounds known to inhibit rat PEPCK-C – oxalic acid (3, CID 971), phosphonoformic acid (4, CID 3415), phosphoglycolic acid (5, CID 529), 3-phosphonopropionic acid (6, CID 1682), 1,2-ethanediphosphonic acid (7, CID 80247), sulfoacetic acid (8, CID 31257), 3-sulfopropanoic acid (9, CID 409694) – and 3-mercaptopicolinic acid (10, CID 119070), known to inhibit HsPEPCK-M,15,18 were obtained from the PubChem database (Figure 2).37 In addition, the compounds screened from ZINC database using the pharmacophore hypothesis were also retrieved. All molecules were prepared with the Ligprep module from Maestro (Schrödinger, LLC, New York, NY, 2014). The preparation included the generation of tautomers and the ionization states at pH 7. The chirality of all compounds was maintained as in the original files.
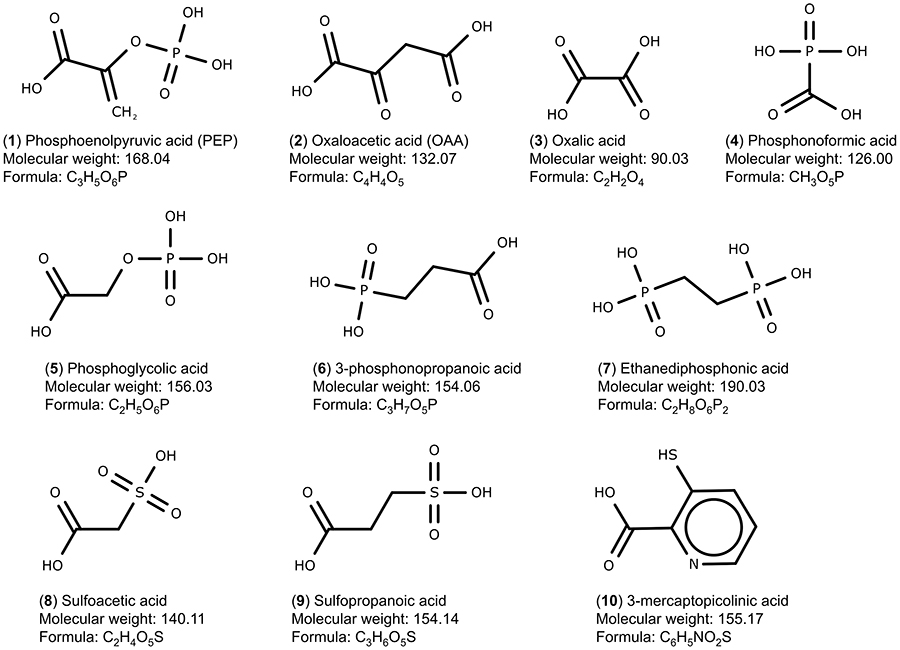 |
Figure 2 Set of PEPCK substrates (1–2), PEPCK-C inhibitors (3–9), and PEPCK-M inhibitor (10) used in the docking study. The molecular weight is in g/mol. |
Molecular docking and virtual screening
Molecular docking experiments were performed aiming to predict the binding poses and scoring of the selected compounds for this study against the modeled HsPEPCK-M conformations. To test the molecular docking software and the ideal preparation for the HsPEPCK active site, a redocking experiment was carried out using the PDB 1KHF and the PEP substrate (from the original complex) with the Glide Extra Precision (Glide XP) protocol (version 6.2, Schrödinger, LLC, New York, NY, 2014).
The docking experiments used grid centered at the ligands present in the active sites of the models (OAA or PEP). An exception was the model u_HsPEPCK-M which had no substrates; for this reason, the centroid of PEP was used after the superposition of the models. The grids were generated with a 10 Å edge.
The known rat PEPCK-C inhibitors were also docked using the HsPEPCK-C models u_HsPEPCK-M, GTP_HsPEPCK-M and GDP_HsPEPCK-M with Glide in the XP protocol. The library of natural compounds obtained from the structure-based pharmacophore screening was also submitted to molecular docking assay using the HsPEPCK-M models (u_HsPEPCK-M, GTP_HsPEPCK-M, and GDP_HsPEPCK-M) employing a hierarchical scheme: Glide high-throughput virtual screening (HTVS), to screen the initial set of compounds (total of 7,485); Glide standard precision (SP), to evaluate the top 50% compounds obtained from the previous step; and Glide XP, to screen the top 30% compounds retrieved by Glide SP protocol. The SP mode is suitable for testing a large number of compounds with high accuracy, while XP mode allows a more significant elimination of false positives, reflecting a higher enrichment of the final set of ligands. The best-ranked compounds in Glide XP screening were submitted to the next steps of this study.
Virtual screening validation
The ability to differentiate active compounds and known inactive compounds was evaluated by the molecular docking of two sets of decoys: DUD-E and DecoyFinder 2.0.38,39 For both programs, the set of PEPCK inhibitors (oxalic acid, phosphonoformic acid, phosphoglycolic acid, 3-phosphonopropionic acid, 1,2-ethanediphosphonic acid, sulfoacetic acid, 3-sulfopropionate, pyrophosphate, 3-mercaptopicolinic acid)15,18 and the two substrates (OAA and PEP) were used as queries. Program DUD-E generates 50 molecules for each known active, accounting for a total of 550 compounds. For the DecoyFinder program, a total of 396 decoys were generated using the molecular weight-based decoys option.
The two sets of decoys, the substrates along with the active compounds described for PEPCKs, were docked in the three configurations of HsPEPCK-M using Glide XP protocol. All molecules (decoys and inhibitors) were prepared following the ligand preparation protocol (described earlier). From the dockings, enrichment factor (EF) and area under the curve (AUC) values of receiver operating characteristic (ROC) curves were calculated to evaluate the model’s performance.
Clustering and chemical classification
The set of potential inhibitors obtained from virtual screening based on molecular docking was evaluated and clustered with Scaffold Hunter 2.6.3 software.40 For each compound, a corresponding fingerprint (size of 1024 bits) was calculated based on the Daylight method. From this, hierarchical clustering of the compounds was performed using Ward linkage method and Tanimoto coefficient as the similarity metric. Therefore, at each stage of the clustering process, the two clusters that resulted in a minimum increase of similarity variance were merged.
The chemical classification of the compounds was done using the taxonomy procedure with ClassyFire (http://classyfire.wishartlab.com/).41 Afterward, the four selected compounds – (i) hydroxycitrate (ZINC01656421), (ii) tartaric acid (ZINC895296), (iii) danshensu (ZINC00895535) and (iv) 2-(2-Phenylhydrazono)malonic acid (ZINC02571340) – along with the known rat PEPCK-C inhibitors, were subjected to ADME-TOX prediction using the server FAFDrugs4.42
Results and discussion
Sequence and structure comparison
A sequence alignment and analysis of HsPEPCK-M in relation to other PEPCKs were carried out to evaluate and compare the key/essential aspects of these primary structures and extrapolate the information to our target enzyme. The global alignment of PEPCK sequences is presented in Figure S1, as well as the important residues described for RnPEPCK-C compared to the human PEPCK-M and PEPCK-C sequences. The sequence comparisons showed that the identity and similarity between the sequences of HsPEPCK-M and HsPEPCK-C are 69% and 82%, respectively. The N-terminal amino acid sequence of HsPEPCK-M (residues 1–32) has a region indicated (by similarity) as a transit peptide in UniProt (www.uniprot.org). This section would be responsible for addressing the protein to the mitochondria. A cleavage region was also identified by the PrediSi server (www.predisi.de) at residue 26.43 The transit peptide is the region with the greatest dissimilarity between HsPEPCK-M and the other sequences. When comparing just active site’s residues, the identity increased to 100%. A similar degree of conservation is observed when comparing the HsPEPCK-M sequence with other PEPCKs: (i) identity of 65% and similarity of 75% to GgPEPCK-M (ii) 69% and 82% to RnPEPCK-C. The sequences corresponding the Ω-loop, P-loop (Kinase-1a) and R-loop, described by Carlson and collaborators as important in induced fit aspects of the PEPCK-C active sites, showed just one substitution of valine to isoleucine in the R-loop of PEPCK-Ms in relation to PEPCK-Cs.13 Moreover, two known domains described for PEPCK-C enzymes were also observed in the HsPEPCK-M sequence with no substitutions: (i) the PCK-Specific Domain, (ii) Kinase-2.
The 3D models of the three conformations of HsPEPCK-M, including GTP_HsPEPCK-M, GDP_HsPEPCK-M, and u_HsPEPCK-M, were generated by comparative modeling based on HsPEPCK-C and GgPEPCK-M structures. The overall 3D structure of GTP_HsPEPCK-M and the prepared templates HsPEPCK-C with GTP (PDB ID 1KHE) and GgPEPCK-M (PDB ID 2FAH) are represented in Figure 3A along with the active site poses of GTP_HsPEPCK-M, GDP_HsPEPCK-M, and u_HsPEPCK-M (Figure 3B–D, respectively). The described queries identities and similarities were satisfactory for the assurance of the quality of the models. These are emphasized by the results obtained with the evaluation tools that indicated that all generated models presented at least 94.4% of amino acids in favored regions, 99.3% in allowed regions, and ERRAT with an overall quality factor higher than 90.1 (Figure S2). The Ramachandran plot of GTP_PEPCK-M, GDP_PEPCK-M, and u_PEPCK-M are represented in Figure S2 A–C, respectively. The overall quality factor, according to ERRAT, of GTP_PEPCK-M, GDP_PEPCK-M, and u_PEPCK-M are represented in Figure S2 D–F, respectively. Besides the ligands (except for u_HsPEPCK-M), these models contain two manganese ions with their 3D coordinates derived from their corresponding HsPEPCK-C template structures.
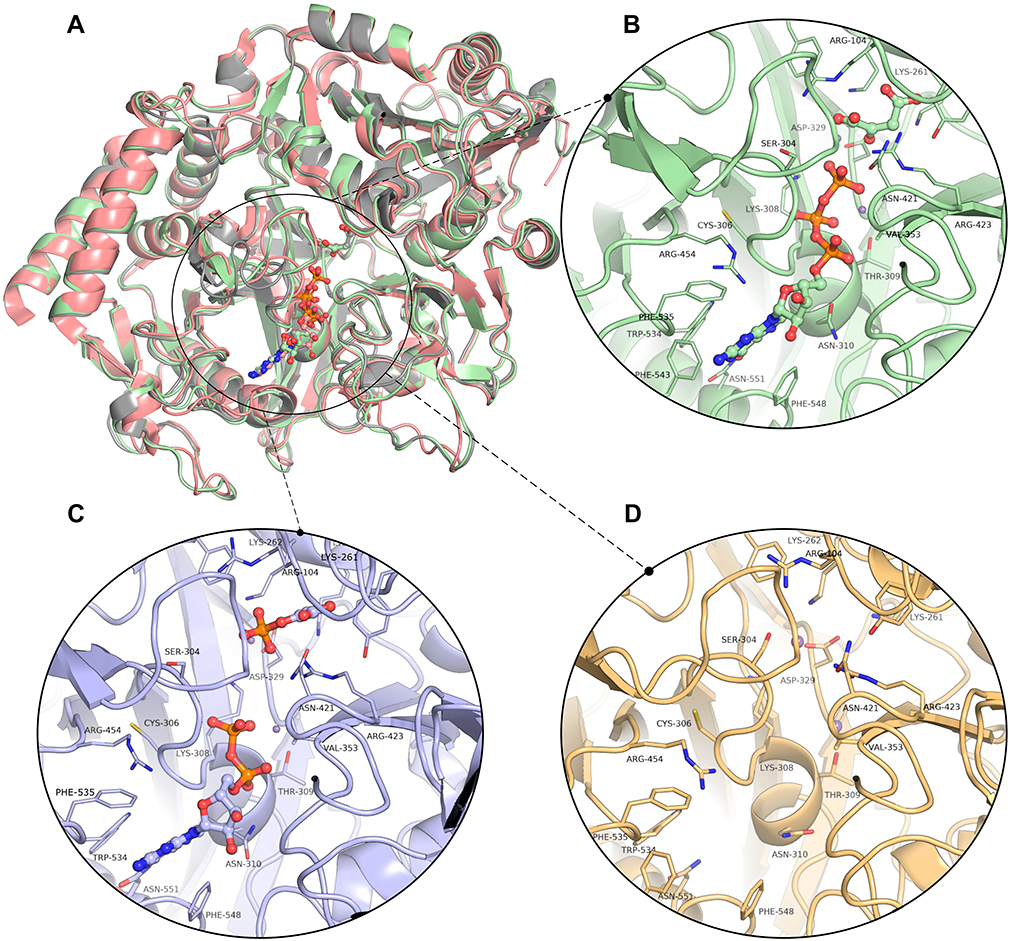 |
Figure 3 The three-dimensional structures of HsPEPCK-M models and the templates. (A) The structural superposition of GTP_HsPEPCK-M (green), the prepared template HsPEPCK-C with GTP (PDB ID 1KHE – grey) and GgPEPCK-M (PDB ID 2FAH – pink). The active site poses of (B) the model GTP_HsPEPCK-M complexed with GTP and OAA (C) GDP_HsPEPCK-M complexed with GDP and PEP and (D) u_HsPEPCK-M without ligands. All ligands are represented as ball and sticks, and residues’ sidechains are represented as sticks. The two manganese ions are represented as purple spheres. Protein structural figures were generated using Pymol 2.1.0. |
As expected, the global structure of the models for HsPEPCK-M resembles the structure of other known PEPCKs – a monomer following the P-loop-containing nucleoside triphosphate hydrolase fold.44 The HsPEPCK-M structure is divided into two lobes, one N-terminal and the other C-terminal with the active site situated in the middle of them. The N-terminal lobe contains the R-loop (residues 102–109) while the C-terminal contains both P-loop/Kinase-1a domain (302–309) and Ω-loop (482–492). Besides the characteristic loops, the models also included the domains PCK-Specific Domain and Kinase-2.
The differences between the three models (u_HsPEPCK-M, GDP_HsPEPCK-M, and GTP_HsPEPCK-M), vital to explore the different states of the active site, were subtle and involved mainly changes in the P-loop and Ω-loop. These changes, together with the different substrates (or no substrate for u_HsPEPCK-M), allows for the probing of a range of conformations in the active site.
Structure-based pharmacophore modeling and ZINC database virtual screening
Structure-based pharmacophore modeling was carried out to identify complementary chemical features between PEPCK’s active site and the ligand as well as the spatial arrangements of these features. The goal was to determine and select the pharmacophoric features essential for biological activity of ligands on HsPEPCKs. To this end, we used the crystal structure of HsPEPCK-C bound to PEP as reference and crystal structures of RnPEPCK-C complexed with inhibitors (with structures derived from PEP and OAA) to identify critical interactions. As a result, two pharmacophore hypotheses were achieved: PH1 and PH2 (Figure 4A and B). PH1 included three pharmacophoric features: one negative ionizable center and two hydrogen-bond acceptors (Figure 4A). The feature representing a negative ionizable group was considered since the phosphate group of PEP, and its equivalents in the analyzed inhibitors (except for PDB ID 2RKE) had a negative charge that interacts with the two Mn2+ and active site residues with a positive charge – Arg87, Lys243, Lys244, and Arg405 (HsPEPCK-C numbering). The two hydrogen-bond acceptors, both corresponding to the carboxyl group of PEP, were selected due to the interaction already described with residues Gly237 and Asn403. These hydrogen-bond acceptors were also observed in the inhibitors present on 2RKA and 2RKE structures at precisely the same positions and on the inhibitor from 2RKD structure with a similar interaction with residue Gly237. The second hypothesis, PH2, was constructed with the pharmacophoric features already described for PH1 and two more features: one hydrogen-bond acceptor and a hydrophobic group as highlighted in Figure 4B. These additional selections considered mainly the complementarity of features between HsPEPCK-C active site and PEP in the 1KHF structure: (i) hydrogen-bond interaction of PEP with the side chain of Arg405 and (ii) the hydrophobic region of PEP (highlighted in green) complementary with the hydrocarbon chain of Lys244 side chain.
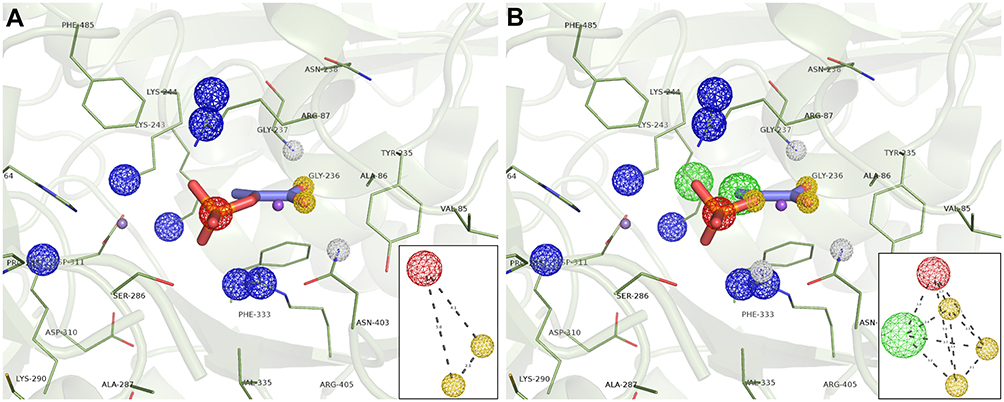 |
Figure 4 Structure-based pharmacophore hypothesis (PH). Important pharmacophoric features in the active site of HsPEPCK-C in the 1KHF structure are represented in (A) PH1 and (B) PH2. In the insets are the schematic representations of PH1 and PH2 with the distances between features in angstroms (Å). Pharmacophore features are colored according to their characteristic: red=negative ionic, blue=positive ionic, yellow=H-bond acceptor and white=H-bond donor. |
These two pharmacophore hypotheses were used as query to screen the natural compounds set from the ZINC Database (totaling 179,816 compounds) to recover potential biologically active compounds presenting the pharmacophoric features and architecture of the queries. The radius of the features was 0.75 Å for hydrogen-bond acceptor, 1 Å for the hydrophobic group and 1.5 Å for the negatively charged group. This pharmacophore-based virtual screening retrieved 7,124 compounds (after excluding duplications) that were submitted to tautomer generation and set to suitable ionization states. As a result, a total of 7,485 compounds characterized a library enriched with potential mammalian PEPCKs inhibitors.
Docking validation
The HsPEPCK-M models in combination with the Glide XP protocol were validated for their ability to distinguish active compounds from a set of decoys (putative inactive compounds). From the ranking of the compounds, ROC curves were generated (Figure S3), and enrichment factors to a defined sample size and AUC values of the models (Table 1) were calculated.
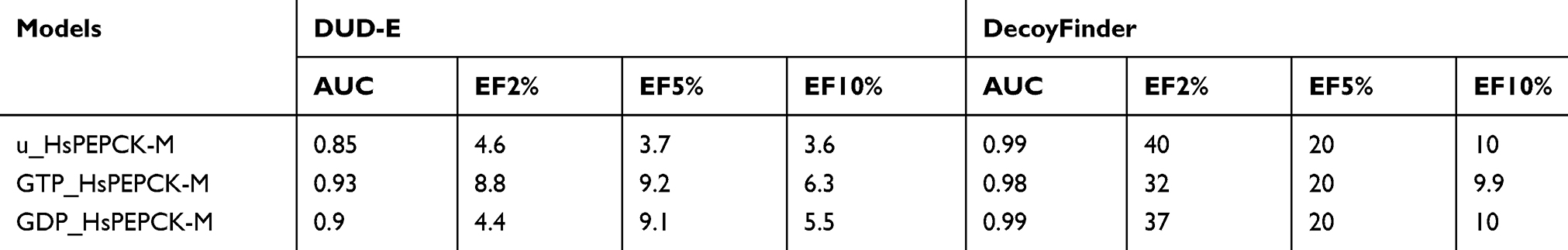 |
Table 1 Area under the curve and enrichment factors for decoy dockings |
The enrichment factors values indicate the number of times the evaluated models and docking protocol outperformed a random selection of ligands. In all cases, considering EF of 2%, 5%, and 10%, the values were equal or higher than 3.6. The ROC curve allowed to show the sensitivity about the specificity of the method (models and docking protocol). From this, the calculation of the AUC was able to quantify the performance of the Glide XP scoring function about the models. ROC curves for decoy docking with DecoyFinder (Figure S3 A–C) and DUD-E (Figure S3 D–F) were generated, for GTP_HsPEPCK-M, GDP_HsPEPCK-M, and u_HsPEPCK-M, respectively. These six ROC curves obtained good AUC values above 0.85. Traditionally, a classification algorithm is considered excellent if 0.9≤ AUC ≤1, good if 0.80≤ AUC <0.9, fair if 0.70≤ AUC <0.8, poor if 0.50≤ AUC <0.7, and an AUC <0.5 is the same as a random classifier.45 In general, the model GTP_HsPEPCK-M outperformed the others using DUD-E decoys while the model u_HsPEPCK-M was the best one using DecoyFinder decoys. These AUC and EF values indicate that all the models can successfully discriminate positives from false positives (decoys). Likewise, the redocking experiment, performed with the template structure PDB ID 1KHF, was also used to validate the protocol for docking. The structure for HsPEPCK-C co-crystallized with PEP (PDB ID 1KHF) showed a root mean square deviation (RMSD) of 0.74 Å from the best redocking pose with Glide XP (Figure S4 A). These results suggest the suitability of Glide XP software and the parameters used for further structure-based analyses.
Molecular docking
With the aim of identifying new potential inhibitors for HsPEPCK-M, the three models generated, and the set of ZINC natural compounds screened by the pharmacophore hypotheses were used in a hierarchical virtual screening based on molecular docking. From this approach, it was possible to access the potential binding poses and scoring of these selected compounds. As a positive control, the substrates (PEP and OAA) and seven substrate analogs are known to inhibit PEPCK-Cs and 3-mercaptopicolinic acid (CID 119070), an inhibitor of both PEPCK-Cs and PEPCK-Ms, were used in the docking experiment Figure S5). The result of the docking with the substrates and inhibitors along with the best ten ligands obtained with the virtual screening based on molecular docking are shown in Table 2.
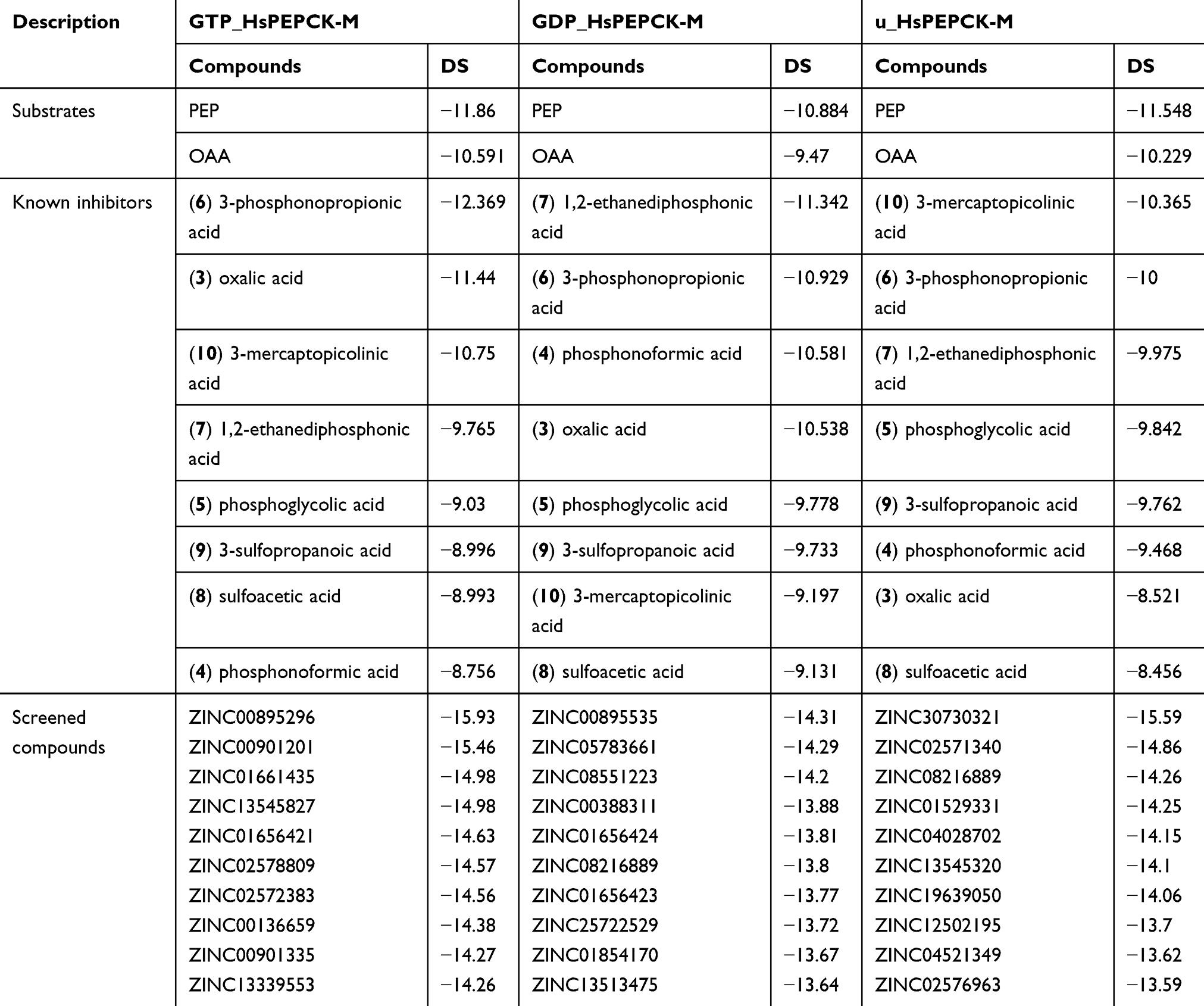 |
Table 2 Docking Scores of the positive controls and the ten best scoring compounds for HsPEPCK-M in all configurations |
The analyses of scoring of these substrates and known inhibitors and their biding poses evaluation are important as a point of comparison for the virtual screening outcomes. In general, the detailed analysis of the binding mode of the substrates showed good agreement with that observed on template poses. From the dockings of PEP substrate on the three HsPEPCK-M models, the best pose was observed in GDP_PEPCK-M enzyme showing root mean square deviation (RMSD) of 1.356 Å when compared to that pose in HsPEPCK-C (PDB ID 1KHF). The interaction pattern was also similar Figure S5 A and B: (i) the phosphate group of PEP coordinates with manganese ion (Mn2+) while making a salt bridge with Arg423; (ii) oxygen from PEP’s carboxylate makes a hydrogen bond with the amide backbone of Gly255 and Arg104. The substrate OAA, on the best pose using GTP_PEPCK-M, showed an RMSD of 1.796 Å from the experimentally determined pose in the RnPEPCK-C (PDB ID 2QF2) (Figure S5 C and D). It is important to note that the 2QF2 structure was not used directly as a template for a model generation – the OAA substrate was added to the model by overlapping the structures in the comparative modeling step. Despite this, the docking pose preserved important interactions that are observed in the crystallographic pose: (i) similar to the interaction observed in PEP, OAA also interacts with the Mn2+ through one of its carboxylate group; (ii) a salt bridge between Arg104 guanidine and the OAA carboxylate; (iii) OAA carboxylate also makes a salt bridge with Arg423 sidechain. However, the docked pose of OAA loses an important interaction between its carbonyl group and Mn2+ – it interacts instead with the guanidine group of Arg423.
Furthermore, 3-mercaptopicolinic acid (MPA) returned a docking score of −10.750 in GTP_HsPEPCK-M, −9.197 in the GDP_HsPEPCK-M and −10.365 for u_HsPEPCK-M. The DS of MPA is an important positive control because it is known to inhibit HsPEPCK-M. The binding mode of MPA, determined in the structure of RnPEPCK-C (PDB ID 4YW9), showed interactions similar to the ones from the docking pose of GTP_HsPEPCK-M (Figure S5 E and F): (i) the carboxylate group of the ligand accepts a hydrogen bond from Arg104; (ii) Arg423 makes a pi-cation interaction with the pyridine ring of the ligand; (iii) the carboxylate group of the ligand coordinates with the Mn2+. In the docking pose, the orientation of the thiol group of MPA was inverted from the one in the crystallographic pose – it is coordinating with Mn2+. In the experimental pose, the nitrogen of the pyridine ring has this role.
The docking score values of the known PEPCK-M inhibitor, 3-mercaptopicolinic acid, on the three HsPEPCK-M models were used as cut-offs to select the top-scored compounds from the three molecular docking virtual screenings. Therefore, the compounds with DS values equal or less than −10.75, −9.197 and −10.581 on the dockings with GTP_HsPEPCK-M, GDP_HsPEPCK-M, and u_HsPEPCK-M, respectively, were considered as potential HsPEPCK-M inhibitors, since they present better DS values than 3-mercaptopicolinic acid. As an outcome, a total of 612 natural compounds were potentially able to inhibit at least one of the three conformations of HsPEPCK-M, of which 62 have the potential to inhibit all of them.
The chemical classification of these 612 potential inhibitors was achieved with a structure-based chemical taxonomy procedure performed with ClassyFire software.41 The superclasses and classes distributions of the chemical classifications are represented in Figure 5A and B, respectively. From these, organic acids and derivates, more specifically, carboxylic acids and derivates are the main superclass and class observed in the HsPEPCK-M potential inhibitors pool. Another significant superclass is the benzenoid category, with most of them being of benzene and substituted derivatives class. Additionally, fatty acyls, organooxygen compounds, phenylpropanoic acids, hydroxy acids and derivatives, phenols, pyridines and derivatives, organic phosphonic acids and derivatives are other classes with an expressive number of compounds. In contrast, most of the known PEPCK inhibitors (3-phosphonopropanoic acid, 2-phosphonoethylphosphonic acid, phosphonoformic acid, and phosphoglycolate) and substrates (phosphoenolpyruvate) are predominantly from the class organic phosphonic acids and derivatives. These analyses may indicate that most of the putative inhibitors have new chemical scaffolds when compared to the known inhibitors, as expected by a pharmacophore virtual screening approach.
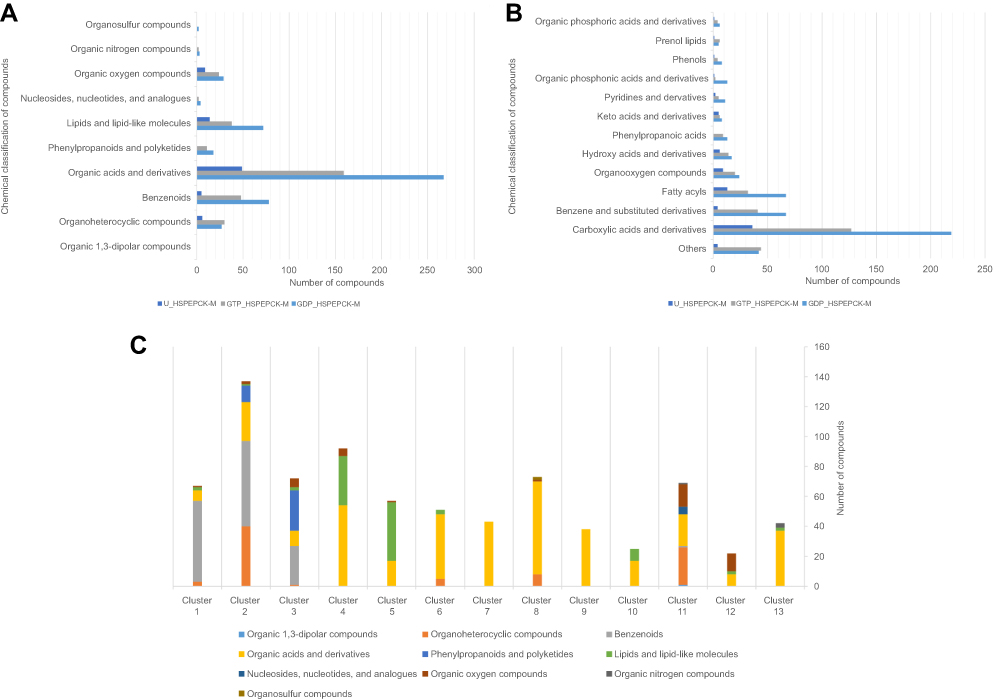 |
Figure 5 Chemical classification and distribution of compounds on classes based on ClassyFire software. (A) Chemical classes of the HsPEPCK-M potential inhibitors. (B) Chemical superclasses of the HsPEPCK-M potential inhibitors. (C) The chemical superclasses and distribution of the compounds on 13 clusters obtained in the clusterization procedure. |
Virtual screening clusterization
Generally, at the end of a virtual screening protocol, many identified compounds somewhat resemble one another. Therefore, it is sound to select representative compounds for more detailed evaluation steps. In these cases, grouping the set of compounds into clusters containing similar chemical structures and selecting representative compounds of each cluster is a common procedure. Considering this, the compounds retrieved from the molecular docking virtual screening step were hierarchically clustered based on their structural similarities measured by Tanimoto coefficient. As a result, a total of 13 clusters were obtained, as shown in dendrogram representation in Figure 6. The heat map, together with the dendrogram, allowed to evaluate the distribution of the DS values for all the three HsPEPCK-M modeled conformations. The representative compounds for each cluster are also depicted in Figure 6, and the distribution of superclasses among the clusters are represented in Figure 5C.
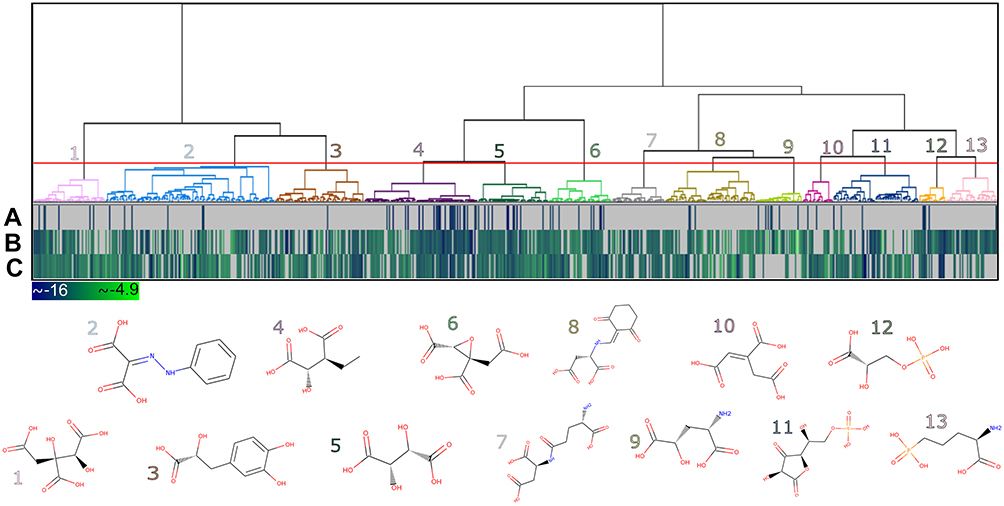 |
Figure 6 Heat map and dendrogram for the clusters of the 779 potential HsPEPCK-M inhibitors. The selected cut-off point generated 13 groups – an illustrative chemical structure is presented for each cluster (1–13). The heat map represents the docking score for (A) GTP_HsPEPCK-M, (B) GDP_HsPEPCK-M and (C) u_HsPEPCK-M. The best scores are depicted in dark blue while the worst are in light green. |
The clusters contained 22 to 137 compounds each, and cluster 2 presented the highest number of compounds (Table S2). In this cluster, benzenoids were the most frequent, followed by organoheterocyclic compounds, like pyridines and derivatives – the same as observed in MPA, HsPEPCK-M known inhibitor. Phenylpropanoids and polyketides compounds were also observed in cluster 2 but were present in a higher number on cluster 3. Other groups with benzenoids compounds were the clusters 1 and 3. Lipids and lipid-like molecules were expressive on clusters 4 and 5. It is important to note, that all clusters presented compounds like carboxylic acids, organic phosphonic acids and derivatives, and peptidomimetics. This large distribution of compounds of these classes over all clusters reflects their structural diversity. The high number of organic acids, in general, is probably explained by two aspects: (i) the anionic feature selected by the pharmacophore hypotheses used on the screening of the initial set of natural compounds; (ii) and the electrostatic interactions of the negatively charged moieties of these compounds with the highly positive microenvironment in the HsPEPCK-M’s active site (mostly with the manganese ion and the Arg423, Arg105 Lys262, and Lys261 residues), observed in molecular docking. This is important since this type of electrostatic interactions are also observed in the known PEPCK inhibitors as described before by Stiffin and collaborators.18
Molecular interaction analyses of the selected representative compounds
As a result of the virtual screening by docking with the three conformations, and from the clustering protocol, a set of four compounds presenting promising characteristics were selected for further analysis: (i) hydroxycitrate (ZINC01656421), (ii) tartaric acid (ZINC895296), (iii) danshensu (ZINC00895535) and (iv) 2-(2-Phenylhydrazono)malonic acid (ZINC02571340).
The compound hydroxycitrate (ZINC01656421), a tricarboxylic acid derivative representative of the cluster 4 (Figure 6), found in the list of best scored in the 3 conformations (DS −14.62, −11.699 and −13.234 for GTP_HsPEPCK-M, GDP_HsPEPCK-M and u_HsPEPCK-M, respectively), was already studied in the literature for the treatment against cancer.46 In this study, the authors found that a combination of hydroxycitrate, α-lipoic acid, and octreotide, when tested in NCI-H69 cells (small cell lung cancer model), had a “strong inhibitory effect on tumor development” being equivalent to treatment with cisplatin.46 It was shown in the literature that hydroxycitrate is a strong inhibitor of ATP citrate lyase (EC 2.3.3.8).47 ATP citrate lyase and PEPCK both have as substrates oxaloacetate and a triphosphate (GTP in the case of PEPCK). Our study also suggests that hydroxycitrate is a good inhibitor of PEPCKs. The docking pose of hydroxycitrate in the GTP_HsPEPCK-M is presented in Figure 7A and B. The docking reveals interactions akin to the ones presented by the substrates and the inhibitor MPA: (i) one of the carboxylate groups and the hydroxyl group of the ligand coordinates with the Mn2+ ion; (ii) a second carboxylate in hydroxycitrate makes salt bridges with Arg104; (iii) the third carboxylate of the ligand makes a hydrogen bond with the amide backbone of Gly255.
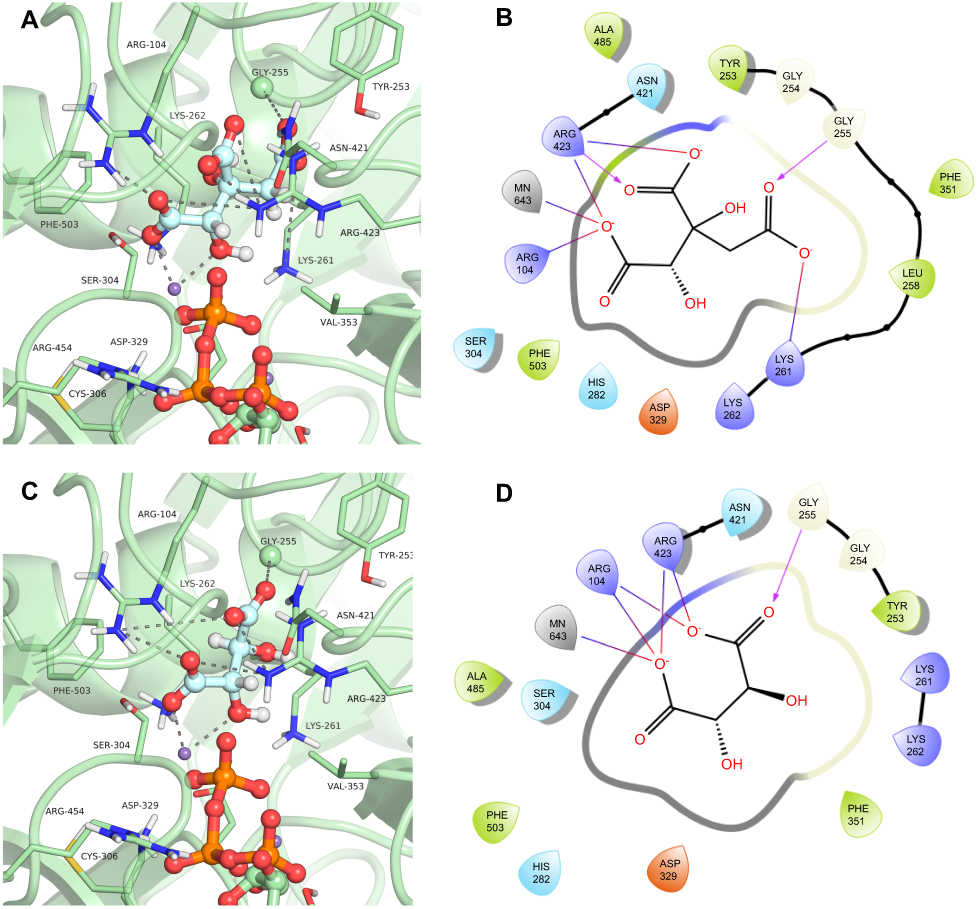 |
Figure 7 Binding mode of the hydroxycitrate (ZINC01656421) and tartaric acid (ZINC895296) in GTP_HsPEPCK-M active site obtained with the molecular docking assay. (A) and (C) are the 3D binding modes of hydroxycitrate and tartaric acid, respectively. The inhibitors are represented as ball and sticks, the protein represented in light green, and the intermolecular interactions are represented as gray dashes; (B) and (D) are the 2D interactions diagram of molecular docking results of hydroxycitrate and tartaric acid, respectively. The 2D residues are illustrated as negatively charged (red), positively charged (purple), polar (blue), hydrophobic (green). The hydrogen bonds are represented as purple arrows and salt bridges are represented as blue/red lines. The 3D and 2D images were generated using Pymol 2.1.0 and Maestro, respectively. |
Another well-scored compound was tartaric acid (ZINC895296), found in cluster 5 (Figure 6). The compound was the best ranked in the virtual screening with GTP_HsPEPCK-M and the best score overall (DS −15.925, −13.234 and −12.049 for GTP_HsPEPCK-M, GDP_HsPEPCK-M, and u_HsPEPCK-M, respectively). In the literature, tartaric acid is shown to inhibit both rats lysosomal and prostatic acid phosphatases with a Ki of 1 µM and also human prostatic acid phosphatase with a Ki of 29 µM.48,49 Human prostatic acid phosphatase in serum is used to diagnose prostate cancer, but we could not find evidence relating it to PEPCK.50 The binding mode o tartaric acid in the model GTP_HsPEPCK-M is presented in Figure 7C and D. This examination reveals many of the same interactions presented by other well-scored ligands described previously: (i) tartaric acid coordinates with the Mn2+ ion by one carboxylate groups and a hydroxyl group; (ii) the ligand makes salt bridges from both carboxylate groups with Arg104 sidechain; (iii) both carboxylate groups also make interactions with Arg423 sidechain.
The compound 3-(3,4-Dihydroxyphenyl)lactic acid, also known as danshensu (ZINC00895535) found in cluster 3, was also selected (Figure 6). This compound had the best docking score in the virtual screening with GDP_HsPEPCK-M (DS −14.306), although had not good scores in the other two models (ie, GTP_HsPEPCK-M and u_HsPEPCK-M). Danshensu was found to decrease the radioresistance and enhance apoptosis induced by radiation in NSCLC cells.51 This activity was connected to the inhibition of monoamine oxidase B, which then, in turn, decreased the activation of the nuclear factor-κB pathway (NF-κB).51 The supposed inhibition of HsPEPCK-M mediated by danshensu could also be one of the causative agents of apoptosis NSCLC. Similarly, inhibition of HsPEPCK-M by MPA induced apoptosis in glucose-depleted tests with human NSCLC cell lines A549 and H23.7 The binding mode of danshensu in GDP_HsPEPCK-M model is presented in Figure 8A and B. The molecular framework of danshensu allows it to make new interactions plus some also observed in the substrates: (i) the Mn2+ ion is coordinated with the carboxylate group and the adjacent hydroxyl group of the ligand; (ii) the carbonyl group of Ala485 makes an H-bond with one of the hydroxyl groups of the catechol ring; (iii) the catechol ring of the ligand also makes a pi-cation interaction with Arg423.
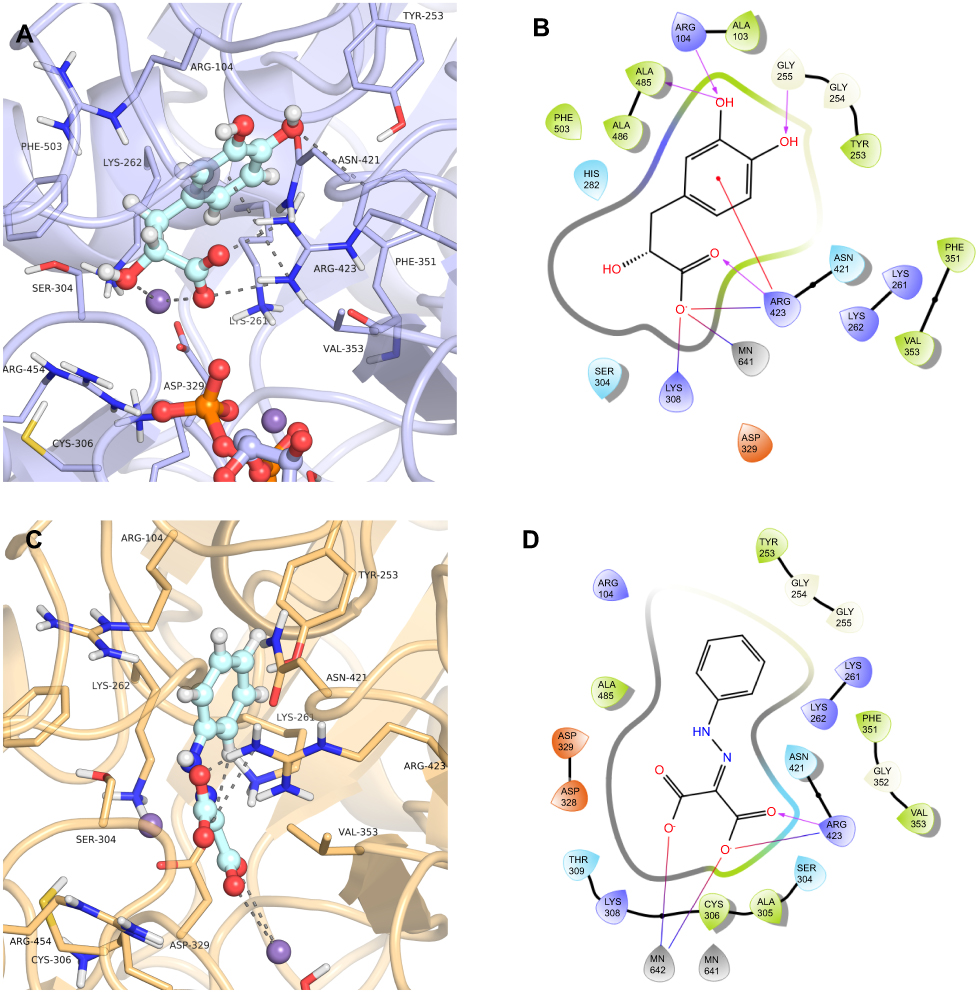 |
Figure 8 Binding mode of the danshensu (ZINC00895535) and 2-(2-Phenylhydrazono)malonic acid (ZINC02571340) in GDP_HsPEPCK-M and u_HsPEPCK-M active site, respectively, obtained with the molecular docking assay. (A) and (C) are the 3D binding modes of danshensu and 2-(2-Phenylhydrazono)malonic acid, respectively. The inhibitors are represented as ball and sticks, the protein represented in light purple (GDP_HsPEPCK-M) and light orange (u_HsPEPCK-M), and the intermolecular interactions are represented as gray dashes; (B) and (D) are the 2D interactions diagram of molecular docking results of danshensu and 2-(2-Phenylhydrazono)malonic acid, respectively. The 2D residues are illustrated as negatively charged (red), positively charged (purple), polar (blue), hydrophobic (green). The hydrogen bonds are represented as purple arrows and salt bridges are represented as blue/red lines. The 3D and 2D images were generated using Pymol 2.1.0 and Maestro, respectively. |
The fourth compound selected was 2-(2-Phenylhydrazono)malonic acid (ZINC02571340), a benzenoid from cluster 2 (Figure 6). The benzenoids are the second most abundant class in the virtual screening. The ligand had a DS of −14.862 in u_HsPEPCK-M, being the third best compound for this conformation. The docking pose of ZINC02571340 in the model u_HsPEPCK-M is presented in Figure 8C and D. The pocket of the model u_HsPEPCK-M is bigger than the other three conformations, because of the absence of GTP or GDP, and allowed a ligand pose with new interactions: (i) one of the carboxylate groups of ZINC02571340 coordinates with one of the Mn2+ ions – located where the γ-phosphate of GTP would be; (ii) the other carboxylate group makes a salt-bridge with the side chain of Arg423.
The compounds presented here are examples of interesting scaffolds selected among the representative clusters derived from the virtual screening protocol. These compounds have similar interaction patterns with those observed in the inhibitor MPA and in the substrates, especially the interaction with the manganese ion found to be important in previous studies of inhibition with substrate analogs.18 These interaction patterns and favorable docking scores suggest that these are promising compounds for future optimization and inhibition assays.
The PEPCK-M has an active site with positively charged residues (Arg104, Lys261, Lys262, and Arg423) and two manganese ions – these characteristics help stabilize the reactive intermediate enolate.13 However, because of these characteristics, several of the selected compounds present chemical groups with negative charges. In general, charged compounds have difficulty penetrating the lipid bilayer. To try to predict the overall in vivo behavior and identify potentially druggable compounds, we perform ADME-TOX predictions for the selected compounds using FAFDrugs4. Table 3 summarizes the observed results. In general, the compounds had good scores for the available indicators: all of them were approved in the Lipinski rules of five and had good predicted oral bioavailability by Veber and Egan rules.52–54 The PAINS filter was also accepted in the compounds except for 3-mercaptopicolinic acid (CID 119070) that had a topological polar surface area (tPSA) slightly lower than 75 Å2 raising a warning in the Pfizer 3/75 Rule. It is important to note that the compounds tartaric acid (ZINC895296) and oxalic acid (CID 971) were labeled by FAFDrugs as salts and were not evaluated. These predictions suggest that these compounds have good oral bioavailability and low toxicity indicating that they could be good starting points for drug optimization.
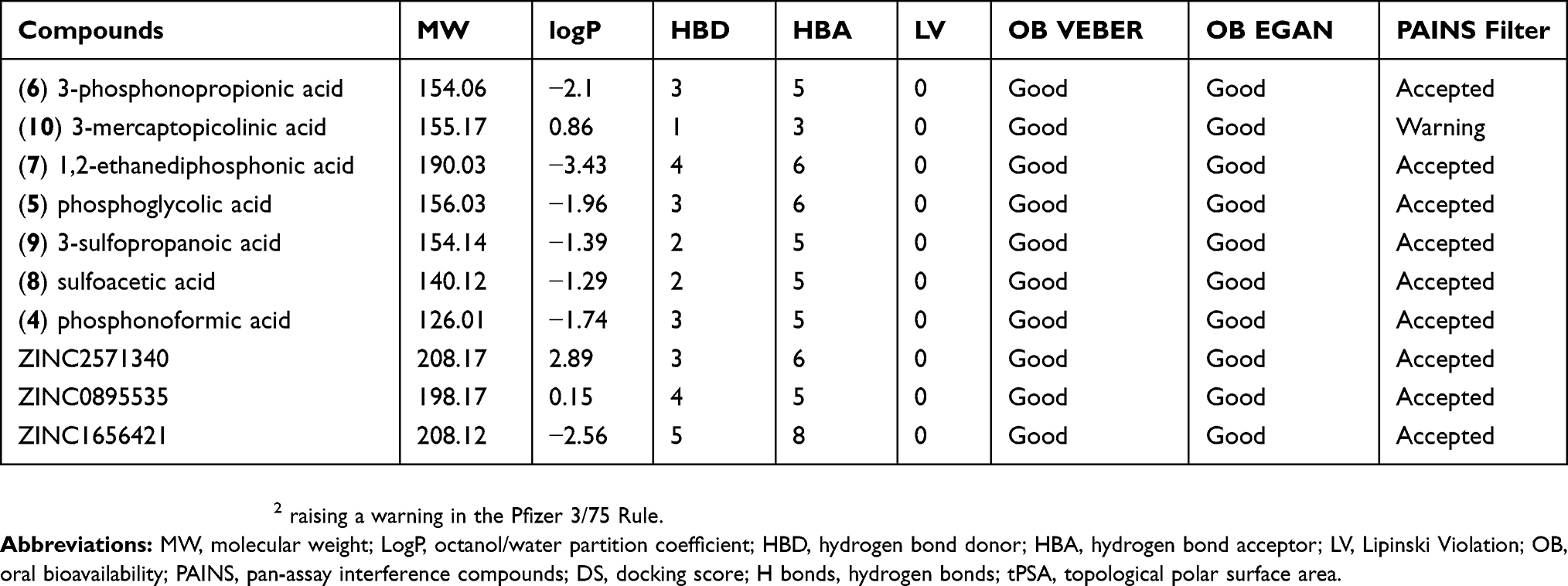 |
Table 3 ADME properties, PAINS filters for PEPCK known inhibitors and selected putative HsPEPCK-M inhibitors |
The search for drugs against cancer must also consider the high chance of side effects on healthy tissues. Recently, advances in technologies such as aptamers – synthetic oligonucleotides of RNA or single-strand DNA capable of binding with great specificity – have the promise of directing the cytotoxic effect of inhibitors on cells with specific markers.55 In the case of non-small cell lung cancer, aptamers could be bioconjugated with the putative inhibitors found here, reducing their possible side effects.
Conclusion
Here we presented a computer-aided drug design approach for the HsPEPCK-M aiming the proposal of promising potential inhibitors for NSCLC on low-glucose conditions. Among the CADD procedures, a structure-based pharmacophore modeling was carried out with available PEPCK structures complexed with PEP or known inhibitors. Through a pharmacophore-based virtual screening, these constructed models made possible a selection of 7,124 natural compounds with key features for PEPCK inhibition. We also presented the first described comparative models of HsPEPCK-M representing the three theoretical conformations assumed during the catalysis. The comparative models were used on a second virtual screening selection based on molecular docking. As a result, a total of 612 natural compounds were predicted to inhibit at least one of the three HsPEPCK-M conformations, and 62 were expected to inhibit all of them. Between these HsPEPPCK-M potential inhibitors, the carboxylic acids and derivates were the most frequent. The success of these compounds can be attributed to the observed interactions of their negatively charged groups with the manganese ion and with important positively charged residues on HsPEPCK-M’s active site. To select representative compounds for further binding mode analyses, all the retrieved compounds were hierarchically clustered based on structural similarities, resulting in 13 clusters. Four compounds from distinct clusters presented interesting new scaffolds and promising features: Hydroxycitrate (ZINC01656421), Tartaric acid (ZINC895296), Danshensu (ZINC00895535) and 2-(2-Phenylhydrazono)malonic acid (ZINC02571340). These compounds use key interactions observed in inhibitors already described for PEPCKs and have better docking scores values, indicating that they could potentially inhibit HsPEPCK-M and could act as new leads for further drug design studies.
Acknowledgments
The authors are grateful to the Brazilian National Council of Research (CNPq) and the Coordination for the Improvement of Higher Education Personnel (CAPES) for supporting this work.
Disclosure
The authors' report grants from CNPq and CAPES, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Wang H, Naghavi M, Allen C, et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388(10053):1459–1544. doi:10.1016/S0140-6736(16)31012-1
2. Basumallik N, Agarwal M. Cancer, lung, small cell (oat cell); 2018. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29494065. Accessed October 26, 2018.
3. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong K-K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. doi:10.1038/nrc3775
4. Harris AL. Hypoxia — a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi:10.1038/nrc704
5. Birsoy K, Possemato R, Lorbeer FK, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508(1):108–112. doi:10.1038/nature13110
6. Vincent EE, Sergushichev A, Griss T, et al. Mitochondrial phosphoenolpyruvate carboxykinase regulates metabolic adaptation and enables glucose-independent tumor growth. Mol Cell. 2015;60(2):195–207. doi:10.1016/j.molcel.2015.08.013
7. Leithner K, Hrzenjak A, Trötzmüller M, et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene. 2015;34(8):1044–1050. doi:10.1038/onc.2014.47
8. Balsa-Martinez E, Puigserver P. Cancer cells hijack gluconeogenic enzymes to fuel cell growth. Mol Cell. 2015;60(4):509–511. doi:10.1016/j.molcel.2015.11.005
9. Montal ED, Dewi R, Bhalla K, et al. PEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol Cell. 2015;60(4):571–583. doi:10.1016/j.molcel.2015.09.025
10. Beale EG, Harvey BJ, Forest C. PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem Biophys. 2007;48(2–3):89–95. doi:10.1007/s12013-007-0025-6
11. Yang J, Kalhan SC, Hanson RW. What is the metabolic role of phosphoenolpyruvate carboxykinase? J Biol Chem. 2009;284(40):27025–27029. doi:10.1074/jbc.R109.040543
12. Méndez-Lucas A, Hyroššová P, Novellasdemunt L, Viñals F, Perales JC. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. J Biol Chem. 2014;289(32):22090–22102. doi:10.1074/jbc.M114.566927
13. Carlson GM, Holyoak T. Structural insights into the mechanism of phosphoenolpyruvate carboxykinase catalysis. J Biol Chem. 2009;284(40):27037–27041. doi:10.1074/jbc.R109.040568
14. Johnson TA, Holyoak T. The Ω-Loop lid domain of phosphoenolpyruvate carboxykinase is essential for catalytic function. Biochemistry. 2012;51(47):9547–9559. doi:10.1021/bi301278t
15. Balan MD, McLeod MJ, Lotosky WR, Ghaly M, Holyoak T. Inhibition and allosteric regulation of monomeric phosphoenolpyruvate carboxykinase by 3-Mercaptopicolinic acid. Biochemistry. 2015;54(38):5878–5887. doi:10.1021/acs.biochem.5b00822
16. Robinson BH, Oei J. 3-Mercaptopicolinic acid, a preferential inhibitor of the cytosolic phosphoenolpyruvate carboxykinase. FEBS Lett. 1975;58(1–2):12–15. doi:10.1016/0014-5793(75)80214-6
17. Hidalgo J, Latorre P, Carrodeguas JA, Velázquez-Campoy A, Sancho J, López-Buesa P. Inhibition of pig phosphoenolpyruvate carboxykinase isoenzymes by 3-Mercaptopicolinic acid and novel inhibitors. PLoS One. 2016;11(7):1–17. doi:10.1371/journal.pone.0159002
18. Stiffin RM, Sullivan SM, Carlson GM, Holyoak T. Differential inhibition of cytosolic PEPCK by substrate analogues. Kinetic and structural characterization of inhibitor recognition. Biochemistry. 2008;47(7):2099–2109. doi:10.1021/bi7020662
19. Patridge E, Gareiss P, Kinch MS, Hoyer D. An analysis of FDA-approved drugs: natural products and their derivatives. Drug Discov Today. 2016. doi:10.1016/j.drudis.2015.01.009
20. Koch MA, Schuffenhauer A, Scheck M, et al. Charting biologically relevant chemical space: a structural classification of natural products (SCONP). Proc Natl Acad Sci. 2005. doi:10.1073/pnas.0503647102
21. Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat Chem. 2016;8(6):531–541. doi:10.1038/nchem.2479
22. Rawlins MD. Cutting the cost of drug development? Nat Rev Drug Discov. 2004;3(4):360–364. doi:10.1038/nrd1347
23. Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nat Rev Drug Discov. 2005;4(8):649–663. doi:10.1038/nrd1799
24. Aung T, Qu Z, Kortschak R, Adelson D. Understanding the effectiveness of natural compound mixtures in cancer through their molecular mode of action. Int J Mol Sci. 2017;18(3):656. doi:10.3390/ijms18030656
25. Holyoak T, Sullivan SM, Nowak T. Structural insights into the mechanism of PEPCK catalysis †, ‡. Biochemistry. 2006;45(27):8254–8263. doi:10.1021/bi060269g
26. Dunten P, Belunis C, Crowther R, et al. Crystal structure of human cytosolic phosphoenolpyruvate carboxykinase reveals a new GTP-binding site. J Mol Biol. 2002;316(2):257–264. doi:10.1006/jmbi.2001.5364
27. Webb B, Sali A, Comparative protein structure modeling using MODELLER. In: Current Protocols in Bioinformatics. Vol. 2016. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2016. 5.6.1-5.6.37. doi:10.1002/cpbi.3
28. Shen M-Y, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15(11):2507–2524. doi:10.1110/ps.062416606
29. Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356(6364):83–85. doi:10.1038/356083a0
30. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi:10.1002/pro.5560020916
31. Chen VB, Arendall WB, Headd JJ, et al. MolProbity : all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr. 2010;66(1):12–21. doi:10.1107/S0907444909042073
32. Koes DR, Camacho CJ. Pharmer: efficient and exact pharmacophore search. J Chem Inf Model. 2011;51(6):1307–1314. doi:10.1021/ci200097m
33. Irwin JJ, Shoichet BK. ZINC - A free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. doi:10.1021/ci049714+
34. Koes DR, Camacho CJ. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. 2012;40(W1):W409–W414. doi:10.1093/nar/gks378
35. Bas DC, Rogers DM, Jensen JH. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins Struct Funct Genet. 2008;73(3):765–783. doi:10.1002/prot.22102
36. Sullivan SM, Holyoak T. Structures of rat cytosolic PEPCK: insight into the mechanism of phosphorylation and decarboxylation of oxaloacetic acid †, ‡. Biochemistry. 2007;46(35):10078–10088. doi:10.1021/bi701038x
37. Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016;44(D1):D1202–D1213. doi:10.1093/nar/gkv951
38. Cereto-Massagué A, Guasch L, Valls C, Mulero M, Pujadas G, Garcia-Vallvé S. DecoyFinder: an easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics. 2012;28(12):1661–1662. doi:10.1093/bioinformatics/bts249
39. Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of Useful Decoys, Enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. doi:10.1021/jm300687e
40. Wetzel S, Klein K, Renner S, et al. Interactive exploration of chemical space with scaffold hunter. Nat Chem Biol. 2009;5(8):581–583. doi:10.1038/nchembio.187
41. Djoumbou Feunang Y, Eisner R, Knox C, et al. ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J Cheminform. 2016;8(1):1–20. doi:10.1186/s13321-016-0174-y
42. Lagorce D, Sperandio O, Baell JB, Miteva MA, Villoutreix BO. FAF-drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015;43(W1):W200–W207. doi:10.1093/nar/gkv353
43. Hiller K, Grote A, Scheer M, Münch R, Jahn D. PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004. doi:10.1093/nar/gkh378
44. Matte A, Goldie H, Sweet RM, Delbaere LTJ. Crystal structure of Escherichia coli phosphoenolpyruvate carboxykinase: a new structural family with the P-loop nucleoside triphosphate hydrolase fold. J Mol Biol. 1996;256(1):126–143. doi:10.1006/jmbi.1996.0072
45. Hsin K-Y, Matsuoka Y, Asai Y, et al. systemsDock: a web server for network pharmacology-based prediction and analysis. Nucleic Acids Res. 2016;44(W1):W507–W513. doi:10.1093/nar/gkw335
46. Abolhassani M, Guais A, Sanders E, et al. Screening of well-established drugs targeting cancer metabolism: reproducibility of the efficacy of a highly effective drug combination in mice. Invest New Drugs. 2012;30(4):1331–1342. doi:10.1007/s10637-011-9692-7
47. Jena BS, Jayaprakasha GK, Singh RP, Sakariah KK. Chemistry and biochemistry of (−)-hydroxycitric acid from garcinia. J Agric Food Chem. 2002;50(1):10–22. doi:10.1021/jf010753k
48. Lindqvist Y, Schneider G, Vihko P. Three-dimensional structure of rat acid phosphatase in complex with L(+)-tartrate. J Biol Chem. 1993;268(28):20744–20746. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/8407898
49. LaCount MW, Handy G, Lebioda L. Structural origins of l(+)-tartrate inhibition of human prostatic acid phosphatase. J Biol Chem. 1998;273(46):30406–30409. doi:10.1074/jbc.273.46.30406
50. Gutman EB, Sproul EE, Gutman AB. Significance of increased phosphatase activity of bone at the site of osteoplastic metastases secondary to carcinoma of the prostate gland. Am J Cancer. 1936;28(3):485–495. doi:10.1158/ajc.1936.485a
51. Son B, Jun SY, Seo H, et al. Inhibitory effect of traditional oriental medicine-derived monoamine oxidase B inhibitor on radioresistance of non-small cell lung cancer. Sci Rep. 2016;6(1):21986. doi:10.1038/srep21986
52. Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002. doi:10.1021/jm020017n
53. Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000. doi:10.1021/jm000292e
54. Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. doi:10.1016/j.addr.2012.09.019
55. Tan W, Wang H, Chen Y, et al. Molecular aptamers for drug delivery. Trends Biotechnol. 2011;29(12):634–640. doi:10.1016/j.tibtech.2011.06.009
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.