")
Back to Journals » Journal of Inflammation Research » Volume 14
Comprehensive Mechanism, Novel Markers and Multidisciplinary Treatment of Severe Acute Pancreatitis-Associated Cardiac Injury – A Narrative Review
Authors Luo Y , Li Z, Ge P, Guo H, Li L, Zhang G, Xu C, Chen H
Received 23 March 2021
Accepted for publication 15 June 2021
Published 12 July 2021 Volume 2021:14 Pages 3145—3169
DOI https://doi.org/10.2147/JIR.S310990
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
YaLan Luo, 1– 3,* ZhaoXia Li, 2,* Peng Ge, 1– 3,* HaoYa Guo, 1– 3 Lei Li, 4 GuiXin Zhang, 2 CaiMing Xu, 2 HaiLong Chen 2
1Institute (College) of Integrative Medicine, Dalian Medical University, Dalian, Liaoning, People’s Republic of China; 2Department of General Surgery, The First Affiliated Hospital of Dalian Medical University, Dalian, Liaoning, People’s Republic of China; 3Laboratory of Integrative Medicine, The First Affiliated Hospital of Dalian Medical University, Dalian, Liaoning, People’s Republic of China; 4Department of Vascular Surgery, The Second Affiliated Hospital of Dalian Medical University, Dalian, Liaoning, People’s Republic of China
*These authors contributed equally to this work
Correspondence: CaiMing Xu; HaiLong Chen
Department of General Surgery, The First Affiliated Hospital of Dalian Medical University, Zhongshan Road 222, Dalian, 116011, Liaoning, People’s Republic of China
Tel +86-411-83635963
Fax +86-411-83622844
Email [email protected]; [email protected]
Abstract: Acute pancreatitis (AP) is one of the common acute abdominal inflammatory diseases in clinic with acute onset and rapid progress. About 20% of the patients will eventually develop into severe acute pancreatitis (SAP) characterized by a large number of inflammatory cells infiltration, gland flocculus flaky necrosis and hemorrhage, finally inducing systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS). Pancreatic enzyme activation, intestinal endotoxemia (IETM), cytokine activation, microcirculation disturbance, autonomic nerve dysfunction and autophagy dysregulation all play an essential role in the occurrence and progression of SAP. Organ dysfunction is the main cause of early death in SAP. Acute kidney injury (AKI) and acute lung injury (ALI) are common, while cardiac injury (CI) is not, but the case fatality risk is high. Many basic studies have observed obvious ultrastructure change of heart in SAP, including myocardial edema, cardiac hypertrophy, myocardial interstitial collagen deposition. Moreover, in clinical practice, patients with SAP often presented various abnormal electrocardiogram (ECG) and cardiac function. Cases complicated with acute myocardial infarction and pericardial tamponade have also been reported and even result in stress cardiomyopathy. Due to the molecular mechanisms underlying SAP-associated cardiac injury (SACI) remain poorly understood, and there is no complete, unified treatment and sovereign remedy at present, this article reviews reports referring to the pathogenesis, potential markers and treatment methods of SACI in recent years, in order to improve the understanding of cardiac injury in severe pancreatitis.
Keywords: severe acute pancreatitis, cardiac injury, cardiac dysfunction, inflammatory mediators, biomarkers, treatment
Corrigendum for this paper has been published
Introduction
AP is a rapid onset of abdominal inflammation, which is the most common gastrointestinal cause of hospitalization or death in China and many other countries. Most patients with mild AP recover completely after conservative treatment. However, about 20% of patients may advance to SAP,1 with rapid and persistent progression to SIRS and MODS, poor prognosis and mortality as high as 30% (Figure 1).2 The heart is one of the target organs vulnerable to pancreatic inflammation.3 Studies have shown that AP, regardless of its severity, is accompanied by cardiac injury, and the degree of injury is correlated with the clinical type of AP. Mild acute pancreatitis (MAP) is accompanied by slight myocardial injury, which is transient and can recover by itself. SAP is usually complicated with severe myocardial injury and dysfunction. At present, there is no specific or uniform definition of acute pancreatitis-associated cardiac injury. In 1971, Lovett4 first reported that acute pancreatitis could lead to myocardial dysfunction, characterized by impaired myocardial contractile function, decreased ejection fraction, poor contraction response to volume load, decreased peak systolic pressure/end-systolic volume ratio and an enlarged heart. Later, an increasing number of studies have shown that patients with pancreatitis will suffer from cardiac injury. For instance, in a prospective clinical research that recruited 52 consecutive patients with AP, electrocardiographic and echocardiographic changes were observed in more than 50% of patients with AP, and prolonged QTc interval, pericardial effusion and diastolic dysfunction were associated with higher mortality.5 Furthermore, in a study investigating the incidence of organ failure in SAP, it was found that 61.7% of the patients suffer multiple organ failure (MOF), and 26.6% had single organ failure, including the respiratory system (35.1%), the cardiovascular system (22.3%), the gastrointestinal tract (19.1%), the liver (15.9%), the kidney (14.9%), the neurologic and hematological system (8.5%).6 Likewise, in another related study, the incidence of myocardial infarction (MI) induced by SAP was as high as 60.5%.7 Other synchronized manifestations of myocardial dysfunction, such as altered cardiac input/output ratio, troponin imbalance, arrhythmia, cardiogenic shock, myocarditis and other types of MIs have also been reported in patients with SAP.8–10 In addition, Saulea et al11 observed obvious pathological ultrastructure damage such as microcirculation vessels destruction, interstitial edema, matrix collagenization, excessive contraction of myofibrils and cardiomyocytes hypoxia, edema and hypertrophy in the heart of experimental AP rats (Table 1).
 |
Table 1 Characteristics of Severe Acute Pancreatitis-Associated Cardiac Injury |
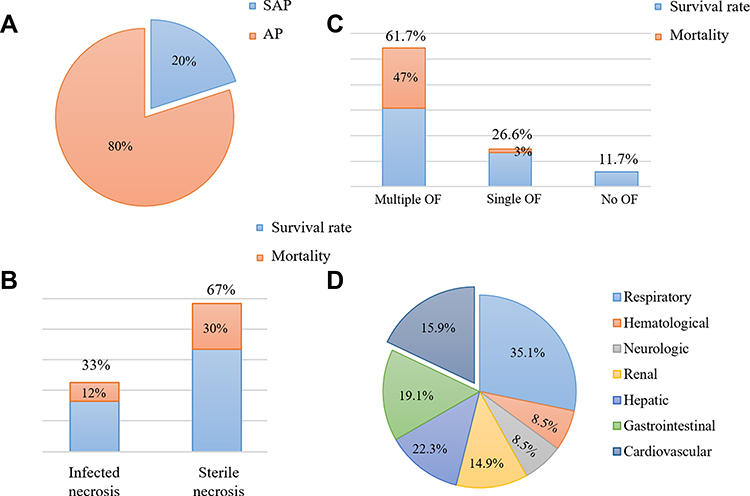 |
Figure 1 Data related to acute pancreatitis. (A and B): proportion and mortality in acute pancreatitis of different severity. (C and D): proportion and mortality of acute pancreatitis complicated with organ failure. |
Worldwide, despite advances in medical treatment, imaging and interventional technology, the incidence of AP continues to increase. The incidence of AP in the United States has risen to approximately 45 per 100,000 people, while that in Britain has increased from 6.9 to 75 per 100,000 men and from 11.2 to 48 per 100,000 women.12 Cardiac injury in AP is challenging to treat, which greatly increases the death risk of AP. A comprehensive and systematic understanding of the occurrence and development of SAP-associated cardiac injury, as well as early and rapid diagnosis, are helpful for effective targeted treatment to reduce mortality. Thus, we summarize the main mechanism of myocardial dysfunction in SAP, evaluate the potential diagnostic markers, and briefly outline the current treatment strategies and potential future methods, to provide guidance of great significance for clinicians to conduct treatment work and scientific research.
The pathogenesis of SACI is hugely complex, and it is a process of inflammatory injury induced by multi-factors, multi-links and multi-organs. We summarized the pathological mechanism of SACI into the following three main links (Figure 2): 1) The local inflammation of the pancreas caused by the acinar cell injury and trypsin release in the early stage of AP give rise to the release of many inflammation-related mediators through cascade effects, leading to SIRS, laying the groundwork for subsequent organ failure (including cardiac injury). 2) The intestinal barrier is damaged, and endotoxin enters the blood, which causes infectious pancreatic necrosis and aggravates the injury of extra-pancreatic organs (including the heart) 3) The mechanism of cardiac vascular and nerve injury in SAP.
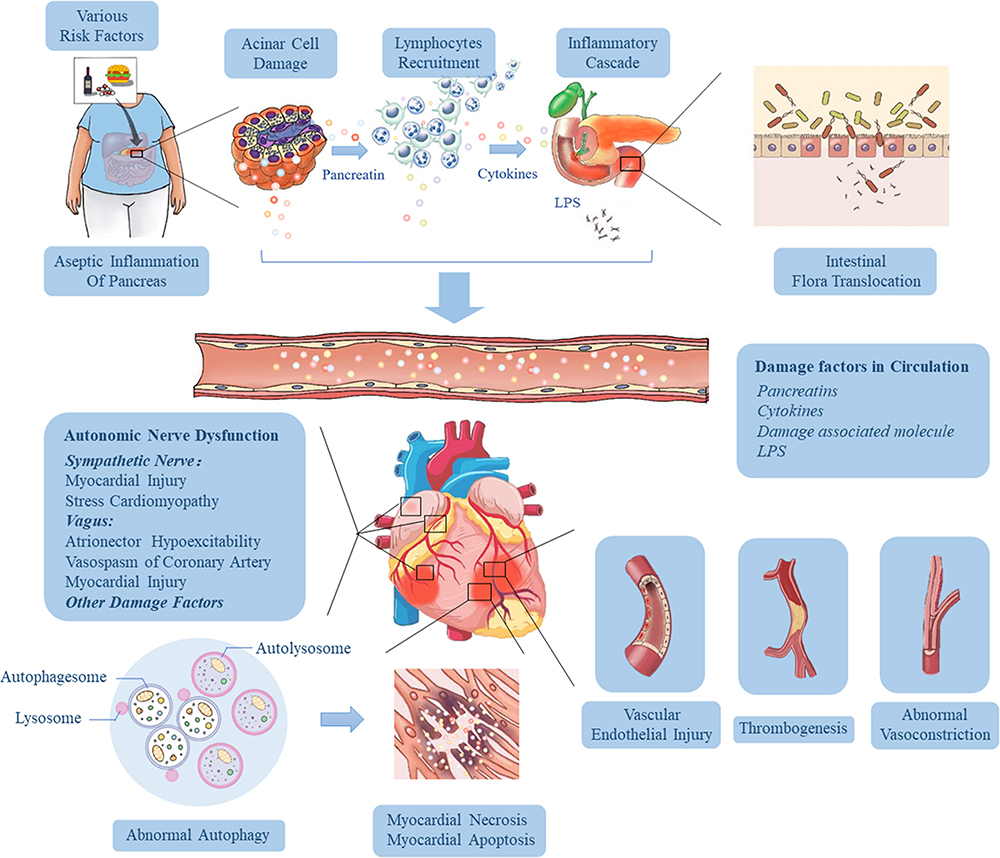 |
Figure 2 Pathophysiological mechanisms influencing the development of cardiac injury in SAP. Various risk factors (gallstones, alcohol, diet and drugs) cause acinar cell damage and the release of pancreatic hydrolase, leading to excessive activation and autocrine of macrophages and neutrophils, resulting in the accumulation of a large number of pro-inflammatory factors. Then the local inflammation at the lesion is amplified through the inflammatory cascade effect, which eventually results in necrosis and hemorrhage of most pancreatic tissue, releasing more and more cytokines, and induces hypercytokinemia (a cytokine storm). As the disease progresses and pancreatic inflammation involves the intestine, it causes dysfunction of intestinal barrier, which leads to the migration of intestinal flora to the pancreas and blood, followed by pancreatic infection and sepsis. These high levels of risk factors (including trypsin, endotoxin and cytokines) in the blood can damage vascular endothelial cells, trigger systemic inflammatory response, lead to myocardial microcirculatory disturbance, autonomic nerve dysfunction and abnormal autophagy, and eventually result in myocardial injury and cardiac dysfunction. |
Systemic Inflammatory Response in SAP
Studies at home and abroad have shown that distant organ failure caused by SAP is closely related to SIRS, in which the homeostasis imbalance caused by excessive production of trypsin, cytokines (TNF-α, IL-1β, IL-18 and IL-6) and inflammatory transmitters (NO, ROS and HMGB1) is the main factor of myocardial injury in AP. These inflammation-related factors regulate and induce each other and jointly cause abnormal electrical activity of cardiomyocytes, mitochondrial damage, energy metabolism disorder, systolic myocardial dysfunction, myocardial hypertrophy, fibrosis and apoptosis by activating complex signal pathway networks (Figure 3).
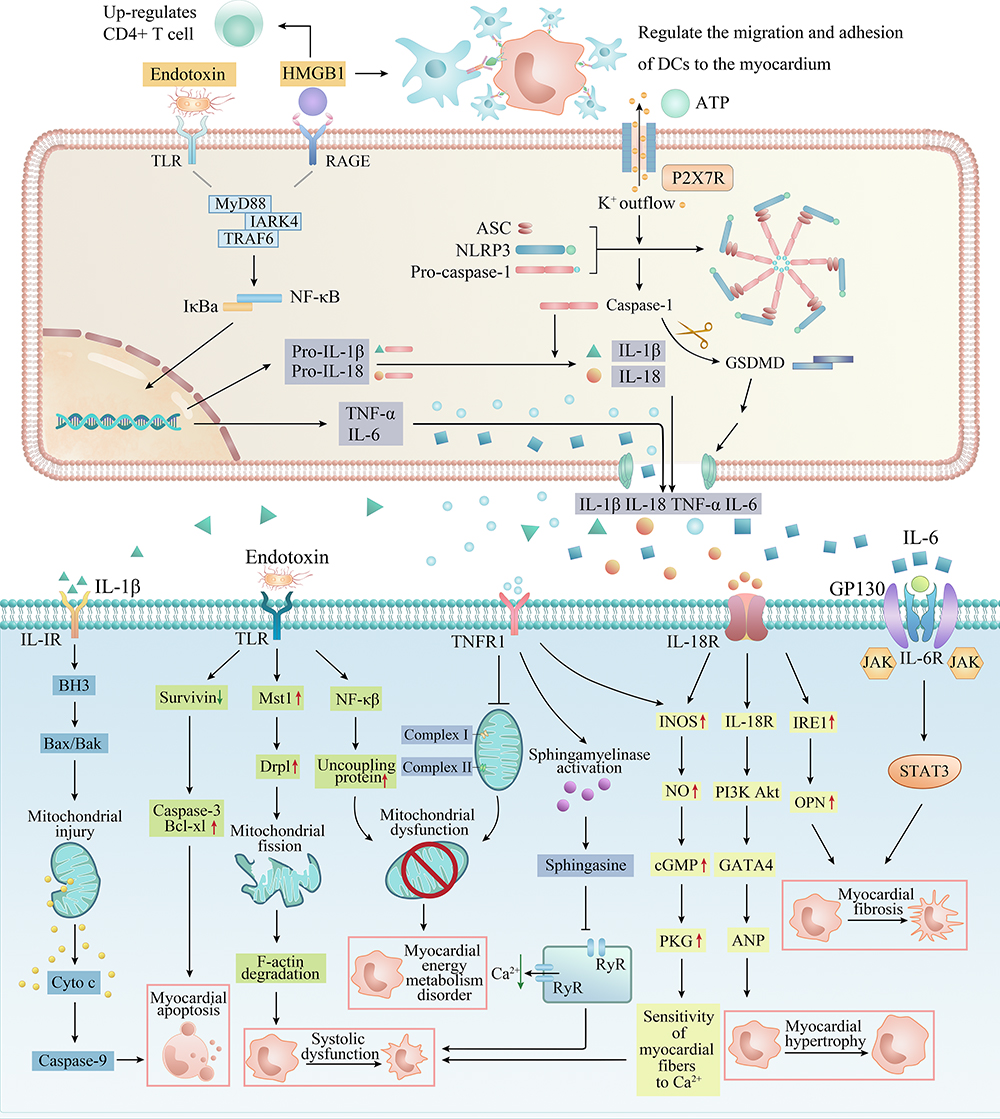 |
Figure 3 The specific pathways of myocardial injury and cardiac dysfunction caused by inflammation-related factors. DAMPs and PAMPs such as HMGB1, ATP and endotoxin from the pancreas and intestine act on membrane receptors such as TLR and P2X7R to recruit inflammatory cells (macrophages, neutrophils and dendritic cells) in serum, activate classical inflammatory pathways such as NF-κB and NLRP3 inflammasome in inflammatory cells, release a large number of pro-inflammatory cytokines, and form a cascade reaction (upper). These inflammatory factors eventually act on cardiomyocytes, causing myocardial energy metabolism disorder, systolic myocardial dysfunction, myocardial hypertrophy, apoptosis and fibrosis through a complex network of signaling pathways (lower). |
Pancreatin
Pancreatin is one of the earliest mediators considered to trigger SIRS by SAP. As early as the 1950s, some scholars put forward that pancreatin could move along the tissue plane to bring about pseudocyst and tissue damage at distal organs. Such movement of pancreatin may cause direct myocardial injury,13 resulting in the increasing of cardiomyocyte permeability, secondary electrical interference and cell necrosis to destroy the structure and function of the normal myocardium. Dan et al14 measured trypsin levels in serum, jejunum, lung and heart of septic model rats, rats with a protease inhibitor (ulinastatin) and control rats. They found that trypsin levels in serum, jejunum, lung and heart of model rats were significantly higher than those in the control group and decreased significantly after administration of ulinastatin. The level of serum trypsin was negatively correlated with the expression of two intestinal barrier function-associated proteins (mucin-2 and E-cadherin) but positively correlated with inflammation indices such as TNF α, IL-6 and neutrophil elastase. These data suggest that trypsin is closely related to intestinal barrier dysfunction, lung injury and heart injury during systemic inflammation. In addition, trypsin has been reported to alter platelet adhesion and affect the coagulation system, leading to coronary thrombosis and myocardial ischemia.13,15 Masuda et al16 also found that utilizing urinary trypsin inhibitor (UTI) may contribute to the recovery of cardiac function after ischemia/reperfusion (I/R) by reducing the severity of mitochondrial dysfunction and maintaining energy production during a state of shock. Similarly, Mauro et al17 proved that plasma-derived α-1 anti-trypsin offers cardioprotective effects by inhibiting inflammasome activation. Besides trypsin, pancreatic lipase may also be a factor that cannot be ignored in SAP-associated cardiac injury. The presence of mature adipocytes in the myocardium is a sufficient substrate for pancreatic lipase-induced steatonecrosis. Roncati et al18 observed steatonecrosis in areas of lipomatous infiltration of the heart following acute exacerbation of latent chronic pancreatitis, but the specific injury mechanism still needs much research to support.
TNF-α
Relevant research found that TNF-α increased obviously in serum of patients with AP, reaching the peak on the first day of MAP while on the fourth day in patients with SAP.19 Therefore, dynamic monitoring of TNF-α is supposed to evaluate the severity of AP. Many experimental studies have revealed that TNF-α has a negative inotropic effect on the heart in vivo,20 and this decreased contractility may be related to abnormal energy metabolism of the myocardium. Hofmann et al21 found that TNF-α not only showed immediate negative inotropic effects but also increased oxygen demand in the myocardium of human right atrial. They suggest that TNF-α impairs myocardial contraction by affecting the chemico-mechanical energy transduction of cardiomyocytes. On the one hand, TNF-α could immediately suppress the chemico-mechanical energy transduction through the sphingosine-mediated pathway.22 On the other hand, Gellerich and Trumbeckaite23 also showed that TNF-α could cause mitochondrial dysfunction and energy metabolic disorder in cardiomyocytes by inhibiting complex I and complex II activity in the mitochondrial intima. Jude et al24 further verified that TNF-α has a negative inotropic effect on isolated rat hearts and can induce PKC activation, resulting in impaired cardiac contractility. More importantly, some studies19 have shown that TNF-α could lead to abnormal myocardial electrical activity through NO-dependent pathway.
Interleukin (IL) Family
IL-1β belongs to IL-1 family (IL-1F), mainly produced by activated monocytes-macrophages and endothelial cells, and is involved in many systemic and acute injury reactions. Serum IL-1β levels in patients with AP were significantly increased and continued to elevate with the aggravation of AP.25 The high level of IL-1β in serum of patients with AP in the later stage may be the main cause of various cardiac injuries. Studies of Chamberlain26 and Kirii27 have demonstrated the atherogenic property of IL-1β, leading to ischemic myocardial injury. Lack of IL-1β can reduce the severity of atherosclerosis in model mice, possibly achieved by decreasing the expression of vascular cell adhesion molecule 1 (VCAM-1) and monocyte chemoattractant protein 1 (MCP-1) in the aorta.28 Khoury et al29 have shown that IL-1β can also down-regulate L-type calcium channels on myocardial cell membranes, which is a determinant of action potential duration and is related to the excitation-contraction coupling of cardiac myocytes. Therefore, IL-1β can influence systolic cardiac function through calcium channels. Another study indicated that IL-1β activates caspase-3 to promote cardiomyocyte apoptosis by releasing cytochrome C (Cyt C) into the cytoplasm.30 In addition, a growing number of studies have observed a significant up-regulation of IL-1β and IL-18 in infarcted myocardium.31 This phenomenon is probably due to the release of endogenous danger signals (such as ATP) derived from injured and necrotic cardiomyocytes, which activates a strong inflammatory reaction that exacerbates and prolongs cardiac damage while removing necrotic cells and matrix debris. This intense inflammatory reaction is mediated by a protein complex called NLRP3 inflammasome.32 Nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome is composed of NLRP3 protein, apoptosis-associated speck-like protein containing CARD (ASC) and pro-caspase-1. When NLRP3 inflammasome is activated in response to extracellular signals, pro-caspase-1 can be cleaved and activated. Activated caspase-1 can induce cell pyroptosis and release inflammatory factors by cleaving GSDMD protein, pro-IL-1β and pro-IL-18.33
The animal study by Toldo et al34 showed that IL-1β caused a remarkable increase in IL-18, and the myocardial contractile dysfunction induced by IL-1β was attenuated after applicating IL-18 receptor blockers, suggesting that the effect of IL-1β on myocardial contractile function may be realized by up-regulating the level of IL-18. IL-18 is also a member of IL-1F, which mainly comes from monocyte-macrophages. According to the research of Janiak et al,35 the level of IL-18 in serum of patients with AP on the 1st, 3rd and 5th day after admission was significantly higher than that in the healthy control group, and the concentration of IL-18 was positively correlated with C-reaction protein (CRP). IL-18 is therefore considered to be an early indicator of the severity of AP. Mallat et al36 observed an increase in plasma IL-18 concentrations of patients with acute coronary syndromes related to the severity of myocardial dysfunction. The pathological role of IL-18 in unstable coronary artery disease and ischemic myocardial dysfunction mainly refers to the following reasons: (1) promotes atherosclerotic plaque instability and systemic inflammatory responses by activating monocytes/macrophages, lymphocytes and endothelial cells. (2) IL-18 up-regulates expression of endothelial cell adhesion molecules and generation of pro-inflammatory mediators such as IL-1β, IL-8, TNF-α and inducible nitric oxide synthase (iNOS), enlarging the scope of myocardial inflammatory injury.37 (3) enhances the exogenous apoptosis pathway by inducing the expression of death receptor Fas on cardiomyocyte membrane. Yoshida et al38 further verified that IL-18 could lead to myocardial necrosis and myocardial vascular endothelial injury by enhancing the effect of TNF-α. Furthermore, the activated IL-18 combines with its receptors (α and β subunits) to form a ligand-receptor complex which could activate the PI3K-Akt-GATA4 pathway, which leads to hypertrophy and increases osteopontin (OPN), which leads to fibrosis,39 playing a crucial role in triggering myocardial inflammation and remodeling.
Sternby et al40 analyzed 232 patients with AP, showing that the serum IL-6 concentration of SAP patients was significantly higher than that of MAP patients. Another study indicated that plasma IL-6 is implicated in the incidence of cardiovascular events in AP patients complicated with peritonitis treated by peritoneal dialysis,41 which further confirms that IL-6 may play a certain role in AP-associated cardiac injury. The bulk of the evidence suggested that IL-6 is continuously up-regulated in various experimental models of cardiac injury and heart failure.42–44 IL-6 binds to the IL-6R on the surface of cardiomyocytes and subsequently associates with gp130, which dimerizing and activating Janus kinases and triggering STAT3 phosphorylation. Continuous gp130/STAT3 signaling enhanced the inflammation and myocardial hypertrophy of remodeling infarcted hearts, characterized by increased myocardial stiffness and decreased contractile force.45 Wang et al46 uncovered that IL-6 might induce cardiac fibroblast proliferation, differentiation and fibrosis by activating MMP2/MMP3 signal mediated by TGF-1. Also, IL-6 has a vital role in myocardial hypertrophy, fibrosis and apoptosis induced by angiotensin II (AngII).47
IL-10 is a natural anti-cytokine and the most important known anti-inflammatory cytokine, which can inhibit the production of TNF-α, IL-1, IL-8, IL-12 and other cytokines, and the release of oxygen-free radicals.48 Zhang49 found that compared with normal control rats, the pro-inflammatory factor TNF-α increased significantly, and the anti-inflammatory factor IL-10 decreased significantly in rats with SAP. After heme treatment, the oxidative stress and TNF-α levels in plasma and tissues were significantly reduced, while IL-10 was significantly increased, suggesting that induction of heme oxygenase −1 (HO-1) in the early stage of SAP can regulate systemic inflammatory response by inhibiting TNF-α and increasing IL-10, and prevent pancreatic and distant organs from injury. Van Laethem JL and MarchantA50 also verified the potential protective effect of IL-10 in rats with acute necrotizing pancreatitis (ANP). They found that recombinant IL-10 can reduce the severity of experimental acute pancreatitis by reducing acinar cell necrosis and TNF-α gene expression, suggesting potential treatment strategies. TGF-β1 has both pro-inflammatory and anti-inflammatory effects and exerts immune enhancement activity in some tissues while showing immune suppression in systemic circulation.51 Kulkarni et al52 have demonstrated that TGF-β1 plays an important role in homeostatic regulation of immune cell proliferation and extravasation into tissues. They observed excessive inflammatory response in TGF-β1 null mice, and a large number of lymphocytes and macrophages infiltrated many organs, especially in the heart and lungs.
NO and NOS
TNF-α and IL-1β exert enormous function on SAP-associated cardiac injury, while nitric oxide (NO) and nitric oxide synthase (NOS) are secondary agents of cardiac inhibition in SAP.53,54 Andican et al55 found NO increased in both pancreas and circulation in AP, and NO contributed to the pathogenesis of AP under oxidative stress. Extensive experimental studies have revealed the specific mechanism of NO-induced myocardial injury. According to the study of Ichinose & Buys,56 NO could induce the generation of a large number of cytotoxic substances (eg, peroxynitrite) in vivo, leading to myocardial dysfunction. Besides, DavidL57 discovered that high NO could also promote the release of inflammatory factors and the accumulation of intracellular Na+/Ca2+ in cardiomyocytes through cyclic guanosine monophosphate (cGMP) signaling pathway, thus inhibiting myocardial contraction/relaxation function. It is worth noting that experimental studies on NO’ role in cardiac injury also produced conflicting findings. NO can also restrict cardiac remodeling after an infarction, thus promoting cardiac protection under post-ischemic conditions. This dual effect of NO on the heart may be due to the dose-dependent effect and the regulatory effect of different NOS subtypes of NO. A low concentration of NO and a small amount of cGMP inhibited phosphodiesterase III to prevent the hydrolysis of cAMP.58 Subsequently, the activation of protein kinase A (PKA) followed by accumulated cAMP leads to the opening of Ca2+ channel in the sarcoplasmic reticulum and depolarization of myocyte membrane potential, thereby enhancing myocardial contractility. However, high NO concentration caused a corresponding increase in cGMP, activating protein kinase G (PKG) and leading to cardiac depression by blocking calcium channels in the myocardium.59 NO is mainly produced by three kinds of NOS, including inducible NOS (iNOS), neuronal NOS (nNOS) and endothelial NOS (eNOS).60 iNOS is generally believed to take part in myocardial injury under various pathological conditions such as ischemia-reperfusion injury, sepsis, aging, myocardial infarction and heart failure through cGMP-dependent and/or independent pathways. nNOS is constitutively expressed in cardiomyocytes,61 which is involved in modulating β adrenergic receptor pathway.62 Myocardial nNOS expression and activity increased in failing hearts, raising the possibility of left ventricular (LV) remodeling progression and functional deterioration after myocardial infarction (MI).63 eNOS plays a potential role in regulating cardiomyocytes homeostasis, especially in the early stage of myocardial suppression. eNOS in the myocardium produces NO, which modifies calcium channel to inhibit calcium entry and induce myofibril relaxation, which has great effects on preventing myocardial dysfunction induced by sepsis.64 Likewise, alprostadil relieved myocardial ischemia/reperfusion injury by promoting antioxidant activity and eNOS activation in rats.65 All above research supported the notion that NO have different regulatory and dose-dependent effects, and there is a physiological balance among NO, superoxide and peroxynitrite in subcellular compartments. Possibly, a holistic understanding of the biological complexity and properties of NO and its derived active nitrogen species could provide a more specific and effective treatment targets for myocardial injury.
ROS and Mitochondria Dysfunction
Overproduction of reactive oxygen species (ROS) is also one of the critical causes of cardiac injury in SAP. Braganza et al66 found that oxidative stress has pervaded the vascular compartment by the time of admission in patients with acute pancreatitis by analyzing admission blood samples. Wen et al67 found that the protein level and protein activity of Nox2 and Nox4 in myocardial tissue of SAP rats were significantly higher than sham-operated rats, suggesting that Nox2 and Nox4 were abnormally activated in myocardial injury induced by SAP. This elevation of these enzymes directly mediates ROS production, increases oxidative stress, promotes apoptosis and finally leads to myocardial injury. 1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine (tempol), a membrane-permeable radical scavenger, could significantly reduce the intensity of inflammation and oxidative stress and improve the morphological damage and exhibits anti-inflammatory and cardioprotective effects in the cerulein-induced pancreatitis rats.68 Excessive oxidation of lipid by ROS leads to serious membrane damage of cardiomyocytes. The main mechanism is that ROS attacks the unsaturated double bonds of polyunsaturated fatty acids in phospholipid of cell membrane, arouses the chain and proliferation reaction of free radicals to form a series of lipid-free radicals and the degradation products malondialdehyde, which will further reduce the fluidity and increase the permeability of cardiomyocytes membrane.69 Meanwhile, ROS-mediated lipooxidative damage is also one of the crucial mechanisms causing Ca2+ channel dysfunction. ROS can selectively act on L-type calcium channels, preventing the flow of extracellular Ca2+ into cells.70 The IP3 receptor, Ryanodine receptor and Na+-Ca2+ exchanger in endoplasmic reticulum, proteins associated with Ca2+ channel, are also regulated by redox.71–73 Studies have shown that the calcium sensitivity of myocardial fibers decreases during sepsis, which may be related to the activation of ROS-related apoptosis signal-regulating kinase-1 (ASK-1) and troponin T phosphorylation.74,75 In addition, ROS initiates the transcription of many inflammatory factors by participating in the regulation of NF-κB signaling pathway, which aggravates the local inflammatory reaction of myocardium.76–78
Myocardial mitochondrial membranes contain more unsaturated fatty acids than other plasma membranes and are the most sensitive to lipid peroxidation.79,80 When the isolated heart tissue was exposed to the solution that produces ROS, it was observed that mitochondria seriously swelled and even disintegrated. Mitochondria dysfunction leads to impaired ATP synthesis, and this energy disorder of cardiomyocytes induces decreased contractile function.81 Furthermore, mitochondrial dysfunction resulted in long-term accumulation of oxygen-derived-free radicals, which eventually caused catastrophic destruction and functional loss of mitochondrial DNA (mtDNA), and subsequently induce cell hypertrophy, apoptosis and interstitial fibrosis by activating matrix metalloproteinases.82 Yao et al has found that a specific antioxidant targeting mitochondrion, mitochondria-targeted vitamin E (Mito-Vit-E), could significantly maintain the integrity of mtDNA, reduced the damage of mtDNA, inhibited the leakage of mtDNA into the cytoplasm, and suppressed the up-regulation of TLR9 pathway factors, MyD88 and RAGE, thus alleviating cardiac inflammation, which is a potential cardioprotective agent.83 Moreover, in myocardial injury and dysfunction, regardless of the underlying etiology, the mitochondrium will undergo significant structural and functional changes, including the opening of mitochondrial membrane permeability transition hole,84,85 the loss of membrane potential,86 the imbalance of Ca2+ homeostasis, the generation of ATP and oxygen-derived-free radicals and the release of various pro-apoptotic proteins. All the associated pro-inflammatory mediators, including mitochondrial reactive oxygen species (mtROS), mtDNA fragments, ATP and cytochrome C, enter the circulatory system to mediate inflammatory cascade reaction through different signal pathways,87 resulting in myocardial cell injury even apoptosis88 and necrosis.89
HMGB1
High mobility group box-1 (HMGB1) is a highly conserved non-histone protein that widely exists in the nuclei of eukaryotic cells.90 As a pro-inflammatory factor, HMGB1 has been proved to play an important role in various diseases.91,92 Under certain conditions, HMGB1 is released outside the cell to recruit and initiate innate immune cell through TLR2, TLR4 and RAGE,93 and induce TNF-α, IL-1β, IL-6 and other pro-inflammatory factors to promote inflammatory cell aggregation, thus amplifying local inflammation.94 In a research investigating the spatial and temporal differential expression of HMGB1 and inflammatory cytokines (TNF-α, IL-1 and IL-6) in experimental SAP rats, it was found that the expression of HMGB1, as a late inflammatory factor, was later than the early inflammatory factors IL-1, TNF-α and IL-6. Serum level of IL-1, TNF-α and IL-6 increased rapidly at 3 h after model induction, peaked at 6 h, then decreased at 12 h, and was lower at 24 h than those at 12 h. However, the level of HMGB1 in pancreatic tissue did not change significantly at 3 h and 6 h, but increased remarkably at 12 h and maintained up to 24 h, indicating that HMGB1 may play a key role in maintaining the development of SAP.95 Many studies have demonstrated a close relationship between HMGB1 and cardiac injury in the late stage of AP. The experimental results of Xue et al96 indicate that HMGB1/TLR4 signaling pathway can induce the maturation and activation of dendritic cells (DCs), regulate the migration, adhesion and aggregation of DCs to the myocardium, stimulate the expression of surface costimulatory molecules, thus participating in myocardial ischemia/reperfusion (I/R) injury. The utilization of HMGB1 specific neutralizing antibody significantly inhibited these responses. Wang et al97 concluded through gene knockout experiment that HMGB1 changes the spleen’s immune microenvironment through TLR4, up-regulates CD4+ T cell ratio, CD4+/CD8+ T cell ratio and Th17 cells in the spleen, and induces myocardial inflammatory injury. Ren et al98 proposed that miR-29a-3p transferred by mesenchymal stem cells-derived extracellular vesicles plays a cardioprotective role in myocardial injury induced by SAP via down-regulating HMGB1/TLR4/Akt axis. Tzeng et al99 also discussed the effects of HMGB1 on cardiac myocytes. They found that HMGB1 had a direct negative inotropic effect on isolated cardiomyocytes, which showed that after HMGB1 (100ng/mL) treatment, the sarcomere shortening of contractile cardiomyocytes decreased by 70%, and the height of the peak Ca2+ decreased by 50% within 5 minutes (P < 0.01). The negative effects of HMGB1 on cell contractility and calcium homeostasis were partially reversible. In addition, HMGB1 induced PKC-epsilon translocation, which leads to significant inhibition of inward I-type calcium currents, indicating that HMGB1 impairs sarcomere shortening by regulating PKC-epsilon translocation to reduce membrane calcium influx and thus reduce calcium availability in cardiomyocytes. These studies suggest that HMGB1 may be a new myocardial inhibitory factor in the process of cardiac injury.
Other Factors
In addition to the above common inflammatory mediators, platelet-activating factor (PAF), triglyceride, and free fatty acids (FFA) also play important roles in SIRS and MOF induced by SAP. PAF is a cell surface secretion of bioactive lipid, combined with a cell surface receptor called PAF-R to produce physiological and pathological effects. Studies have shown that AP causes the release of PAF, which leads to systemic damage such as circulatory system disorders and MOF. PAF is involved in the occurrence and development of AP and is considered the key mediator of SAP, giving rise to various serious complications and high mortality. The use of platelet-activating factor receptor antagonist (PAF-RAs) can signally reduce local and systemic events after AP.100,101
Hypertriglyceridemia-associated acute pancreatitis (HTGAP) gradually increased and showed a younger, more serious trend. As early as the 1980s, it was reported that patients with HTGAP died of ischemic heart disease.102 A retrospective study also noted a significant increase in the incidence of cardiovascular events one year later in AP patients with TG level > 1000mg/dL), compared with control patients (TG < 200mg/dL).103 In addition, it has been observed that the significant increase of serum FFA in patients with acute hyperlipidemic pancreatitis is positively correlated with the severity of the disease, which is an independent risk factor for the disease to progress to SAP.104 Under aerobic conditions, long-chain FFA is the preferred metabolic substrate of the myocardium. It has been proven to be harmful both experimentally and clinically under the condition of ischemia/hypoxia, and the serum FFA level of patients with myocardial ischemia is significantly high. The possible harmful mechanisms include (1) accumulation of toxic intermediates of fatty acid metabolism, such as long-chain acyl-coenzyme, a thioester, and long-chain acylcarnitine; (2) inhibition of glucose utilization, especially glycolytic ischemia and/or reperfusion; (3) inhibition of oxidative metabolism and decoupling of electron transfer. Although the relative importance of these mechanisms is still controversial, experiments have proven that FFA metabolism inhibitors can indeed reduce the size of myocardial infarction and relieve post-ischemic cardiac dysfunction in animal models of local and systemic ischemia.105
Calcineurin is a central calcium responsive signal molecule, which can mediate the occurrence of AP. Recently, researchers have revealed its new role in AP. Wen et al106 found that calcineurin derived from hematopoiesis and neutrophils could activate immune cells to localize to the lungs and cause SIRS in AP mice. In addition, Calcineurin is also in the pancreas and promotes local inflammation of the pancreas during AP. These findings suggest a potential strategy for preventing pancreatitis and extra-pancreatic inflammation by blocking or deleting calcineurin.
Intestinal Endotoxemia in SAP
The intestinal tract is one of the earliest damaged extra-pancreatic organs by SAP. Increasing evidence has highlighted the relevance of increased intestinal permeability and consequent bacterial translocation in SAP development, which leads to intestinal endotoxemia.107 Animal studies showed that compared with negative control rats, SAP rats suffered intestinal flora disorder, intestinal mucosal barrier damage, and increased plasma endotoxin concentration.108,109 In clinical trials, J Martínez,110 M A von der Möhlen111 and Anne J van der Meer112 observed that serum endotoxin level in SAP patients was significantly higher than that in MAP patients, and its level was correlated with the peak of inflammatory factors such as TNF-α, IL-1β, IL-6 and IL-10. Excessive production of pro-inflammatory factors may be an important factor of myocardial injury caused by endotoxin during SAP. In addition, endotoxin can also directly damage cardiomyocytes. Through injecting endotoxin into male C57BL/6 wild-type (WT) mice, Shang et al113 showed that myocardial injury induced by endotoxin might be related to the degradation of F-actin. Their data indicated that Mst1 expression was up-regulated rapidly in lipopolysaccharide (LPS)-treated heart and elevated expression of Mst1 up-regulated Drp1, which initiated mitochondrial fission. Excessive mitochondrial fission leads to mitochondrial oxidative injury, mitochondrial membrane potential reduction, mitochondrial proapoptotic elements translocation into the cytoplasm/nucleus, dysfunction of mitochondrial energy function and activation of mitochondrial apoptosis. F-actin degradation acted as an apparent downstream event of mitochondrial fission activation. Therefore, Mst1 expression, mitochondrial fission modification and F-actin stabilization may be potential therapeutic targets for endotoxin-induced myocardial injury. Wang et al114 found in an endotoxin-induced animal model that the expression of uncoupling protein in myocardial tissue increased, while myocardial energy metabolism and cardiac function decreased significantly, showing a negative correlation, suggesting that endotoxin may also accentuate myocardial injury by affecting myocardial energy metabolism. The research of Kawaguchi et al115 also supports this view. They discussed the role of β3-adrenergic receptor (β3AR) in endotoxin-induced myocardial dysfunction. β3AR is closely related to energy metabolism and participates in lipolysis, glucose transport and insulin secretion. They observed cardiac dysfunction in mice within 6–12 hours of endotoxin injection, and the expression of β3AR in myocardial tissue increased obviously. After the administration of β3AR antagonist SR59230A, the cardiac function of mice was significantly improved, the expression levels of mitochondrial membrane protein complex and ATP were preserved, the expression of genes related to fatty acid oxidation and glucose metabolism increased significantly, while the expression of inducible nitric oxide synthase (iNOS) and nitric oxide content decreased significantly. These results indicate that blocking β3AR could improve impaired energy metabolism of myocardial tissue by inhibiting iNOS expression and restore cardiac function of endotoxin-induced heart failure. Nežić et al116 also found that impaired cardiomyocyte survival signaling played a crucial role in endotoxin-induced cardiac injury. They observed a significant decrease in survivin protein and a significant increase in the expressions of cleaved caspase-3 and Bcl-xL in endotoxin-induced rat myocardial tissue and further speculated that myocardial apoptosis might play a role in endotoxin-induced myocardial injury. Therefore, endotoxin can damage myocardium by enhancing the expression of inflammatory factors and leading to myocardial death via directly affecting myocardial metabolism, interfering with apoptosis and other ways.
In addition, according to a study of Shanbhag et al,117 acute pancreatitis conditioned mesenteric lymph causes cardiac dysfunction in rats independent of hypotension. They put forward a hypothesis of “gut-lymph”, pointing out that multiple organ dysfunction syndrome is due to release of toxic factors from the intestine into the mesenteric lymph. Additionally, they established a normotensive SAP rat model with lymphatic intervention or thoracic duct ligation to prove that mesenteric lymph from normotensive acute pancreatitis animals caused significant cardiac dysfunction, which could be prevented by thoracic duct ligation and external drainage of mesenteric lymph.
Cardiac Injury in SAP
Damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), including trypsin, inflammation-related factors and endotoxin, derived from the pancreas and intestine damage vascular endothelial cells, induce autonomic nervous dysfunction and abnormal autophagy, directly or indirectly cause structural damage and cardiac dysfunction (through the blood supply, nerves and cardiomyocytes themselves).
Coronary Vascular Dysfunction
Under various pathological conditions, myocardial microvascular injury, abnormal vasomotor regulation mechanism and increased blood viscosity will lead to changes in cardiac microcirculation and then affect myocardial perfusion, resulting in cardiomyocytes injury and cardiac dysfunction.
In the early stage of SAP, NF-κB was over-activated, which triggered mononuclear phagocyte and granulocyte system, and released a large number of inflammatory factors, leading to vascular endothelial injury.38 Besides, activated neutrophils release microparticles-small plasma membrane vesicles containing myeloperoxidase during SAP, which could catalyze the generation of large amounts of oxygen-derived-free radicals and damage vascular endothelial cells widely.118
Kahrau et al119 compared the pulmonary capillary blood flow, permeability, leukocyte adhesion and alveolar septum thickness in rats with mild/edematous pancreatitis (EP) and severe/necrotizing (NP) by intravital microscopic. Compared with EP, the capillary permeability of NP was significantly increased while the blood flow was significantly decreased. The results suggest that the deterioration of AP to SAP, especially organ failure, is accompanied by severe microcirculation disturbance.
Under physiological conditions, the vasomotor function is regulated by various humoral factors, among which endothelin-1 (ET-1) secreted by vascular endothelial cells has a strong vasoconstrictor effect, and coronary artery may be more susceptible than peripheral artery.120 Also, thromboxane A2 (TXA2) with vasoconstrictive effect keeps dynamic balance with prostacyclin with vasodilating effect and regulates vascular tension together. Milnerowicz121 and Foitzik122 noticed that the average concentration of plasma ET-1 in SAP patients was higher than that in MAP patients, and therapy with a new endothelin A receptor antagonist (ET-RA) significantly reduced mortality rates in SAP, improved microcirculation and stabilized capillary permeability, reduced intravascular fluid loss and extravascular fluid sequestration and improved distant organ function (including heart, lung and kidney). Kiviniemi123 and Closa124 showed that the ratio of thromboxane A2/prostacyclin in serum of SAP experimental animals was significantly higher than that of normal control animals. Taken together, we can see that in SAP, the imbalance of various vasomotor substances may lead to coronary artery spasm and a decrease of myocardial blood flow.
In SAP, under the effects of inflammatory factors (such as TNF-α), ischemia-reperfusion, and endothelin-1 mentioned above, capillary endothelium is damaged, permeability is increased, resulting in capillary leak syndrome,125 and a large amount of tissue fluid and inflammatory mucous exudates enter the third space, leading to a decrease in blood volume. Also, studies have demonstrated that thromboxane A2 (TXA2) and serum platelet-activating factor (PAF) obviously increased during SAP, and the number of platelets adhered in capillaries and venules is significantly increased, and the flow speed of platelets in capillaries is remarkably decreased.126 These changes in blood volume, vascular endothelial injury and hemodynamics contribute to microthrombus. Kellner and Robertson13 discovered the formation of coronary thrombosis in acute pancreatitis in their research which could be due to the increased levels of fibrinogen, coagulation factor VIII, D- dimer127 and platelets128 in plasma. Also possibly, following the release of trypsin into the blood, kallikrein is converted into its activated form, triggering the kinin system, resulting in the activation and supplementation of the 12 factor.129
Autonomic Nerve Dysfunction
Previous studies have confirmed that autonomic nervous dysfunction in the early stage of SAP is mainly manifested as increased sympathetic nerve activity.130 Abdominal pain is the most common symptom of pancreatitis. Because of the stimulation of pain and the decrease of blood volume, sympathetic nerve is excited. When sympathetic nerve is excited, it releases many catecholamines such as adrenaline and norepinephrine. Animal studies showed that plasma adrenergic levels were 25 times higher in rats with acute pancreatitis than those without acute pancreatitis.131 A high concentration of catecholamine could cause reversible damage to myocardial cells. Wallner et al132 have confirmed that acute catecholamine exposure could cause reversible damage to 10% of mouse cardiomyocytes. Moreover, it has been widely accepted that high cyclic catecholamines will inevitably lead to stress cardiomyopathy. In addition, the excitement of sympathetic nerve causes the activity of renin-angiotensin system to increase, and the produced Ang II plays an important role in myocardial injury and remodeling.133 Frolkis et al134 also authenticated that Ang II and TNF-α may act synergistically and promote the occurrence of myocardial injury.
Moreover, Vagal reflex, which is also common in acute pancreatitis, can cause heart injury by directly acting on myocardium, indirectly changing coronary blood flow, or increasing secretion of pancreatic proteolytic enzyme. It is well known that the heart is innervated by T2-8 spinal nerve, while the gallbladder and common bile duct are innervated by T4-9 spinal nerve, and both intersect at T4-5 spinal nerve. Therefore, when there is pressure increases in the bile duct and adjacent tissues due to inflammation, it will cause coronary artery contraction spasm, sinus node excitability decrease, slow down of conduction velocity of specific systems, cardiac output decrease, blood pressure decrease, cardiac arrest and a series of severe consequences.135,136
Autophagy Dysregulation
Under normal physiological conditions, autophagy removes aging cells and proteins in vivo, and it is a self-protection mechanism of the body. Essandoh’s study137 found that tumor susceptibility gene 101 plays a protective role in endotoxin-induced myocardial injury by promoting autophagy of damaged mitochondria induced by Parkin/PTEN-induced kinase 1 (PINK1). However, excessive autophagy can aggravate cardiac inflammation. When severe acute pancreatitis appears systemic inflammatory syndrome, the organism is in a stress state. Strong evidence indicated138 that autophagy could lead to the death of cardiomyocytes under stress, which may be related to the over-decomposition of proteins and organelles (such as mitochondria), the degradation of survival factors and other forms of autophagy cross-talk or the over-formation of autophagic body. This dual effect of autophagy suggests that it may be closely related to cardiac injury associated with severe acute pancreatitis, but further study is needed.
Markers of Myocardial Injury in SAP
Traditional myocardial injury markers such as cardiac troponin I (cTnI), brain natriuretic peptide (BNP) and creatine kinase (CK)-MB have good sensitivity in diagnosing myocardial function. However, during the occurrence and development of SAP, it may lead to multiple organ failure. The above indexes are easily affected by other complications and the diagnostic specificity of myocardial injury caused by SAP decreases. Hence, it is necessary to combine other biomarkers that can evaluate the severity of SAP to improve the directivity of these markers to myocardial injury caused by acute pancreatitis (Table 2).
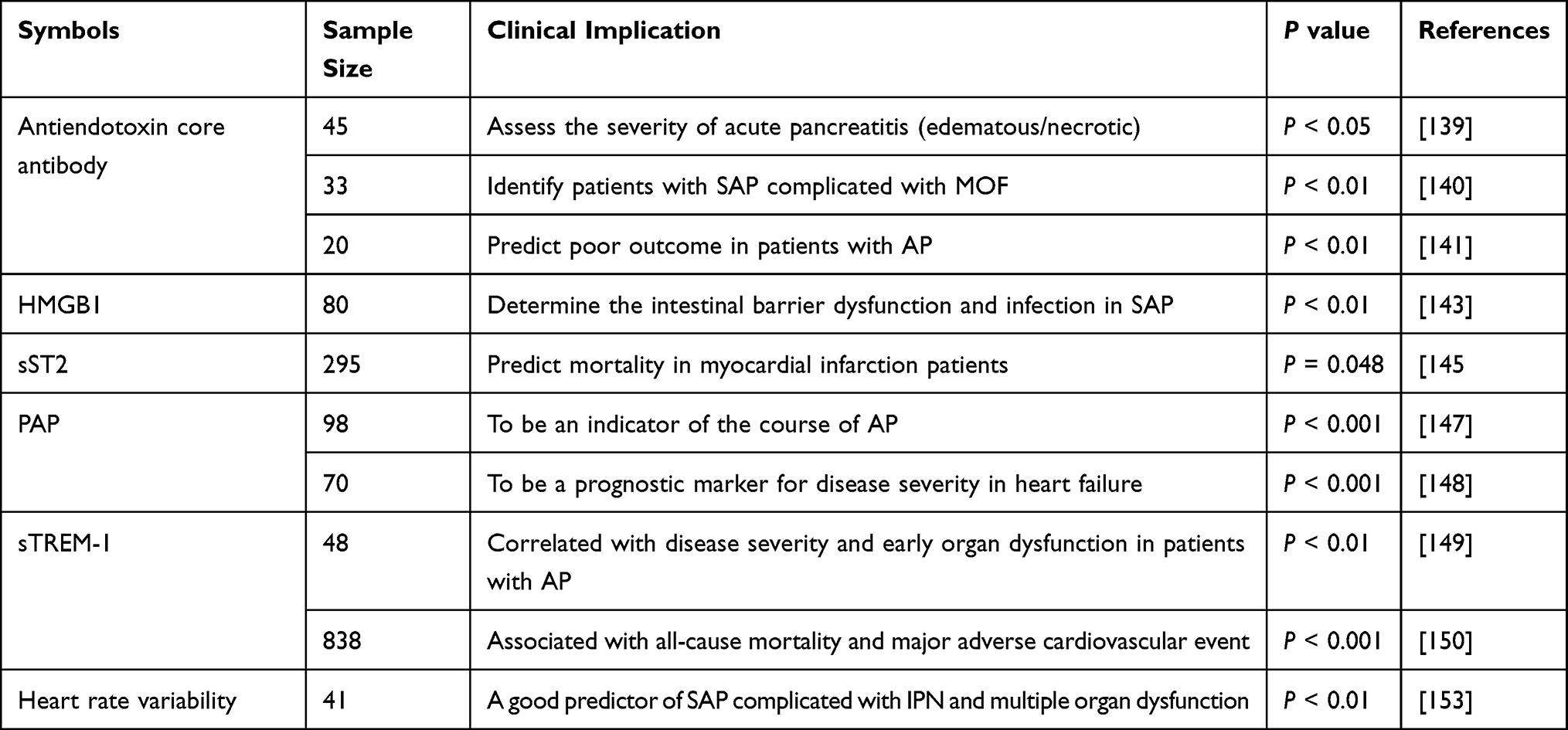 |
Table 2 Markers with Predictive or Prognostic Value for SAP-Associated Cardiac Injury |
Antiendotoxin Core Antibody
It has been mentioned that endotoxin contributes to multiple organ failure (MOF) in acute pancreatitis. Endotoxemia is transient and may not be detected by intermittent blood sampling. On the contrary, changes of the patient’s endogenous antiendotoxin core antibody pool last for many days, and the depletion of this pool may be a pivotal event to determine the physiological significance of endotoxemia.
In a prospective clinical study, Buttenschoen et al139 observed that necrotising pancreatitis was accompanied by persistent endotoxemia with an extended rise in antiendotoxin antibodies. Patients with oedematous pancreatitis suffer from transient endotoxemia with a temporary increase of Ig specific for endotoxin. And Windsor et al140 measured daily the Acute Physiology Score (APS) and levels of CRP, IL-6, endotoxin, immunoglobulin (Ig) G and IgM antiendotoxin core antibodies in a series of 33 patients with acute pancreatitis and recorded the complications prospectively to explore the role of serum endotoxin and antiendotoxin core antibody levels in predicting the development of multiple organ failure in acute pancreatitis. Results showed that endotoxin was detected in the serum of 13 patients, while a significant change in levels of endogenous antiendotoxin core antibodies was detected in all patients with severe acute pancreatitis and in 28 overall. MOF developed in 7 patients, 5 of whom died. The combination of a rising APS over the first 48 h of admission and a significant fall in endogenous IgG antibody level was observed in all patients who developed MOF (7 of 7), but in only one of 16 without MOF (P = 0.00003; overall predictive value 91%). This study indicated that measuring the initial trend of APS and the concentration of endogenous IgG antiendotoxin core antibody could provide a method to identify patients with severe acute pancreatitis at high risk of developing MOF. This group may benefit from passive immunotherapy with antiendotoxin antibodies. In addition, Bose et al141 correlated endotoxemia with the severity and complications of acute pancreatitis as graded by contrast-enhanced computed tomography and Blamey’s criteria. The results showed that endotoxemia with a fall in antiendotoxin antibody titer predicted a poor prognosis of patients with acute pancreatitis.
HMGBI
High Mobility Group Box 1 (HMGB1) was discovered as a nuclear protein, but it has “second life” outside the cell, where it is regarded as a damage-associated molecular pattern. Extracellular HMGB1 triggers and maintains inflammatory response by inducing cytokine release and recruiting leukocytes. These characteristics make extracellular HMGB1 a key molecular target in many diseases. Noteworthy, high levels of serum HMGB1, especially the hyper-acetylated and disulfide isoforms, are sensitive biomarkers of diseases and are related to different disease stages.142
Xu et al143 discussed the correlation of HMGB1 with intestinal barrier injury and infection in patients with severe acute pancreatitis (SAP). They measured the serum levels of HMGB1, amylase, lipase and biochemical indexes in 80 SAP patients at admission. Moreover, the relationship between serum HMGB1 level and intestinal barrier injury and other clinical factors was analyzed. The results showed that the average level of serum HMGB1 in SAP patients (6.02±2.42 ng/mL) was significantly higher than that in healthy volunteers (1.87±0.63 ng/mL). Serum HMGB1 level was positively correlated with the Ranson score. The HMGB1 level in patients with infection during the clinical course, the HMGB1 levels in non-survivors were higher than those in survivors, and positively correlated with DAO activity, L/M ratio and endotoxin concentration (R = 0.484, P <0.01). All these figures indicated that HMGB1 level in patients with SAP increased significantly, which can be used as an important indicator to determine the intestinal barrier dysfunction and infection, and may have indirect implications for subsequent systemic response syndrome and multiple-organ dysfunction syndrome. Further explorations may prove the advantage of this biomarker in predicting cardiovascular complications.
sST2
ST2, a member of IL-1 receptor superfamily, has two main isoforms–transmembrane ST2 (ST2L) and soluble ST2 (sST2). It is mainly expressed in Th2 cells, mast cells and fibroblasts, participating in multiple inflammatory processes and playing an immunomodulatory role. In recent years, sST2 has received extensive attention as a new marker of heart failure.
According to the study of Li,144 the serum levels of traditional myocardial injury markers cTnI, CK-MB and lactate dehydrogenase (LDH1) in SAP-associated cardiac injury model animals were markedly increased, serum sST2 level was significantly reduced. The anomalies, including serum amylase and cardiac-related enzymes, were reversed following adenovirus overexpression of sST2. Therefore, we speculate that combined measurement of serum sST2, cTnI, LDH and LDH1 could be used to evaluate the severity of myocardial injury in SAP, and overexpression of sST2 could reduce the severity of SAP and related cardiac injury. However, in another prospective clinical trial to evaluate the correlation between sST2 and the prognosis of STEMI patients receiving primary percutaneous coronary intervention, an opposite result was observed. Liu et al145 included 295 patients, and the data showed that biomarkers of myocardial cell damage and inflammation were positively correlated with sST2 levels. Higher sST2 was continuously associated with a higher risk of major adverse cardiovascular events in patients. This paradox suggests that the role of sST2 in myocardial injury is still controversial, and more clinical data are needed further to analyze the sensitivity and specificity of its diagnosis.
PAP
Pancreatitis-associated protein (PAP) was first found in pancreatic juice of rats with AP by French Keim, and it is related to the occurrence and development of AP warranted the named PAP.146 PAP plays a role in various physiological or pathological processes such as cellular immunity, cell proliferation and differentiation, anti-apoptosis, inflammation, and tissue repair.
In the study of Iovanna et al,147 98 patients with acute pancreatitis were classified into three groups according to the severity of the disease, and the serum PAP was retrospectively monitored during hospitalization to discuss whether serum PAP could be used as an indicator of the course of acute pancreatitis. Results showed that patients with mild pancreatitis had normal PAP values (<10 µg/L). PAP level in necrotic pancreas was 8 times higher than that in non-necrotic pancreas. The PAP level in serum further increased when the disease worsened. It is indicated that monitoring serum PAP in patients with AP would provide a dynamic assessment of severity of the disease and anticipate the patient’s recovery. Furthermore, Fitzgibbons et al148 found that PAP concentrations also correlate with the severity of heart failure (HF) in a prospective clinical study. ROC curves revealed that PAP had similar sensitivity and specificity for HF admission at 6 months as BNP and equivalent predictive value for 12-month and 24-month all-cause mortality. Based on the ROC curve analysis, patients were grouped into those with a serum PAP <24 or ≥24 μg/L. Patients with PAP ≥24 μg/L had significantly greater BNP and CRP levels and greater 6- and 24-month all-cause mortality (p <0.05). In conclusion, PAP levels correlate with disease severity in patients with SAP and HF and are a novel prognostic marker for disease severity in the patients.
sTREM-1
Myeloid cell trigger receptor-1 (TREM-1) belongs to a family-related natural killer cell receptor, which is basically expressed on the surface of neutrophils, monocytes and macrophages of myeloid cells. Soluble TREM-1 (sTREM-1) is a soluble form of TREM-1. The level of sTREM-1 is increased in body fluids of patients with various infections and diseases, and its increase is related to the amplification of inflammatory reaction.
Yasuda et al149 explored the role of sTREM-1 in acute pancreatitis by analyzing the clinical data of 48 patients with AP and 7 healthy controls. The results showed that serum sTREM-1 levels increased significantly in AP (63 ± 11 pg/mL) and correlated with Ranson score (R=0.628, p < 0.001) and Acute Physiology and Chronic Health Evaluation II score (R =0.504, p <0.001). Serum sTREM-1 levels were higher in patients with early organ dysfunction than those without early organ dysfunction (101 ± 19 vs 25 ± 4 pg/mL, p <0.001). Incidences of early organ dysfunction in patients whose serum sTREM-1 levels were < or = 40 and >40 pg/mL were 17% and 83%, respectively (p <0.001). The usefulness of serum sTREM-1 in detecting early organ dysfunction was superior to CRP, IL-6, IL-8, Ranson score, and Acute Physiology and Chronic Health Evaluation II score. All these studies suggested that serum sTREM-1 level may be a useful marker and potential therapeutic target for early organ dysfunction in AP. In addition, by measuring sTREM-1 levels in blood samples of 838 patients with acute myocardial infarction, Wang et al150 found that patients with high sTREM-1 had an increased risk of all-cause mortality and major adverse cardiovascular event (MACE) compared with those with low sTREM-1 (P<0.001). High sTREM-1 was an independent predictor of all-cause mortality (hazard ratio, 1.978; 95% CI, 1.462 to 2.675; P<0.001) and MACE (hazard ratio, 2.413; 95% CI, 2.022 to 2.879; P<0.001). Therefore, we believe that sTREM-1 may have a certain predictive value in the prognosis of severe acute pancreatitis and associated cardiac injury.
Heart Rate Variability
Heart rate variability (HRV) is considered the pathological changes of sinoatrial node reflected in cardiac cycle changes and is an important index for non-invasive and quantitative evaluation of cardiac autonomic neuropathy.151 Studies have shown HRV analysis could help early diagnosis and disease prognosis in critically ill patients.152
Zhang et al153 investigated the value of admission HRV as a marker of infected pancreatic necrosis (IPN) or MODS in patients with SAP in a prospective observational study. They found that levels of low frequency (LF), low-frequency norm (nLF) and low frequency/high frequency (LF/HF) were significantly lower in SAP patients with IPN and MODS than those in patients without complications, while high-frequency norm (nHF) levels were significantly higher (P < 0.01). nHF is a good predictor of IPN and MODS, and areas under ROC curves of nHF in prediction of IPN and MODS were 0.927 and 0.821, superior to procalcitonin (AUC=0.709 and 0.722) and APACHEII (AUC=0.785 and 0.899). These results suggest that patients with SAP have sympathetic inhibition in the early stage, and HRV has been proved to be a good predictor of SAP infective pancreatic necrosis and multiple-organ dysfunction.
Prevention and Treatment of SAP-Associated Cardiac Injury
The treatment of SAP affects the prognosis of extra-pancreatic organs, so the treatment of primary SAP and the treatment of myocardial damage should be paid equal attention to (summary of treatment strategies can be seen in Table 3).
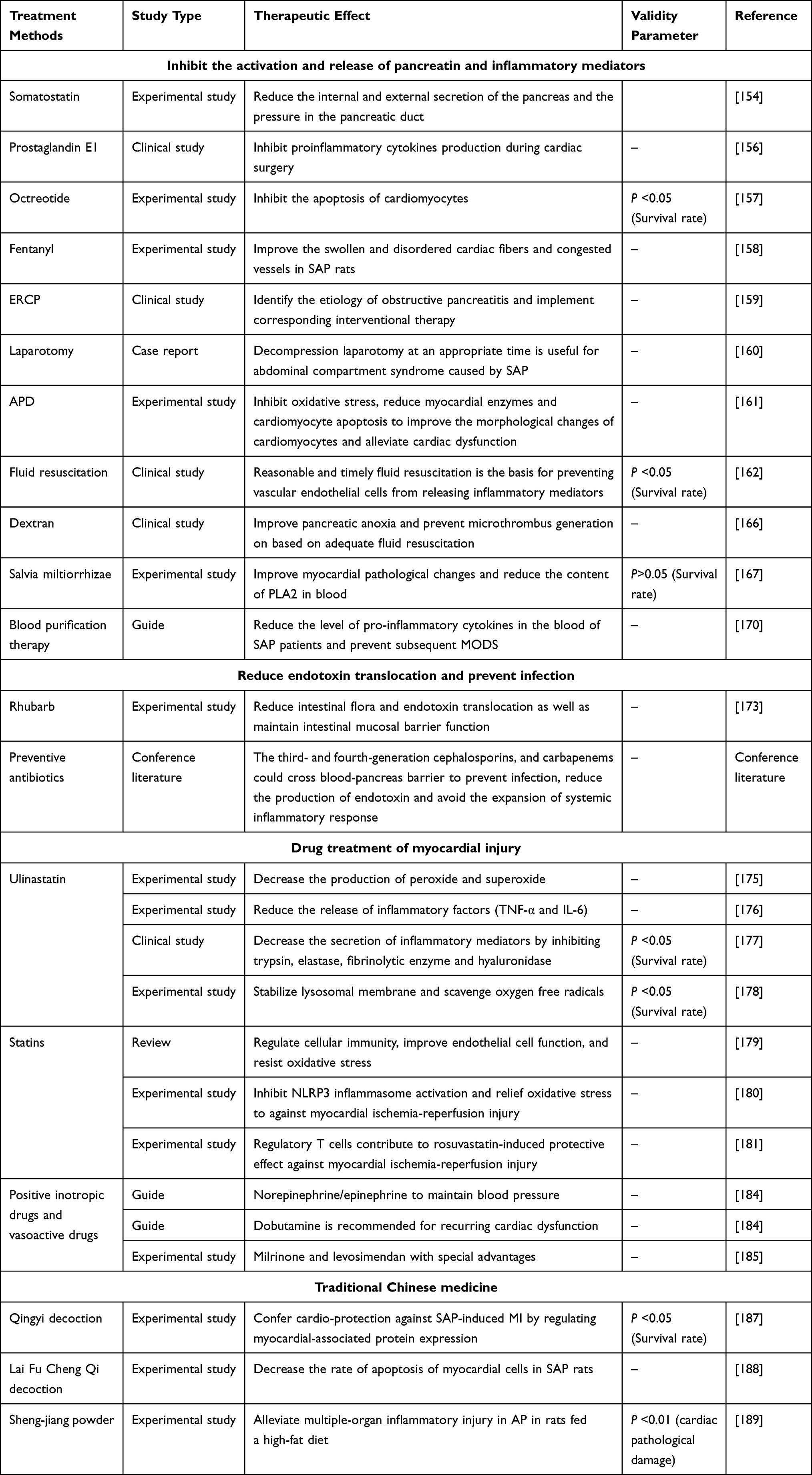 |
Table 3 Prevention and Treatment of SAP-Associated Cardiac Injury |
Inhibit the Activation and Release of Pancreatin and Inflammatory Mediators
Drugs such as somatostatin, octreotide, prostaglandin E1 (PGE1) and fentanyl could directly or indirectly inhibit the secretion of pancreatin and reduce the toxic effect of pancreatin on myocardium. Somatostatin can effectively reduce the internal and external secretion of the pancreas and the pressure in the pancreatic duct,154 and somatostatin pretreatment can improve the hemodynamic state of the body by reducing the release of cytokines.155 PGE1 can strongly inhibit pancreatic exocrine secretion, stabilize lysosomal membrane in acinar cells, restrict intracellular pancreatin activation, and prevent the dissolution and destruction of pancreatic cells.156 Zhang et al157 discovered the protective effect of octreotide on SAP rat heart by comparing the concentration of inflammatory indexes in blood, and pro-apoptotic proteins (Bax, Bcl-2 and Caspase-3) in myocardium and the pathological changes of the heart of SAP rats and octreotide treated rats. Wang et al158 found that fentanyl could significantly improve the swollen and disordered cardiac fibers and congested vessels in SAP rats by regulating NF-κB signaling pathway and markedly inhibit the up-regulation of IL-1β/ IL-6 induced by SAP.
Although the concept of early non-surgical treatment of SAP has been widely accepted, surgical interventions and endoscopy required for the etiological treatment are still fundamental measures to prevent the disease and its progress. For example, obstructive biliary pancreatitis and the abdominal compartment syndrome resulting from SAP need timely endoscopic retrograde cholangiopancreatography (ERCP) or surgical treatment.159,160 In addition, some studies proposed that abdominal paracentesis drainage (APD) therapy can play a protective role in SAP-induced myocardial injury via inhibiting HMGB1-mediated oxidative stress. Wen and his team161 found that APD could significantly improve the morphological changes of cardiomyocytes, alleviate cardiac dysfunction, reduce myocardial enzymes and cardiomyocyte apoptosis, decrease the expression of pro-apoptotic Bax and cleaved caspase-3 protein. Moreover, APD remarkably reduced the serum level of HMGB1, inhibited Nox expression, and alleviated cardiac oxidative injury.
Hypoperfusion induced by hypovolemia leads to vascular endothelial cell injury and apoptosis, increasing capillary permeability and releasing many inflammatory mediators that aggravate systemic organ damage. Reasonable and timely fluid resuscitation is the basis for preventing vascular endothelial cells from releasing inflammatory mediators. Studies have shown that combining different resuscitation fluids (saline, hydroxyethyl starch and glutamine) is more efficient in SAP resuscitate by reducing inflammation and sustaining the intestinal barrier.162 It is worth noting that the symptoms of AP are complex. In the choice of fluid therapy, we need to implement targeted treatment according to the different conditions of patients to prevent patients from MODS. Excessive fluid therapy will aggravate the fluid retention of patients, give rise to cardiac overload, resulting in acute heart failure. Huang et al163 discussed possible factors causing abnormal heart failure after successful fluid resuscitation on AP patients. They found that older patients with AP had more severe organ failure or more organ involvement and had more interstitial fluid accumulation during fluid resuscitation. Furthermore, aggressive resuscitation is associated with the development of acute kidney injury in AP.164 In recent years, it has been proposed that fluid resuscitation via the rectum can effectively improve hemodynamic disorders and organ dysfunction (jejunum, liver and lung) in SAP compared with intravenous fluid resuscitation, which may be a potential supplementary fluid management in the early stage of SAP.165 In addition, pancreatic hypoxia, microcirculation disorder and even microthrombus formation can aggravate the release of cytokines, so it is necessary to improve pancreatic anoxia and prevent microthrombus generation based on adequate fluid resuscitation. Dextran,166 Salvia miltiorrhizae167 and PGE1 can increase pancreatic blood flow, reduce hemorrhage and necrosis of the pancreas, decrease pancreatic capillary permeability, alleviate endothelial cell injury and inhibit cytokine release.156 Pancreatitis complicated with oxidative stress results in the rapid accumulation of cytokines in the body, so antioxidant therapy is important.168 Drugs commonly clinically include reduced glutathione, high-dose of vitamin C, N- acetamide cysteine, selenium, vitamin E and others.
Recently, blood purification therapy has been applied clinically to treat cytokine-induced pathological effects, but this must be done within 72 hours of onset to achieve good curative efficacy. Early continuous blood purification can reduce pro-inflammatory cytokines, prevent MODS and significantly improve the prognosis of patients with severe sepsis.169 In addition, high-flow short-time hemofiltration should be used for SAP because long-time hemofiltration can cause excessive release of IL-10 to inhibit immune function and increase the susceptibility of patients to infection. However, high-flow continuous hemofiltration is recommended for fulminant pancreatitis.170
Reduce Endotoxin Translocation and Prevent Infection
Pancreatitis, a retroperitoneal inflammation, damaged intestinal mucosal barrier function, leading to migration of bacteria and endotoxin into the liver, heart, lung and other parts of the body, which exacerbate the release of extra-pancreatic cytokines.171 Unclipping the intestinal tract, removing toxins accumulated in the intestinal tract and nourishing the intestinal tract after dredging can significantly reduce infectious complications (IPN and sepsis) and improve survival rate.172 Rhubarb or decoction containing rhubarb, such as Dachengqi decoction and Qingyi decoction, has a certain effect on reducing intestinal flora and endotoxin translocation.173
Although the early application of prophylactic antibiotics is still controversial, most studies report that preventive antibiotics can effectively reduce infectious complications and mortality. Because early infection with pancreatitis is more likely to cause intestinal bacterial translocation, it is suggested to choose antibiotics with a high concentration in the pancreas that can cross the blood-pancreas barrier to prevent infection and reduce endotoxin production avoid the expansion of systemic inflammatory response.174 Such drugs include the third- and fourth-generation cephalosporins, and carbapenems.
Drug Treatment of Myocardial Injury
Ulinastatin
With the development of critical care medicine, ulinastatin has been widely recognized in preventing MOF. Wang et al demonstrated in a rat model of myocardial infarction that high-dose ulinastatin could reduce total cholesterol, catalase, glutathione peroxidase and superoxide dismutase in myocardial tissue, thereby relieving inflammation and preventing rats from myocardial infarction.175 Similarly, Tian et al showed that ulinastatin combined with rhubarb could reduce the release of inflammatory factors such as TNF-α and IL-6, change the percentage of lymphocytes to reduce organ damage induced by sepsis, improve the prognosis of patients.176 In addition, ulinastatin can decrease the secretion of inflammatory mediators by inhibiting trypsin, elastase, fibrinolytic enzyme and hyaluronidase,177 and has the functions of stabilizing lysosomal membrane and scavenging oxygen-free radicals.178
Statins
In acute pancreatitis, various direct and indirect factors cause coronary artery contraction and spasm, resulting in myocardial ischemia/reperfusion injury. Statins function as an anti-inflammation and anti-coagulation. It also regulates cellular immunity, improves endothelial cell function and resists oxidative stress.179 According to Yu et al,180 rosuvastatin can ameliorate myocardial histopathological changes and reduce MI area in isoproterenol-induced MI model in rats. Compared with the model group, the antioxidant level in the drug group was significantly increased, and the activity of NLRP3 inflammasome was decreased. These results indicate that rosuvastatin can alleviate myocardial injury by inhibiting NLRP3 inflammasome and relieving oxidative stress. In a related study, Ke et al181 showed that regulatory T cells contribute to the rosuvastatin-induced protective effect against myocardial ischemia-reperfusion injury.
Diuretics
Heart failure is the final stage of heart disease caused by various factors (including pancreatitis), and volume overload is the core of its pathophysiology. Clinically, diuretics (including loop diuretics, thiazide diuretics and glucocorticoid receptor antagonists) are often used to promote the excretion of water and sodium in the kidney to alleviate symptoms. They play pharmacological effects by dilating the renal artery, increasing the renal blood flow, inhibiting the reabsorption of sodium and chlorine in the renal tubules, and maintaining the balance of water and salt in the body.182 Attention, it has been reported that diuretics could induce acute pancreatitis (DAP), of which thiazides are the most common. The direct toxic effects of furosemide and pentamidine on pancreatic acinar cells lead to degeneration and necrosis of glandular cells and trypsin activation.183 The common clinical inducing doses were chlorothiazide 25~100mg/d, chlortalidone 50mg/d and furosemide 40mg~80mg/d. The corresponding symptoms usually occur within 3–5 weeks after medication. Most people have typical AP manifestations, but a few people only have a slight increase in amylase activity (1.5~2 times). Very few patients may have sudden shock or death. Therefore, the clinical application of diuretics in heart failure caused by acute pancreatitis needs more standardized management and more evidence-based medicine.
Positive Inotropic Drugs and Vasoactive Drugs
The Surviving Sepsis Campaign recommended norepinephrine as the first choice of vasopressor to correct septic shock. If patients need more vasoconstrictors to maintain adequate blood pressure, epinephrine can be added or replaced. Dobutamine is recommended for recurring cardiac dysfunction.184 It has been reported that milrinone,185 a second-generation dipyridone cardiotonic drug with high efficiency and low toxicity, can reduce the levels of TNF-α and IL-1 in rat cardiomyocytes and regulate inflammatory mediators and cytokines. Additionally, because of its dual effects of anti-inflammatory and anti-apoptotic functions, it can improve myocardial perfusion and promote hemodynamic stability. Levosimendan186 is a non-digitalis positive inotropic drug, which can improve hemodynamics and has the advantages of not causing calcium overload, myocardial oxygen consumption and arrhythmia.
Treatment with Traditional Chinese Medicine
Many Traditional Chinese Medicines have the characteristics of antagonizing endotoxin and inflammatory mediators, so they play an important role in various inflammatory, metabolic and autoimmune diseases. The efficacy of various Chinese Medicines has been demonstrated in a model of rat AP induced by retrograde injection of 5% sodium taurocholate into the pancreaticobiliary duct, which resulted in an obvious injury of tissue structure and infiltration of inflammatory cells. Salvia miltiorrhizae,167 baicalin,157 Qingyi decoction,187 Lai Fu Cheng Qi decoction188 and other Chinese herbal medicine preparations significantly reduced serum inflammatory factors and myocardial injury-related indicators, and effectively improve the peripheral microcirculation, promote fluid resuscitation, heighten myocardial contractility, and reduced the mortality SAP-induced MI. Moreover, Sheng-jiang powder189 is supposed to have a protective effect on multiple-organ inflammatory damage in AP in rats fed with a high-fat diet.
Abbreviations
AP, acute pancreatitis; SAP, severe acute pancreatitis; ANP, acute necrotizing pancreatitis; SIRS, systemic inflammatory response syndrome; MODS, multiple-organ dysfunction syndrome; MOF, multiple-organ failure; CI, cardiac injury; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; I/R, ischemia/reperfusion; LPS, lipopolysaccharide; HMGB1, high mobility group box-1; DCs, dendritic cells; IL, interleukin; VCAM-1, vascular cell adhesion molecule 1; MCP-1, monocyte chemoattractant protein 1; Cyt C, cytochrome C; NLRP3, nucleotide-binding oligomerization domain-like receptor protein 3; ASC, apoptosis-associated speck-like protein containing CARD; CRP, C-reaction protein; iNOS, inducible nitric oxide synthase; NO, nitric oxide; AngII, angiotensin II; HO-1, heme oxygenase-1; cGMP, cyclic guanosine monophosphate; PKA, protein kinase A; PKG, protein kinase G; ECG, electrocardiogram; LV, left ventricular; ROS, reactive oxygen species; ASK-1, apoptosis signal-regulating kinase-1; mtDNA, mitochondrial DNA; PAF, platelet-activating factor; FFA, free fatty acids; ET-1, endothelin-1; TXA2, thromboxane A2; cTnI, cardiac troponin I; BNP, brain natriuretic peptide; CK, creatine kinase; TREM-1, myeloid cell trigger receptor-1; PGE1, prostaglandin E1; ERCP, endoscopic retrograde cholangiopancreatography; APD, abdominal paracentesis drainage; HRV, heart rate variability; IPN, infected pancreatic necrosis; HTGAP, hypertriglyceridemia-associated acute pancreatitis.
Summary
The pathophysiology of myocardial dysfunction caused by SAP is unclear, and scientific investigations on its mechanism are still very scarce. However, some inspiration and guidance can be obtained from the pathogenesis of myocardial injury caused by sepsis. Trypsin, inflammatory mediators, ROS, endotoxin, vascular disorders, and autonomic nerves play important roles in the “pancreas-heart axis”. At present, there are no specific drugs used clinically to prevent SAP from progressing to MI, and the focus of treatment remains supportive and targets improving systemic inflammatory response. Currently, timely, diverse and combined treatment methods have been adopted for the myocardial injury caused by SIRS. However, due to the complexity of SAP-related MI (not only mediated by SIRS), these treatment methods and strategies are not fully applicable, and the mortality rate remains high. Therefore, it is extremely urgent to explore the pathogenesis of myocardial injury associated with acute pancreatitis and develop specific drugs targeting its molecular level.
Funding
This work was supported by The key project of the National significant R&D Program “Intergovernmental Cooperation in International Science and Technology Innovation” (grant number 2019YFE0119300) and National Natural Science Foundation of China (grant number 82074158).
Disclosure
Yalan Luo, Zhaoxia Li and Peng Ge are co-first authors. The authors declare that there are no conflicts of interest.
References
1. Thoeni RF. The revised Atlanta classification of acute pancreatitis: its importance for the radiologist and its effect on treatment. Radiology. 2012;262:751–764. doi:10.1148/radiol.11110947
2. Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62(1):102–111. doi:10.1136/gutjnl-2012-302779
3. Yegneswaran B, Kostis JB, Pitchumoni CS. Cardiovascular manifestations of acute pancreatitis. J Crit Care. 2011;26:
4. Lovett WL, Wangensteen SL, Glenn TM, Lefer AM. Presence of a myocardial depressant factor in patients in circulatory shock. Surgery. 1971;70:223–231.
5. Nadkarni N, Bhasin DK, Rana SS, et al. Diastolic dysfunction, prolonged QTc interval and pericardial effusion as predictors of mortality in acute pancreatitis. J Gastroenterol Hepatol. 2012;27(10):1576–1580. doi:10.1111/j.1440-1746.2012.07229.x
6. Kong L, Santiago N, Han TQ, Zhang SD. Clinical characteristics and prognostic factors of severe acute pancreatitis. World J Gastroenterol. 2004;10(22):3336–3338. doi:10.3748/wjg.v10.i22.3336
7. Calleja GA, Barkin JS. Acute pancreatitis. Med Clin North Am. 1993;77(5):1037–1056. doi:10.1016/S0025-7125(16)30209-7
8. Vasantha Kumar A, Mohan Reddy G, Anirudh Kumar A. Acute pancreatitis complicated by acute myocardial infarction - a rare association. Indian Heart J. 2013;65(4):474–477. doi:10.1016/j.ihj.2013.06.009
9. Zhang JW, Zhang GX, Chen HL, et al. Therapeutic effect of Qingyi decoction in severe acute pancreatitis-induced intestinal barrier injury. World J Gastroenterol. 2015;21(12):3537–3546. doi:10.3748/wjg.v21.i12.3537
10. Collins HE, Zhu-Mauldin X, Marchase RB, Chatham JC. STIM1/Orai1-mediated SOCE: current perspectives and potential roles in cardiac function and pathology. Am J Physiol Heart Circ Physiol. 2013;305(4):H446–58. doi:10.1152/ajpheart.00104.2013
11. Saulea A, Costin S, Rotari V. Heart ultrastructure in experimental acute pancreatitis. Rom J Physiol. 1997;34(1–4):35–44.
12. Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1252–1261. doi:10.1053/j.gastro.2013.01.068
13. Kellner A, Robertson T. Selective necrosis of cardiac and skeletal muscle induced experimentally by means of proteolytic enzyme solutions given intravenously. J Exp Med. 1954;99(4):387–404. doi:10.1084/jem.99.4.387
14. Dan Z, Liao XL, Hou CS, Xu BB, Yang J, Kang Y. [The expression of trypsin in serum and vital organs of septic rats]. Zhonghua Nei Ke Za Zhi. 2018;57(7):505–510. (Chinese). doi:10.3760/cma.j.issn.0578-1426.2018.07.007
15. Tejada JG, Hernández F, Chimeno J, Alonso MA, Martin R, Bastante T. Acute pancreatitis mimicking acute inferior myocardial infarction. Angiology. 2008;59(3):365–367. doi:10.1177/0003319707304533
16. Masuda T, Sato K, Noda C, et al. Protective effect of urinary trypsin inhibitor on myocardial mitochondria during hemorrhagic shock and reperfusion. Crit Care Med. 2003;31(7):1987–1992. doi:10.1097/01.CCM.0000057037.44171.BA
17. Mauro AG, Mezzaroma E, Marchetti C, et al. A Preclinical Translational Study of the cardioprotective effects of plasma-derived Alpha-1 anti-trypsin in acute myocardial infarction. J Cardiovasc Pharmacol. 2017;69(5):273–278. doi:10.1097/FJC.0000000000000474
18. Roncati L, Gualandri G, Fortuni G, Barbolini G. Sudden death and lipomatous infiltration of the heart involved by fat necrosis resulting from acute pancreatitis. Forensic Sci Int. 2012;217(1–3):e19–22. doi:10.1016/j.forsciint.2011.10.038
19. Yang YZ, Xiang Y, Chen M, Xian LN, Deng XY. Clinical significance of dynamic detection for serum levels of MCP-1, TNF-α and IL-8 in patients with acute pancreatitis. Asian Pac J Trop Med. 2016;9:1111–1114. doi:10.1016/j.apjtm.2016.09.001
20. Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47(4):378–386. doi:10.1016/j.ceca.2010.02.002
21. Hofmann U, Heuer S, Meder K, et al. The proinflammatory cytokines TNF-alpha and IL-1 beta impair economy of contraction in human myocardium. Cytokine. 2007;39(3):157–162. doi:10.1016/j.cyto.2007.07.185
22. Hofmann U, Domeier E, Frantz S, et al. Increased myocardial oxygen consumption by TNF-alpha is mediated by a sphingosine signaling pathway. Am J Physiol Heart Circ Physiol. 2003;284(6):H2100–5. doi:10.1152/ajpheart.00888.2002
23. Gellerich FN, Trumbeckaite S, Hertel K, et al. Impaired energy metabolism in hearts of septic baboons: diminished activities of Complex I and Complex II of the mitochondrial respiratory chain. Shock. 1999;11(5):336–341. doi:10.1097/00024382-199905000-00006
24. Jude B, Vetel S, Giroux-Metges MA, Pennec JP. Rapid negative inotropic effect induced by TNF-α in rat heart perfused related to PKC activation. Cytokine. 2018;107:65–69. doi:10.1016/j.cyto.2017.11.015
25. Li D, Li J, Wang L, Zhang Q. Association between IL-1β, IL-8, and IL-10 polymorphisms and risk of acute pancreatitis. Genet Mol Res. 2015;14:6635–6641. doi:10.4238/2015.June.18.6
26. Chamberlain J, Francis S, Brookes Z, et al. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS One. 2009;4(4):e5073. doi:10.1371/journal.pone.0005073
27. Kirii H, Niwa T, Yamada Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23(4):656–660. doi:10.1161/01.ATV.0000064374.15232.C3
28. Frangogiannis NG. Interleukin-1 in cardiac injury, repair, and remodeling: pathophysiologic and translational concepts. Discoveries (Craiova). 2015;3(1):e41. doi:10.15190/d.2015.33
29. El Khoury N, Mathieu S, Fiset C. Interleukin-1β reduces L-type Ca2+ current through protein kinase Cϵ activation in mouse heart. J Biol Chem. 2014;289:21896–21908. doi:10.1074/jbc.M114.549642
30. Shen Y, Qin J, Bu P. Pathways involved in interleukin-1β-mediated murine cardiomyocyte apoptosis. Tex Heart Inst J. 2015;42:109–116. doi:10.14503/THIJ-14-4254
31. Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A. 2001;98(5):2871–2876. doi:10.1073/pnas.041611398
32. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15(4):203–214. doi:10.1038/nrcardio.2017.161
33. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi:10.1146/annurev-immunol-031210-101405
34. Toldo S, Mezzaroma E, O’Brien L, et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1025–1031. doi:10.1152/ajpheart.00795.2013
35. Janiak A, Leśniowski B, Jasińska A, Pietruczuk M, Małecka-Panas E. Interleukin 18 as an early marker or prognostic factor in acute pancreatitis. Prz Gastroenterol. 2015;10:203–207. doi:10.5114/pg.2015.50993
36. Mallat Z, Henry P, Fressonnet R, et al. Increased plasma concentrations of interleukin-18 in acute coronary syndromes. Heart. 2002;88:467–469. doi:10.1136/heart.88.5.467
37. Bartekova M, Radosinska J, Jelemensky M, Dhalla NS. Role of cytokines and inflammation in heart function during health and disease. Heart Fail Rev. 2018;23(5):733–758.
38. Yoshida T, Friehs I, Mummidi S, et al. Pressure overload induces IL-18 and IL-18R expression, but markedly suppresses IL-18BP expression in a rabbit model. IL-18 potentiates TNF-α-induced cardiomyocyte death. J Mol Cell Cardiol. 2014;75:141–151. doi:10.1016/j.yjmcc.2014.07.007
39. O’Brien LC, Mezzaroma E, Van Tassell BW, et al. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol Med. 2014;20:221–229. doi:10.2119/molmed.2014.00034
40. Sternby H, Hartman H, Johansen D, Thorlacius H, Regnér S. Predictive capacity of biomarkers for severe acute pancreatitis. Eur Surg Res. 2016;56:154–163. doi:10.1159/000444141
41. Sikorska D, Pawlaczyk K, Roszak M, et al. Preliminary observations on the association between serum IL-6 and hydration status and cardiovascular risk in patients treated with peritoneal dialysis. Cytokine. 2016;85:171–176. doi:10.1016/j.cyto.2016.06.005
42. Yue P, Massie BM, Simpson PC, Long CS. Cytokine expression increases in nonmyocytes from rats with postinfarction heart failure. Am J Physiol. 1998;275:H250–258. doi:10.1152/ajpheart.1998.275.1.H250
43. Gwechenberger M, Mendoza LH, Youker KA, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi:10.1161/01.cir.99.4.546
44. Baumgarten G, Knuefermann P, Kalra D, et al. Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation. 2002;105:2192–2197. doi:10.1161/01.cir.0000015608.37608.18
45. Rose-John S. Interleukin-6 family cytokines. Cold Spring Harb Perspect Biol. 2018;10:a028415. doi:10.1101/cshperspect.a028415
46. Wang JH, Zhao L, Pan X, et al. Hypoxia-stimulated cardiac fibroblast production of IL-6 promotes myocardial fibrosis via the TGF-β1 signaling pathway. Lab Invest. 2016;96:839–852. doi:10.1038/labinvest.2016.65
47. Chen F, Chen D, Zhang Y, et al. Interleukin-6 deficiency attenuates angiotensin II-induced cardiac pathogenesis with increased myocyte hypertrophy. Biochem Biophys Res Commun. 2017;494:534–541. doi:10.1016/j.bbrc.2017.10.119
48. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi:10.1146/annurev-immunol-031210-101312
49. Zhang FH, Sun YH, Fan KL, et al. Protective effects of heme oxygenase-1 against severe acute pancreatitis via inhibition of tumor necrosis factor-α and augmentation of interleukin-10. BMC Gastroenterol. 2017;17(1):100. doi:10.1186/s12876-017-0651-4
50. Van Laethem JL, Marchant A, Delvaux A, et al. Interleukin 10 prevents necrosis in murine experimental acute pancreatitis. Gastroenterology. 1995;108(6):1917–1922. doi:10.1016/0016-5085(95)90158-2
51. Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J Biochem. 2010;147(6):781–792. doi:10.1093/jb/mvq043
52. Kulkarni AB, Huh CG, Becker D, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90(2):770–774. doi:10.1073/pnas.90.2.770
53. Bandyopadhyay D, Chattopadhyay A, Ghosh G, Datta AG. Oxidative stress-induced ischemic heart disease: protection by antioxidants. Curr Med Chem. 2004;11(3):369–387. doi:10.2174/0929867043456016
54. Zhao Y, Zhao B. Protective effect of natural antioxidants on heart against ischemia-reperfusion damage. Curr Pharm Biotechnol. 2010;11(8):868–874. doi:10.2174/138920110793262105
55. Andican G, Gelisgen R, Unal E, et al. Oxidative stress and nitric oxide in rats with alcohol-induced acute pancreatitis. World J Gastroenterol. 2005;11:2340–2345. doi:10.3748/wjg.v11.i15.2340
56. Ichinose F, Buys ES, Neilan TG, et al. Cardiomyocyte-specific overexpression of nitric oxide synthase 3 prevents myocardial dysfunction in murine models of septic shock. Circ Res. 2007;100(1):130–139. doi:10.1161/01.RES.0000253888.09574.7a
57. Maass DL, White J, Horton JW. Nitric oxide donors alter cardiomyocyte cytokine secretion and cardiac function. Crit Care Med. 2005;33(12):2794–2803. doi:10.1097/01.CCM.0000189747.97541.59
58. Rastaldo R, Pagliaro P, Cappello S, et al. Nitric oxide and cardiac function. Life Sci. 2007;81(10):779–793. doi:10.1016/j.lfs.2007.07.019
59. Mohan P, Brutsaert DL, Paulus WJ, Sys SU. Myocardial contractile response to nitric oxide and cGMP. Circulation. 1996;93(6):1223–1229. doi:10.1161/01.CIR.93.6.1223
60. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):
61. Fagerberg L, Hallström BM, Oksvold P, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406. doi:10.1074/mcp.M113.035600
62. Watts VL, Sepulveda FM, Cingolani OH, et al. Anti-hypertrophic and anti-oxidant effect of beta3-adrenergic stimulation in myocytes requires differential neuronal NOS phosphorylation. J Mol Cell Cardiol. 2013;62:8–17. doi:10.1016/j.yjmcc.2013.04.025
63. Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation. 2005;112(24):3729–3737. doi:10.1161/CIRCULATIONAHA.105.539437
64. Ichinose F. [Impact of nitric oxide synthase 3 on myocardial dysfunction in sepsis]. Masui. 2008;57(3):294–301. (Japanese).
65. Zhang L, Zhang Y, Yu X, Xu H, Sui D, Zhao X. Alprostadil attenuates myocardial ischemia/reperfusion injury by promoting antioxidant activity and eNOS activation in rats. Acta Cir Bras. 2018;33(12):1067–1077. doi:10.1590/s0102-865020180120000004
66. Braganza JM, Scott P, Bilton D, et al. Evidence for early oxidative stress in acute pancreatitis. Clues for correction. Int J Pancreatol. 1995;17:69–81. doi:10.1007/BF02788361
67. Wen Y, Liu R, Lin N, et al. NADPH oxidase hyperactivity contributes to cardiac dysfunction and apoptosis in rats with severe experimental pancreatitis through ROS-mediated MAPK signaling pathway. Oxid Med Cell Longev. 2019;2019:4578175. doi:10.1155/2019/4578175
68. Marciniak A, Walczyna B, Rajtar G, Marciniak S, Wojtak A, Lasiecka K. Tempol, a membrane-permeable radical scavenger, exhibits anti-inflammatory and cardioprotective effects in the cerulein-induced pancreatitis rat model. Oxid Med Cell Longev. 2016;2016:4139851. doi:10.1155/2016/4139851
69. Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115(3):500–508. doi:10.1172/JCI200524408
70. Muralidharan P, Cserne Szappanos H, Ingley E, Hool LC. The cardiac L-type calcium channel alpha subunit is a target for direct redox modification during oxidative stress-the role of cysteine residues in the alpha interacting domain. Clin Exp Pharmacol Physiol. 2017;44(Suppl 1):46–54. doi:10.1111/1440-1681.12750
71. Joseph SK, Booth DM, Young MP, Hajnóczky G. Redox regulation of ER and mitochondrial Ca2+ signaling in cell survival and death. Cell Calcium. 2019;79:89–97. doi:10.1016/j.ceca.2019.02.006
72. Seidlmayer LK, Mages C, Berbner A, et al. Mitofusin 2 is essential for IP3-mediated SR/mitochondria metabolic feedback in ventricular myocytes. Front Physiol. 2019;10:733. doi:10.3389/fphys.2019.00733
73. Dries E, Santiago DJ, Gilbert G, et al. Hyperactive ryanodine receptors in human heart failure and ischaemic cardiomyopathy reside outside of couplons. Cardiovasc Res. 2018;114(11):1512–1524. doi:10.1093/cvr/cvy088
74. Wu LL, Tang C, Liu MS. Altered phosphorylation and calcium sensitivity of cardiac myofibrillar proteins during sepsis. Am J Physiol Regul Integr Comp Physiol. 2001;281(2):R408–16. doi:10.1152/ajpregu.2001.281.2.R408
75. Takeuchi K, Del Nido PJ, Ibrahim AE, et al. Increased myocardial calcium cycling and reduced myofilament calcium sensitivity in early endotoxemia. Surgery. 1999;126(2):231–238. doi:10.1016/S0039-6060(99)70160-1
76. Lin B, Xu J, Feng DG, Wang F, Wang JX, Zhao H. DUSP14 knockout accelerates cardiac ischemia reperfusion (IR) injury through activating NF-κB and MAPKs signaling pathways modulated by ROS generation. Biochem Biophys Res Commun. 2018;501(1):24–32. doi:10.1016/j.bbrc.2018.04.101
77. Liang Y, Ip M, Mak J. (-)-Epigallocatechin-3-gallate suppresses cigarette smoke-induced inflammation in human cardiomyocytes via ROS-mediated MAPK and NF-κB pathways. Phytomedicine. 2019;58:152768. doi:10.1016/j.phymed.2018.11.028
78. Lei Q, Yi T, Chen C. NF-κB-gasdermin D (GSDMD) axis couples oxidative stress and NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome-mediated cardiomyocyte pyroptosis following myocardial infarction. Med Sci Monit. 2018;24:6044–6052. doi:10.12659/MSM.908529
79. Sullivan EM, Pennington ER, Sparagna GC, et al. Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome. J Biol Chem. 2018;293(2):466–483. doi:10.1074/jbc.M117.812834
80. Pepe S. Effect of dietary polyunsaturated fatty acids on age-related changes in cardiac mitochondrial membranes. Exp Gerontol. 2005;40(8–9):751–758. doi:10.1016/j.exger.2005.03.013
81. Shao H, Li J, Zhou Y, et al. Dose-dependent protective effect of propofol against mitochondrial dysfunction in ischaemic/reperfused rat heart: role of cardiolipin. Br J Pharmacol. 2008;153(8):1641–1649. doi:10.1038/bjp.2008.45
82. Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res. 2009;81(3):449–456. doi:10.1093/cvr/cvn280
83. Yao X, Carlson D, Sun Y, et al. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS One. 2015;10(10):e0139416. doi:10.1371/journal.pone.0139416
84. Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both. Biochim Biophys Acta. 2011;1813(4):616–622. doi:10.1016/j.bbamcr.2010.09.013
85. Larche J, Lancel S, Hassoun SM, et al. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol. 2006;48(2):377–385. doi:10.1016/j.jacc.2006.02.069
86. Abdallah Y, Kasseckert SA, Iraqi W, et al. Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med. 2011;15(11):2478–2485. doi:10.1111/j.1582-4934.2010.01249.x
87. Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi:10.1038/nature08780
88. Hüttemann M, Helling S, Sanderson TH, et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012;1817(4):598–609. doi:10.1016/j.bbabio.2011.07.001
89. Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–658. doi:10.1038/nature03317
90. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi:10.1038/nri1594
91. Ugrinova I, Pasheva E. HMGB1 protein: a therapeutic target inside and outside the cell. Adv Protein Chem Struct Biol. 2017;107:37–76. doi:10.1016/bs.apcsb.2016.10.001
92. Martinotti S, Patrone M, Ranzato E. Emerging roles for HMGB1 protein in immunity, inflammation, and cancer. Immunotargets Ther. 2015;4:101–109. doi:10.2147/ITT.S58064
93. Saïd-Sadier N, Ojcius DM. Alarmins, inflammasomes and immunity. Biomed J. 2012;35(6):437–449. doi:10.4103/2319-4170.104408
94. Wakefield D, Gray P, Chang J, Di Girolamo N, McCluskey P. The role of PAMPs and DAMPs in the pathogenesis of acute and recurrent anterior uveitis. Br J Ophthalmol. 2010;94(3):271–274. doi:10.1136/bjo.2008.146753
95. Yu C, Huang L, Li X, Zhu H, Li Z, Yu X. Spatial and temporal differences of HMGB1 expression in the pancreas of rats with acute pancreatitis. Int J Clin Exp Pathol. 2015;8:6928–6935.
96. Xue J, Ge H, Lin Z, et al. The role of dendritic cells regulated by HMGB1/TLR4 signalling pathway in myocardial ischaemia reperfusion injury. J Cell Mol Med. 2019;23(4):2849–2862. doi:10.1111/jcmm.14192
97. Wang R, Wang P, Du G. [HMGB1 promotes myocardial ischemic injury and regulates the proportion of CD4+, CD8+T cells and Th17 cells in spleen through TLR4]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2018;34(9):794–799. (Chinese).
98. Ren S, Pan L, Yang L, et al. miR-29a-3p transferred by mesenchymal stem cells-derived extracellular vesicles protects against myocardial injury after severe acute pancreatitis. Life Sci. 2021;272:119189. doi:10.1016/j.lfs.2021.119189
99. Tzeng HP, Fan J, Vallejo JG, et al. Negative inotropic effects of high-mobility group box 1 protein in isolated contracting cardiac myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H1490–1496. doi:10.1152/ajpheart.00910.2007
100. Chen C, Xia SH, Chen H, Li XH. Therapy for acute pancreatitis with platelet-activating factor receptor antagonists. World J Gastroenterol. 2008;14(30):4735–4738. doi:10.3748/wjg.14.4735
101. Foitzik T, Hotz HG, Eibl G, Hotz B, Kirchengast M, Buhr HJ. Therapy for microcirculatory disorders in severe acute pancreatitis: effectiveness of platelet-activating factor receptor blockade vs. endothelin receptor blockade. J Gastrointest Surg. 1999;3(3):244–251. doi:10.1016/S1091-255X(99)80066-3
102. Lithell H, Vessby B, Walldius G, Carlson LA. Hypertriglyceridemia--acute pancreatitis--ischemic heart disease. A case study in a pair of monozygotic twins. Acta Med Scand. 1987;221(3):311–316. doi:10.1111/j.0954-6820.1987.tb00900.x
103. Copeland LA, Swendsen CS, Sears DM, MacCarthy AA, McNeal CJ. Association between triglyceride levels and cardiovascular disease in patients with acute pancreatitis. PLoS One. 2018;13(1):e0179998. doi:10.1371/journal.pone.0179998
104. Tsuang W, Navaneethan U, Ruiz L, Palascak JB, Gelrud A. Hypertriglyceridemic pancreatitis: presentation and management. Am J Gastroenterol. 2009;104:984–991. doi:10.1038/ajg.2009.27
105. Hendrickson SC, St Louis JD, Lowe JE, Abdel-aleem S. Free fatty acid metabolism during myocardial ischemia and reperfusion. Mol Cell Biochem. 1997;166(1–2):85–94. doi:10.1023/A:1006886601825
106. Wen L, Javed TA, Dobbs AK, et al. The protective effects of calcineurin on pancreatitis in mice depend on the cellular source. Gastroenterology. 2020;159(3):1036–1050.e8. doi:10.1053/j.gastro.2020.05.051
107. Ammori BJ, Leeder PC, King RF, et al. Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg. 1999;3:252–262. doi:10.1016/s1091-255x(99)80067-5
108. Tan C, Ling Z, Huang Y, et al. Dysbiosis of intestinal microbiota associated with inflammation involved in the progression of acute pancreatitis. Pancreas. 2015;44:868–875. doi:10.1097/MPA.0000000000000355
109. Schietroma M, Pessia B, Carlei F, Mariani P, Sista F, Amicucci G. Intestinal permeability and systemic endotoxemia in patients with acute pancreatitis. Ann Ital Chir. 2016;87:138–144.
110. Martínez J, Palazón JM, Muñoz C, et al. Endotoxin and anti-endotoxin antibodies in the prognosis of acute pancreatitis. Rev Esp Enferm Dig. 2002;94:406–416.
111. von der Möhlen MA, Kimmings AN, Wedel NI, et al. Inhibition of endotoxin-induced cytokine release and neutrophil activation in humans by use of recombinant bactericidal/permeability-increasing protein. J Infect Dis. 1995;172:144–151. doi:10.1093/infdis/172.1.144
112. van der Meer AJ, Scicluna BP, Moerland PD, et al. The selective sirtuin 1 activator SRT2104 reduces endotoxin-induced cytokine release and coagulation activation in humans. Crit Care Med. 2015;43:e199–202. doi:10.1097/CCM.0000000000000949
113. Shang X, Li J, Yu R, et al. Sepsis-related myocardial injury is associated with Mst1 upregulation, mitochondrial dysfunction and the Drp1/F-actin signaling pathway. J Mol Histol. 2019;50:91–103. doi:10.1007/s10735-018-09809-5
114. Wang X, Liu D, Chai W, Long Y, Su L, Yang R. The role of uncoupling protein 2 during myocardial dysfunction in a canine model of endotoxin shock. Shock. 2015;43:292–297. doi:10.1097/SHK.0000000000000286
115. Kawaguchi S, Okada M, Ijiri E, et al. β(3)-Adrenergic receptor blockade reduces mortality in endotoxin-induced heart failure by suppressing induced nitric oxide synthase and saving cardiac metabolism. Am J Physiol Heart Circ Physiol. 2020;318(2):H283–H294. doi:10.1152/ajpheart.00108.2019
116. Nežić L, Škrbić R, Amidžić L, Gajanin R, Kuča K, Jaćević V. Simvastatin protects cardiomyocytes against endotoxin-induced apoptosis and up-regulates survivin/NF-κB/p65 expression. Sci Rep. 2018;8:14652. doi:10.1038/s41598-018-32376-4
117. Shanbhag ST, Choong B, Petrov M, Delahunt B, Windsor JA, Phillips A. Acute pancreatitis conditioned mesenteric lymph causes cardiac dysfunction in rats independent of hypotension. Surgery. 2018;163(5):1097–1105. doi:10.1016/j.surg.2017.12.013
118. Pitanga TN, de Aragão França L, Rocha VC, et al. Neutrophil-derived microparticles induce myeloperoxidase-mediated damage of vascular endothelial cells. BMC Cell Biol. 2014;15:21. doi:10.1186/1471-2121-15-21
119. Kahrau S, Schneider P, Loddenkemper C, Buhr HJ, Foitzik T. Pulmonary microcirculation in mild and severe experimental pancreatitis. Eur Surg Res. 2003;35(5):402–407. doi:10.1159/000072173
120. Kolettis TM, Barton M, Langleben D, Matsumura Y. Endothelin in coronary artery disease and myocardial infarction. Cardiol Rev. 2013;21:249–256. doi:10.1097/CRD.0b013e318283f65a
121. Milnerowicz S, Milnerowicz H, Nabzdyk S, Jabłonowska M, Grabowski K, Taboła R. Plasma endothelin-1 levels in pancreatic inflammations. Adv Clin Exp Med. 2013;22:361–368.
122. Foitzik T, Eibl G, Hotz HG, Faulhaber J, Kirchengast M, Buhr HJ. Endothelin receptor blockade in severe acute pancreatitis leads to systemic enhancement of microcirculation, stabilization of capillary permeability, and improved survival rates. Surgery. 2000;128:399–407. doi:10.1067/msy.2000.107104
123. Kiviniemi H, Rämö J, Ståhlberg M, et al. Prostacyclin and thromboxane in acute hemorrhagic pancreatitis in dogs. J Surg Res. 1987;42:232–236. doi:10.1016/0022-4804(87)90138-7
124. Closa D, Rosello-Catafau J, Martrat A, et al. Changes of systemic prostacyclin and thromboxane A2 in sodium taurocholate- and cerulein-induced acute pancreatitis in rats. Dig Dis Sci. 1993;38:33–38. doi:10.1007/BF01296770
125. Komara NL, Paragomi P, Greer PJ, et al. Severe acute pancreatitis: capillary permeability model linking systemic inflammation to multiorgan failure. Am J Physiol Gastrointest Liver Physiol. 2020;319(5):G573–G583. doi:10.1152/ajpgi.00285.2020
126. Johnson CD. Platelet-activating factor and platelet-activating factor antagonists in acute pancreatitis. Dig Surg. 1999;16:93–101. doi:10.1159/000018699
127. de Stoppelaar SF, van ‘T Veer C, van der Poll T. The role of platelets in sepsis. Thromb Haemost. 2014;112(4):666–677. doi:10.1160/TH14-02-0126
128. Xu W, Qi X, Chen J, Su C, Guo X. Prevalence of splanchnic vein thrombosis in pancreatitis: a systematic review and meta-analysis of observational studies. Gastroenterol Res Pract. 2015;2015:245460. doi:10.1155/2015/245460
129. Salomone T, Tosi P, Palareti G, et al. Coagulative disorders in human acute pancreatitis: role for the D-dimer. Pancreas. 2003;26(2):111–116. doi:10.1097/00006676-200303000-00003
130. Lindsay TH, Halvorson KG, Peters CM, et al. A quantitative analysis of the sensory and sympathetic innervation of the mouse pancreas. Neuroscience. 2006;137:1417–1426. doi:10.1016/j.neuroscience.2005.10.055
131. Xu J, Wang Z, Ma G, et al. Endogenous catecholamine stimulates alveolar fluid clearance in rats with acute pancreatitis. Respirology. 2009;14:195–202. doi:10.1111/j.1440-1843.2008.01453.x
132. Wallner M, Duran JM, Mohsin S, et al. Acute catecholamine exposure causes reversible myocyte injury without cardiac regeneration. Circ Res. 2016;119:865–879. doi:10.1161/CIRCRESAHA.116.308687
133. Du Toit EF, Nabben M, Lochner A. A potential role for angiotensin II in obesity induced cardiac hypertrophy and ischaemic/reperfusion injury. Basic Res Cardiol. 2005;100:346–354. doi:10.1007/s00395-005-0528-5
134. Frolkis I, Gurevitch J, Yuhas Y, et al. Interaction between paracrine tumor necrosis factor-alpha and paracrine angiotensin II during myocardial ischemia. J Am Coll Cardiol. 2001;37:316–322. doi:10.1016/s0735-1097(00)01055-x
135. Lieberman JS, Taylor A, Wright IS. The effect of intravenous trypsin administration on the electrocardiogram of the rabbit. Circulation. 1954;10(3):338–342. doi:10.1161/01.CIR.10.3.338
136. Manning GW, Hall GE, Banting FG. Vagus stimulation and the production of myocardial damage. Can Med Assoc J. 1937;37(4):314–318.
137. Essandoh K, Wang X, Huang W, et al. Tumor susceptibility gene 101 ameliorates endotoxin-induced cardiac dysfunction by enhancing Parkin-mediated mitophagy. J Biol Chem. 2019;294(48):18057–18068. doi:10.1074/jbc.RA119.008925
138. Nah J, Fernández ÁF, Kitsis RN, Levine B, Sadoshima J. Does autophagy mediate cardiac myocyte death during stress. Circ Res. 2016;119:893–895. doi:10.1161/CIRCRESAHA.116.309765
139. Buttenschoen K, Berger D, Hiki N, et al. Endotoxin and antiendotoxin antibodies in patients with acute pancreatitis. Eur J Surg. 2000;166:459–466. doi:10.1080/110241500750008772
140. Windsor JA, Fearon KC, Ross JA, et al. Role of serum endotoxin and antiendotoxin core antibody levels in predicting the development of multiple organ failure in acute pancreatitis. Br J Surg. 1993;80:1042–1046. doi:10.1002/bjs.1800800840
141. Bose SM, Verma GR, Mazumdar A, Giridhar M, Ganguly NK. Significance of serum endotoxin and antiendotoxin antibody levels in predicting the severity of acute pancreatitis. Surg Today. 2002;32:602–607. doi:10.1007/s005950200109
142. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res. 2016;111:534–544. doi:10.1016/j.phrs.2016.06.031
143. Xu GF, Guo M, Tian ZQ, Wu GZ, Zou XP, Zhang WJ. Increased of serum high-mobility group box chromosomal protein 1 correlated with intestinal mucosal barrier injury in patients with severe acute pancreatitis. World J Emerg Surg. 2014;9:61. doi:10.1186/1749-7922-9-61
144. Li L, Sun Z, Xu C, et al. Adenovirus-mediated overexpression of sST2 attenuates cardiac injury in the rat with severe acute pancreatitis. Life Sci. 2018;202:167–174. doi:10.1016/j.lfs.2018.04.008
145. Liu X, Hu Y, Huang W, et al. Soluble ST2 for prediction of clinical outcomes in patients with ST-segment elevation myocardial infarction receiving primary PCI. Int Heart J. 2019;60(1):19–26. doi:10.1536/ihj.18-020
146. Iovanna J, Orelle B, Keim V, Dagorn JC. Messenger RNA sequence and expression of rat pancreatitis-associated protein, a lectin-related protein overexpressed during acute experimental pancreatitis. J Biol Chem. 1991;266(36):24664–24669. doi:10.1016/S0021-9258(18)54281-2
147. Iovanna JL, Keim V, Nordback I, et al. Serum levels of pancreatitis-associated protein as indicators of the course of acute pancreatitis. Multicentric Study Group on Acute Pancreatitis. Gastroenterology. 1994;106(3):728–734. doi:10.1016/0016-5085(94)90708-0
148. Fitzgibbons TP, Paolino J, Dagorn JC, Meyer TE. Usefulness of pancreatitis-associated protein, a novel biomarker, to predict severity of disease in ambulatory patients with heart failure. Am J Cardiol. 2014;113(1):123–126. doi:10.1016/j.amjcard.2013.09.026
149. Yasuda T, Takeyama Y, Ueda T, et al. Increased levels of soluble triggering receptor expressed on myeloid cells-1 in patients with acute pancreatitis. Crit Care Med. 2008;36(7):2048–2053. doi:10.1097/CCM.0b013e31817b8824
150. Wang YK, Tang JN, Shen YL, et al. Prognostic utility of soluble TREM-1 in predicting mortality and cardiovascular events in patients with acute myocardial infarction. J Am Heart Assoc. 2018;7(12). doi:10.1161/JAHA.118.008985
151. Johansson P, Dahlström U, Broström A. The measurement and prevalence of depression in patients with chronic heart failure. Prog Cardiovasc Nurs. 2006;21(1):28–36. doi:10.1111/j.0197-3118.2006.04644.x
152. Pontet J, Contreras P, Curbelo A, et al. Heart rate variability as early marker of multiple organ dysfunction syndrome in septic patients. J Crit Care. 2003;18(3):156–163. doi:10.1016/j.jcrc.2003.08.005
153. Zhang L, Zhou J, Ke L, et al. Role of heart rate variability in predicting the severity of severe acute pancreatitis. Dig Dis Sci. 2014;59(10):2557–2564. doi:10.1007/s10620-014-3192-5
154. Chen P, Hu B, Tan Q, et al. Role of neurocrine somatostatin on sphincter of Oddi contractility and intestinal ischemia reperfusion-induced acute pancreatitis in macaques. Neurogastroenterol Motil. 2010;22(8):935–41, e240. doi:10.1111/j.1365-2982.2010.01506.x
155. Altimari AF, Prinz RA, Leutz DW, Sandberg L, Kober PM, Raymond RM. Myocardial depression during acute pancreatitis: fact or fiction. Surgery. 1986;100(4):724–731.
156. Kawamura T, Nara N, Kadosaki M, Inada K, Endo S. Prostaglandin E1 reduces myocardial reperfusion injury by inhibiting proinflammatory cytokines production during cardiac surgery. Crit Care Med. 2000;28(7):2201–2208. doi:10.1097/00003246-200007000-00004
157. Xiping Z, Hua T, Hanqing C, et al. The protecting effects and mechanisms of Baicalin and Octreotide on heart injury in rats with SAP. Mediators Inflamm. 2007;2007:19469. doi:10.1155/2007/19469
158. Wang Y, Chen M. Fentanyl ameliorates severe acute pancreatitis-induced myocardial injury in rats by regulating NF-κB signaling pathway. Med Sci Monit. 2017;23:3276–3283. doi:10.12659/MSM.902245
159. Fogel EL, Lehman GA, Tarnasky P, et al. Rectal indometacin dose escalation for prevention of pancreatitis after endoscopic retrograde cholangiopancreatography in high-risk patients: a double-blind, randomised controlled trial. Lancet Gastroenterol Hepatol. 2020;5(2):132–141. doi:10.1016/S2468-1253(19)30337-1
160. Ikeda S, Kagami T, Tani S, et al. Decompressive laparotomy for abdominal compartment syndrome resulting from severe acute pancreatitis: a case report. BMC Gastroenterol. 2019;19(1):141. doi:10.1186/s12876-019-1059-0
161. Wen Y, Sun HY, Tan Z, et al. Abdominal paracentesis drainage ameliorates myocardial injury in severe experimental pancreatitis rats through suppressing oxidative stress. World J Gastroenterol. 2020;26(1):35–54. doi:10.3748/wjg.v26.i1.35
162. Zhao G, Zhang JG, Wu HS, et al. Effects of different resuscitation fluid on severe acute pancreatitis. World J Gastroenterol. 2013;19(13):2044–2052. doi:10.3748/wjg.v19.i13.2044
163. Huang F, Meng H, Sun Y, et al. Factors causing abnormal heart failure after successful fluid resuscitation on acute pancreatitis patients. Pancreas. 2020;49(4):e33–e35. doi:10.1097/MPA.0000000000001532
164. Ye B, Mao W, Chen Y, et al. Aggressive resuscitation is associated with the development of acute kidney injury in acute pancreatitis. Dig Dis Sci. 2019;64(2):544–552. doi:10.1007/s10620-018-5328-5
165. Chen Y, Ma L, Song X, Fei J, Chen E, Mao E. Beneficial effects of fluid resuscitation via the rectum on hemodynamic disorders and multiple organ injuries in an experimental severe acute pancreatitis model. Pancreatology. 2015;15(6):626–634. doi:10.1016/j.pan.2015.09.001
166. Gombocz K, Beledi A, Alotti N, et al. Influence of dextran-70 on systemic inflammatory response and myocardial ischaemia-reperfusion following cardiac operations. Crit Care. 2007;11(4):R87. doi:10.1186/cc6095
167. Zhang XP, Feng GH, Zhang J, et al. Protective effects of Salvia miltiorrhizae on the hearts of rats with severe acute pancreatits or obstructive jaundice. J Zhejiang Univ Sci B. 2009;10(3):193–202. doi:10.1631/jzus.B0820179
168. Nagao S, Taguchi K, Sakai H, et al. Carbon monoxide-bound hemoglobin vesicles ameliorate multiorgan injuries induced by severe acute pancreatitis in mice by their anti-inflammatory and antioxidant properties. Int J Nanomedicine. 2016;11:5611–5620. doi:10.2147/IJN.S118185
169. Zhou R, Weng F, Dai W, Yan J. [The organ protective effects and timing of continuous blood purification in the treatment of severe sepsis: a double-blind randomized controlled trial]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2016;28(3):241–245. (Chinese).
170. Hirota M, Takada T, Kitamura N, et al. Fundamental and intensive care of acute pancreatitis. J Hepatobiliary Pancreat Sci. 2010;17(1):45–52. doi:10.1007/s00534-009-0210-7
171. Tani T, Fujino M, Hanasawa K, Shimizu T, Endo Y, Kodama M. Bacterial translocation and tumor necrosis factor-alpha gene expression in experimental hemorrhagic shock. Crit Care Med. 2000;28(11):3705–3709. doi:10.1097/00003246-200011000-00028
172. Oláh A, Romics L
173. Yao P, Cui M, Li Y, Deng Y, Wu H. Effects of rhubarb on intestinal flora and toll-like receptors of intestinal mucosa in rats with severe acute pancreatitis. Pancreas. 2015;44(5):799–804. doi:10.1097/MPA.0000000000000339
174. Bateman RM, Sharpe MD, Jagger JE, et al. 36th International Symposium on Intensive Care and Emergency Medicine: Brussels, Belgium. 15-18 March 2016. Crit Care. 2016:20(Suppl 2):94. doi:10.1186/s13054-016-1208-6
175. Wang S, Cheng ZY, Chen XJ, Xue HZ. Ulinastatin protects rats with myocardial infarction by activating Nrf2/NOS pathway. Eur Rev Med Pharmacol Sci. 2018;22(24):8990–8998. doi:10.26355/eurrev_201812_16670
176. Tian H, Zhang M, Du C, et al. Effects of Rhubarb combined with ulinastatin on T-cell subsets in sepsis rats. Int J Clin Exp Med. 2015;8(1):1234–1240.
177. Wang G, Liu Y, Zhou SF, et al. Effect of somatostatin, ulinastatin and gabexate on the treatment of severe acute pancreatitis. Am J Med Sci. 2016;351(5):506–512. doi:10.1016/j.amjms.2016.03.013
178. Htwe T, Suzuki M, Ouchi K, Fukuhara K, Matsuno S. Effects of urinastatin and free radical scavengers on hepatic lipid peroxidation in endotoxemia. J Surg Res. 1996;61(1):206–214. doi:10.1006/jsre.1996.0106
179. Chapman MJ. From pathophysiology to targeted therapy for atherothrombosis: a role for the combination of statin and aspirin in secondary prevention. Pharmacol Ther. 2007;113(1):184–196. doi:10.1016/j.pharmthera.2006.08.005
180. Yu Y, Jin L, Zhuang Y, Hu Y, Cang J, Guo K. Cardioprotective effect of rosuvastatin against isoproterenol-induced myocardial infarction injury in rats. Int J Mol Med. 2018;41(6):3509–3516. doi:10.3892/ijmm.2018.3572
181. Ke D, Fang J, Fan L, Chen Z, Chen L. Regulatory T cells contribute to rosuvastatin-induced cardioprotection against ischemia-reperfusion injury. Coron Artery Dis. 2013;24(4):334–341. doi:10.1097/MCA.0b013e3283608c12
182. Ellison DH, Felker GM. Diuretic treatment in heart failure. N Engl J Med. 2017;377(20):1964–1975. doi:10.1056/NEJMra1703100
183. Nitsche C, Maertin S, Scheiber J, Ritter CA, Lerch MM, Mayerle J. Drug-induced pancreatitis. Curr Gastroenterol Rep. 2012;14(2):131–138. doi:10.1007/s11894-012-0245-9
184. Hollenberg SM, Ahrens TS, Annane D, et al. Practice parameters for hemodynamic support of sepsis in adult patients: 2004 update. Crit Care Med. 2004;32(9):1928–1948. doi:10.1097/01.CCM.0000139761.05492.D6
185. Chanani NK, Cowan DB, Takeuchi K, et al. Differential effects of amrinone and milrinone upon myocardial inflammatory signaling. Circulation. 2002;106(12 Suppl 1):I284–9.
186. Yamashita S, Suzuki T, Iguchi K, et al. Cardioprotective and functional effects of levosimendan and milrinone in mice with cecal ligation and puncture-induced sepsis. Naunyn Schmiedebergs Arch Pharmacol. 2018;391(9):1021–1032. doi:10.1007/s00210-018-1527-z
187. Li L, Li YQ, Sun ZW, et al. Qingyi decoction protects against myocardial injuries induced by severe acute pancreatitis. World J Gastroenterol. 2020;26(12):1317–1328. doi:10.3748/wjg.v26.i12.1317
188. Li N, Tian Y, Wang C, Zhang P, You S. Protective effect of Lai Fu Cheng Qi decoction on severe acute pancreatitis-induced myocardial injury in a rat model. Exp Ther Med. 2015;9(4):1133–1140. doi:10.3892/etm.2015.2250
189. Miao YF, Kang HX, Li J, et al. Effect of Sheng-jiang powder on multiple-organ inflammatory injury in acute pancreatitis in rats fed a high-fat diet. World J Gastroenterol. 2019;25(6):683–695. doi:10.3748/wjg.v25.i6.683
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.