")
Back to Journals » Journal of Experimental Pharmacology » Volume 8
Comprehensive kinase profile of pacritinib, a nonmyelosuppressive Janus kinase 2 inhibitor
Authors Singer J, Al-Fayoumi S , Ma H, Komrokji R, Mesa R, Verstovsek S
Received 16 April 2016
Accepted for publication 21 June 2016
Published 16 August 2016 Volume 2016:8 Pages 11—19
DOI https://doi.org/10.2147/JEP.S110702
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Jack W Singer,1 Suliman Al-Fayoumi,1 Haiching Ma,2 Rami S Komrokji,3 Ruben Mesa,4 Srdan Verstovsek5
1Translational Medicine, CTI BioPharma Corp., Seattle, WA, 2Department of Research and Development, Reaction Biology, Malvern, PA, 3Department of Malignant Hematology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, 4Division of Hematology and Medical Oncology, Mayo Clinic Cancer Center, Scottsdale, AZ, 5Department of Leukemia, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract: Pacritinib, potent inhibitor of Janus kinase 2 (JAK2), JAK2V617F, and fms-like receptor tyrosine kinase 3, is in Phase III development in myelofibrosis. Among type 1 inhibitors, pacritinib shows a lack of myelosuppression at doses that both inhibit JAK2/signal transducer and activator of transcription 3 pathway and demonstrate clinical efficacy. To elucidate these mechanisms and identify other disease targets, a kinome analysis screened 439 recombinant kinases at 100 nM pacritinib concentration. For kinases with >50% inhibition, pacritinib was titrated from 1 to 100 nM. JAK2, JAK2V617F, FLT3, colony-stimulating factor 1 receptor, and interleukin-1 receptor-associated kinase 1 achieved half-maximal inhibitory concentrations <50 nM. Pacritinib did not inhibit JAK1 (82% control at 100 nM). Lack of myelosuppression may stem from inhibiting JAK2 without affecting JAK1 and reducing hematopoietic inhibitory cytokines by suppressing interleukin-1 receptor-associated kinase 1 or colony-stimulating factor 1 receptor. The pacritinib kinome suggests therapeutic utility in acute myeloid leukemia, myelodysplastic syndrome, chronic myelomonocytic leukemia, solid tumors, and inflammatory conditions.
Keywords: kinase analysis, myelofibrosis, hematologic malignancies, Janus kinase 2, JAK2V617F, fms-like receptor tyrosine kinase 3
Introduction
Janus kinase 2 (JAK2) is involved in the signaling cascades critical for maintaining normal hematopoiesis. JAK2 is activated by cytokines that control granulopoiesis (granulocyte-colony stimulating factor, interleukin [IL]-3, granulocyte macrophage-colony stimulating factor), erythropoiesis (erythropoietin), thrombopoiesis (thrombopoietin), eosinopoiesis (IL-5), and T-cell differentiation signaling (IL-12).1 Inhibition of JAK1 and JAK2, such as by ruxolitinib, decreases the activation of signal transducer and activator of transcription (STAT)3/5, and while helping patients with myelofibrosis by improving splenomegaly and quality of life, it suppresses erythropoiesis, myelopoiesis, and thrombopoiesis, resulting in dose-related anemia, neutropenia, and thrombocytopenia in clinical studies.2,3
Pacritinib, a novel inhibitor of both JAK2 and fms-like receptor tyrosine kinase 3 (FLT3), was developed as a selective JAK2/FLT3 inhibitor with minimal suppression of JAK1.4 It has demonstrated promising antitumor activity in lymphoid and myeloproliferative neoplasms in both preclinical studies5,6 and clinical trials.7–11 Evaluation of pacritinib in preclinical models of advanced myeloid malignancies and myelofibrosis demonstrated pharmacological activity. Two Phase I–II studies in patients with primary or secondary myelofibrosis showed that pacritinib reduced splenomegaly and symptom scores and could be used safely in patients regardless of baseline platelet counts. Interestingly, neither evidence of treatment-related decline in platelet counts12,13 nor subsequent increase in anemia was reported. These data were recently confirmed in a Phase III trial.14 Clinical trials of a majority of other JAK inhibitors that are less selective for JAK2 report dose-related anemia and/or thrombocytopenia.
The JAK2 mutation JAK2V617F is frequently found in patients with myeloproliferative neoplasms, occurring in almost all patients with polycythemia vera and in approximately half of the patients with essential thrombocythemia and idiopathic myelofibrosis.1,15 This gain-of-function mutation results in the expression of a constitutively activated JAK2.16 In most of the patients with germ-line JAK2, other mutations that activate this pathway have been recently discovered, including mutations in calreticulin and the thrombopoietin receptor gene (MPL).17
As an inhibitor of FLT3, pacritinib may have utility in the treatment of leukemia. A family of class III receptor tyrosine kinases, including c-fms, c-Kit, FLT3, and platelet-derived growth factor receptors α and β, are important in the maintenance, growth, and development of hematopoietic and nonhematopoietic cells.18 In acute myeloid leukemia (AML), FLT3 mutations are the most frequent genetic mutations and are involved in the signaling pathway of autonomous proliferation and differentiation block in leukemia cells.19 In addition, several clinical studies have confirmed that FLT3 internal tandem duplications are strongly associated with a poor prognosis.19 Because high-dose chemotherapy and stem cell transplantation cannot overcome the adverse effects of FLT3 mutations,19 the development of FLT3 inhibitors is a promising therapeutic strategy. Although JAK2V617F mutation rarely occurs in de novo AML, STAT3 activation is common.20 Since STAT proteins are phosphorylated and activated by JAKs, the frequent pSTAT activation in AML suggests the involvement of JAK2 extrinsic regulators and other proteins in the JAK–STAT pathway. In addition, JAK–STAT represents one alternate pathway by which leukemic cells circumvent FLT3 inhibition. In vitro studies show that FLT3 inhibitors upregulate the JAK–STAT pathway and that JAK2 inhibition may overcome resistance to FLT3 inhibition, suggesting that dual inhibition may improve outcomes in AML.5
To help elucidate the mechanisms underlying pacritinib’s lack of hematopoietic suppression despite its low nanomolar inhibition of JAK2/STAT3 and to identify other target kinases, we performed a kinome-wide screen to evaluate its spectrum of kinase inhibition.
Methods
Materials
Pacritinib was provided by CTI BioPharma, Corp. (Seattle, WA, USA). Other kinase inhibitors outlined in Table S1 were obtained either from Selleckchem (Houston, TX, USA) or Sigma-Aldrich Co. (St Louis, MO, USA), with an average purity of >98%. Kinases were purchased from Thermo Fisher Scientific (Waltham, MA, USA), SignalChem (Richmond, BC, Canada), ProQinase GmbH (Freiburg, Germany), or Carna Biosciences Inc. (Kobe, Japan).
Kinase assay methods
In vitro profiling of the 439-member kinase panel was performed at Reaction Biology Corporation (Malvern, PA, USA) using the HotSpot assay platform.21 Pacritinib was tested through the full kinase panel to confirm both its activities in relation to its primary targets (JAK2, JAK2V617F, and FLT3) and its activities in relation to kinases not in our original conceptualization. The gold standard for kinase profiling, HotSpot technology, is a miniaturized assay platform for radioisotope-based filter binding.
Briefly, kinase–substrate pairs along with the required cofactors were prepared in a reaction buffer containing 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.5), 10 mM magnesium chloride (MgCl2), 1 mM ethylene glycol tetraacetic acid, 0.02% Brij 35® (G-Biosciences, St. Louis, MO, USA), 0.02 mg/mL
bovine serum albumin, 0.1 mM sodium orthovanadate (Na3VO4), 2 mM dithiothreitol, and 1% dimethyl sulfoxide. Small test molecules were then added into the reaction mixture, and approximately 20 minutes later, 10 μM of a mixture of nonradiolabeled adenosine triphosphate (ATP) and 33P-labeled ATP was combined to start the reaction. Reactions were carried out at 25°C for 120 minutes. The reaction mixtures were then spotted onto P81 ion-exchange filter paper. The dry filters were then washed in 0.75% phosphoric acid to remove any unbound phosphate. After subtracting the background from the control reactions, which contained the inactive enzyme, substrate, and radiolabeled ATP, kinase activity was expressed as the percent of kinase activity remaining in the test samples compared to the vehicle (dimethyl sulfoxide) reactions.
For the screening assays, pacritinib was tested at 100 nM. For all kinases that were inhibited by >50% at 100 nM, graduated dosing from 1 to 100 nM was performed. The half maximal inhibitory concentration (IC50) values and curve fits were obtained using Prism® (GraphPad Software, Inc., La Jolla, CA, USA). Kinome tree representations were prepared using Kinome Activity Mapper (Reaction Biology Corporation, Malvern, PA, USA). For reference, mean peak plasma concentrations of free pacritinib at steady state were calculated after administering the standard daily dose of 400 mg.
Statistical methods
Outlier detection
Raw data were measured as the percentage of compound activity for each kinase–substrate pair in duplicate. Negative values were truncated to zero, and any pairs with missing observations or duplicate values were removed from the analysis. For each pair, the coefficient of variation was calculated, and the difference (D) from duplicate observations used kernel density estimation and quantile–quantile plots to be double exponentially distributed.21 Using maximum likelihood methods,22 location and scale parameters were estimated, as were mean and standard deviation. To account for the noise inherent in assay measurements, any observation within 1 standard deviation of the mean distribution of D was further analyzed for compound activity.21
Based on the resulting data set, the method to detect distribution-based outliers was then applied to the coefficient of variation.23 To detect outliers, data (except for the highest and lowest 1%) were positioned onto the quantile–quantile plot for log-normal distribution21 and the data parameters were robustly estimated.22 To determine whether extreme data points were outliers, we computed the threshold to set the expected range of observations. Any result beyond the threshold of the coefficient of variation was considered an outlier and excluded from further analysis.
Hierarchical clustering
For data on kinase activity, hierarchical clustering was conducted as described previously.21
Results
Pacritinib inhibited kinases in several unrelated families in the <50 nM range (Table 1). Pacritinib suppressed all members of the JAK/FLT pathways at low nanomolar concentrations, except for JAK1, on which it was inactive at 100 nM (82% control). For reference, after administering the daily dose of 400 mg, the mean peak plasma concentrations of pacritinib free drug at steady state equaled approximately 200 nM, while the total steady-state levels of pacritinib were in the 5–7 µM range (Figure 1). Hence, the 100 nM concentration of pacritinib evaluated in this study is readily achievable clinically.
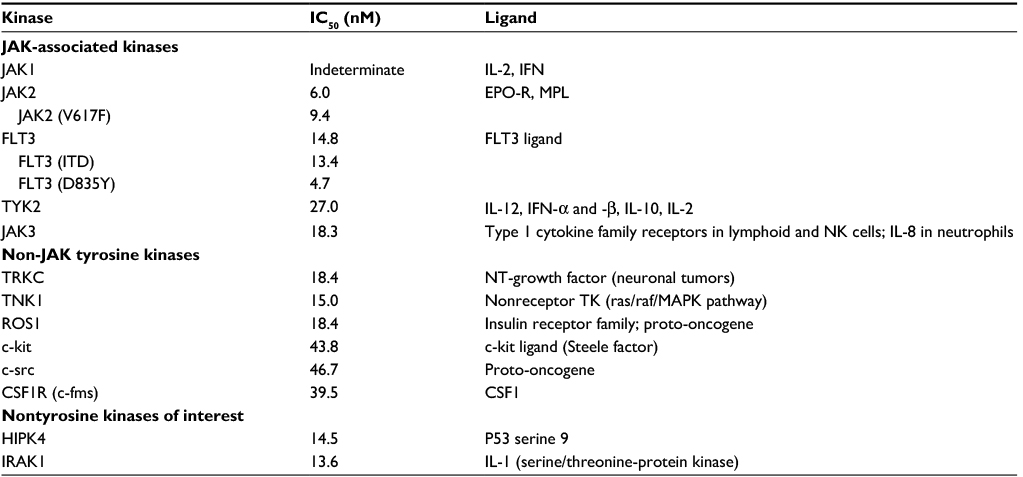 | Table 1 Kinase inhibition profile for pacritinib at 100 nM concentrationa Note: aKinases with IC50 <50 nM are shown. Abbreviations: CSF1R, colony-stimulating factor 1 receptor; EPO-R, erythropoietin receptor; FLT3, fms-like receptor tyrosine kinase 3; HIPK4, homeodomain-interacting protein kinase 4; IC50, half maximal inhibitory concentration; IFN, interferon; IL, interleukin; IRAK1, interleukin-1 receptor-associated kinase 1; ITD, internal tandem duplication; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; MPL, thrombopoietin receptor gene; NK, natural killer; NT, neurotrophin; TK, tyrosine kinase; TYK2, tyrosine kinase 2; TNK1, tyrosine kinase nonreceptor 1; TRKC, Tropomyosin receptor kinase C. |
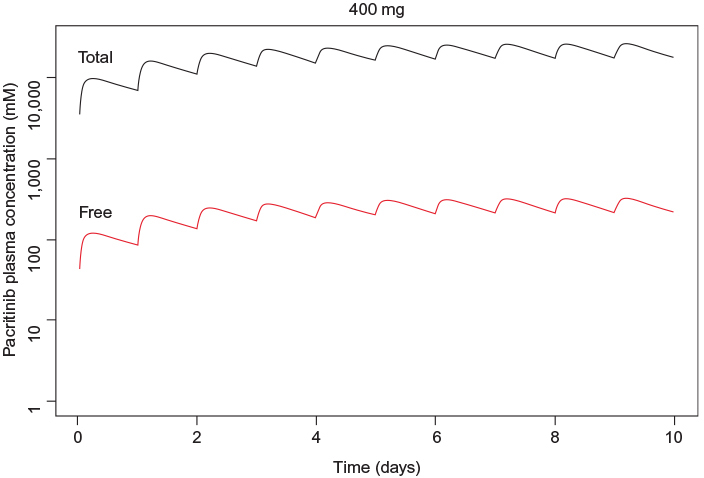 | Figure 1 Simulated steady-state peak plasma concentration (Cmax) for plasma protein-bound and free pacritinib after administration of 400 mg daily dosage of pacritinib in humans. |
Along with the anticipated inhibition of JAK2, FLT3, and their respective common mutations, pacritinib also inhibited several kinases in other families identified as potential therapeutic targets in oncology, including IL-1 receptor-associated kinase 1 (IRAK1) and c-fms (colony-stimulating factor 1 receptor [CSF1R]),24 ROS1,25 and tyrosine kinase nonreceptor 1.26 The kinome map (Figure 2) shows relative pacritinib inhibition, demonstrating a high level of inhibitory activity against specific kinases in disparate families.
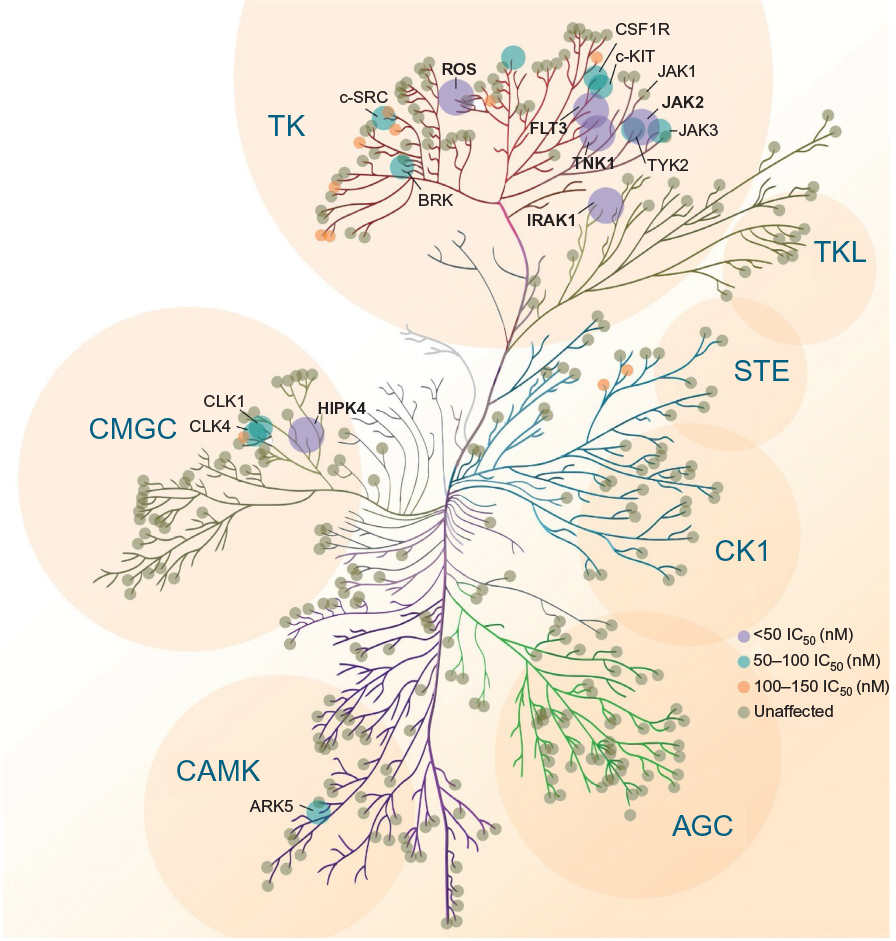 | Figure 2 Pacritinib activity kinome map. Abbreviations: AGC, protein kinase A, G, C group; ARK, Beta-adrenergic receptor kinase; BRK, breast tumor kinase; CK1, casein kinase; CLK, CDC Like Kinase 1; CSF1R, colony-stimulating factor 1 receptor; FLT3, fms-like receptor tyrosine kinase 3; HIPK4, homeodomain-interacting protein kinase 4; IC50, half maximal inhibitory concentration; IRAK1, interleukin-1 receptor-associated kinase 1; JAK, Janus kinase; STE, homolog of sterile; TK, tyrosine kinase; TKL, tyrosine kinase-like group of kinases; TNK1, tyrosine kinase nonreceptor 1; TYK2, tyrosine kinase 2. |
Phosphorylated CSF1R at concentrations <1 µM was inhibited in studies of normal human macrophages and macrophages from patients with chronic lymphocytic leukemia.27 In addition, phosphorylated IRAK1 was inhibited in myeloid leukemia cell lines and fresh samples from patients with acute myelogenous leukemia at concentrations <500 nM.28
Discussion
Since 2001, more than 20 kinase inhibitors have received approval from the US Food and Drug Administration for the treatment of patients with cancers and inflammatory diseases. Most of them are type 1 inhibitors that target the ATP binding site. With the identification of the common JAK2 mutation (V617F), a number of type 1 kinase inhibitors entered development. Ruxolitinib, a JAK1 and JAK2 inhibitor, was the first to receive regulatory approval for the treatment of intermediate- or high-risk myelofibrosis and hydroxyurea-resistant polycythemia vera.29 Treatment with ruxolitinib improves disease-related symptoms, reduces spleen size in both diseases, and appears to be associated with a survival advantage. However, ruxolitinib is associated with dose-related anemia and thrombocytopenia and its use is not recommended in patients with severe thrombocytopenia.2,30 Pacritinib is also a type 1 ATP-binding site inhibitor, but unlike ruxolitinib and most other JAK inhibitors in development (Table 2),31 pacritinib shows specificity for JAK2 and does not affect JAK1. In clinical studies using oral, once-daily dosing at 400 mg, pacritinib achieves steady-state concentrations of approximately 200 nM of free drug (Figure 1) – substantially above the IC50 values of the other recombinant kinases shown – and in most patients, pacritinib does not appear to cause either thrombocytopenia or anemia.
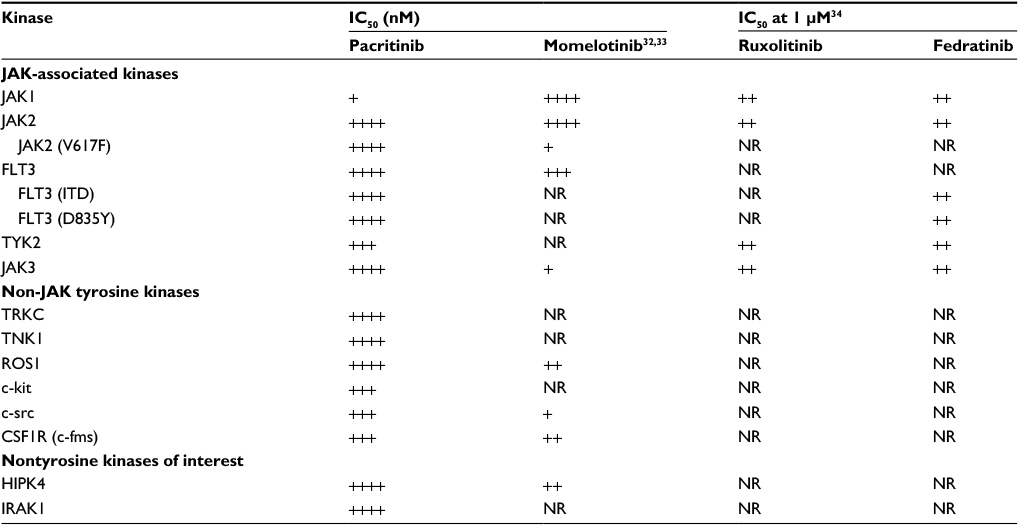 | Table 2 Relative kinase profiles comparing pacritinib, momelotinib, ruxolitinib, and fedratinib Note: Pacritinib scale: ++++, <25 nM; +++, 25 to <50 nM; +, >100 nM. Momelotinib scale: ++++, <25 nM32; +++, <100 nM; ++, <1 µM; +, >1 µM.33 Ruxolitinib and fedratinib scales: ++, IC50 at 1 µM. Abbreviations: CSF1R, colony-stimulating factor 1 receptor; FLT3, fms-like receptor tyrosine kinase 3; HIPK4, homeodomain-interacting protein kinase 4; IC50, half maximal inhibitory concentration; IRAK1, interleukin-1 receptor-associated kinase 1; ITD, internal tandem duplication; JAK, Janus kinase; NR, not reported; TNK1, tyrosine kinase nonreceptor 1; TYK2, tyrosine kinase 2. |
The published results of kinase inhibition profiles of other JAK2 inhibitors in development indicate that except for pacritinib, all inhibit JAK1, JAK2, and JAK3.32,34 Unfortunately, kinome-wide screening has not been published for JAK inhibitors, except for momelotinib. Our kinome analysis discovered important inhibitory effects on these formerly overlooked kinases, which adds dimension to the mechanisms of action and potential targets with therapeutic benefit of pacritinib.
Of the type 1 inhibitors for myeloproliferative disorders, ruxolitinib shows low nanomolar inhibition of JAK1, the kinase triggered by the activation of interferon, IL-6, IL-2, and other lymphokines, which is important in signaling by innate and acquired immune responses.32,35 Suppression of JAK1 phosphorylation is associated with a decreased ability to clear viral infections.32,35 Moreover, suppression of JAK1 signaling decreases natural killer and dendritic cell function, further impairing the immune response.36,38 Suppression of these signaling pathways may be associated with the opportunistic infections that have been associated with ruxolitinib therapy.2,3,38,39 The lack of suppression of signaling mediated by JAK1 may have a role in the lack of myelosuppression associated with pacritinib, although the mechanism remains to be elucidated.
The suppression of inflammatory pathways has been associated with the production of hematopoietic inhibitory cytokines such as members of the tumor necrosis factor family, transforming growth factor beta, and macrophage inflammatory protein 1 alpha.24 Although pacritinib does not suppress signaling through JAK1 but does produce early-onset reduction in symptoms associated with myelofibrosis (data on file), our current results suggest that this may occur due to its inhibition of inflammatory pathways mediated by IRAK1 and CSF1R. Serine–threonine kinase IRAK1 is a critical effector of the myeloproliferative diseases associated with the loss of microRNA-146a and the dysregulation of NF-ϰB.40,41 IRAK1 is activated by Toll-like receptors through a complex with MYD88,42 the main system for recognizing foreign stimuli such as lipopolysaccharide for the innate immune system, and has a critical role in the inflammatory response.36 IRAK1 may prove to be a critical signal in the interaction between oncogenesis and the inflammatory response. Moreover, activating mutations in MYD88 are associated with a number of neoplastic diseases, including Waldenström macroglobulinemia.42 IRAK1 has also been recently identified as a therapeutic target in myelodysplastic syndromes (MDS)24 and AML.43 In AML, IRAK1 may have a role in both stromal–tumor interactions and stromal protection from leukemic cells from AML-directed therapy.24 In primary MDS, inhibiting IRAK1 in marrow or cell lines delayed proliferation and compromised MDS progenitor function.24 Recent results demonstrated that pacritinib inhibits the phosphorylation of IRAK1 in AML cells and reduces both the viability and survival of AML cell lines harboring various mutations.44 IRAK1 has also been shown to be upregulated in chronic myelomonocytic leukemia (CMML); pacritinib was found to have activity in primary CMML cells and synergistic activity when combined with 5-azacitidine.45
Activation of CSF1R, which is also inhibited by pacritinib, may also have an anti-inflammatory effect.46 In addition, CSF1R activation has been shown to have an important role in interactions between nurse-like cells in protecting low-grade lymphomas and chronic lymphocytic leukemia cells from therapeutic interventions and supporting proliferation.47 Recently, a CSF1R inhibitor has shown clinical activity in the treatment of tenosynovial giant cell tumors.48
Along with its lack of myelosuppression, the potential for pacritinib to interfere with microenvironmental tumor interactions through the suppression of IRAK1 and CSF1R suggests its potential for use in combination with established therapies in a variety of liquid and solid tumors. Pacritinib also inhibits the phosphorylation of CSF1R (c-fms), a relevant target in lymphoid and myeloproliferative neoplasms and potentially inflammatory autoimmune diseases.49
Other targets identified in the pacritinib kinome (Table 1) underlie other cancers and may prove important for their treatment. For non-JAK/FLT3 tyrosine kinases, ROS1 showed promising response to pacritinib and its ligand is a proto-oncogene that activates fusions in non-small-cell lung cancer and cholangiocarcinoma. Among non-JAK tyrosine kinases, CSF1R ligand accounts for mutations in CMML and AML and interacts with nurse-like cells in chronic lymphocytic leukemia and mantle cell lymphoma. For nontyrosine kinases of interest, IRAK1 is a proven target in MDS and CMML. These potential indications are under exploration in ongoing clinical trials.50,53 Preliminary results from an ongoing clinical trial of pacritnib monotherapy in relapsed/primary refractory FLT3-mutated AML demonstrated an overall response rate of 17% (4/23), including three complete remissions with incomplete blood count recovery and one partial remission at the 28-day assessment period.54 Most toxicities were low grade (grade 1–2) and the most frequent were nausea/vomiting (53%), diarrhea (33%), and increased alanine aminotransferase (30%).54
The major limitations of this study include the acellular nature of the screening assays and the need to validate that the kinase inhibition identified has similar effects in neoplastic cells. This has been done for two of the kinases of most interest, CSF1R in human monocytes and IRAK1 in myeloid leukemic cell lines and fresh cells from patients with AML,28 both of which will be fully described in other manuscripts. These findings will need to be corroborated through ongoing in vivo models of disease and in clinical trials. In addition, the effect of pacritinib on common mutations involving these kinases would be of interest to validate that the effects demonstrated in the kinase assays are relevant to common activating mutations and gene fusions associated with malignant transformation.
In conclusion, pacritinib inhibits a variety of tyrosine kinases (JAK2, JAK2V617F, FLT3) and nontyrosine kinases (CSF1R, IRAK1, tyrosine kinase nonreceptor 1, and ROS1) at low nanomolar levels. Pacritinib demonstrated combined ability to suppress signaling by kinases involved in microenvironmental tumor interactions as well as by kinases thought to be involved in tumor progression. Pacritinib suppresses known driver mutations in JAK2, FLT3, IRAK1, and CSF1R, and shows clinical tolerability and efficacy in both non-Hodgkin lymphoma and chronic myeloproliferative diseases. Combined with its lack of myelosuppression and potentially less immunosuppressive properties than most other JAK2 inhibitors that also suppress signaling through JAK1, pacritinib has the potential to be used in combination with established therapies for the treatment of patients with a broad variety of neoplastic and inflammatory diseases.
Acknowledgments
The data in this manuscript were initially presented at the 56th American Society of Hematology Annual Meeting, December 6, 2014 in San Francisco, CA, and the meeting abstract was published online as Singer J, et al Blood. 2014;124(21):1874 http://www.bloodjournal.org/content/124/21/1874. The authors of this manuscript received no grant funding for this work.
Disclosure
The authors have the following competing interests: JWS, SA: CTI BioPharma, Corp. (ctibiopharma.com): employment, equity ownership; RSK: Incyte Corporation (incyte.com), Celgene Corp. (celgene.com): research funding, advisory role; RM: Incyte Corporation (incyte.com), CTI BioPharma, Corp., NS Pharma, Inc. (nippon-shinyaku.co.jp), Gilead Science, Inc. (gilead.com), Celgene Corp. (celgene.com): research funding; and SV: CTI BioPharma, Corp.: research funding. Medical writing and editorial assistance was provided, under the direction of the authors, by Hope J Lafferty, AM, ELS. The authors report no other conflicts of interest in this work.
References
Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene. 2000;19(49):5662–5679. | ||
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012; | ||
William AD, Lee AC, Blanchard S, et al. Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6). 1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J Med Chem. 2011;54(13):4638–4658. | ||
Hart S, Goh KC, Novotny-Diermayr V, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011;25(11):1751–1759. | ||
Hart S, Goh KC, Novotny-Diermayr V, et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011;1(11):e44. | ||
Verstovsek S, Deeg HJ, Odenike O, et al. Phase 1/2 study of SB1518, a novel JAK2/FLT3 inhibitor, in the treatment of primary myelofibrosis. Proceedings of the 53rd Annual Meeting of the American Society of Hematology; December 10–13, 2011; Chicago, IL. Abstract 3082. | ||
Verstovsek S, Odenike O, Scott B, et al. Phase 1 dose-escalation trial of SB1518, a novel JAK2/FLT3 inhibitor, in acute and chronic myeloid diseases, including primary or post-essential thrombocythemia/ polycythemia vera myelofibrosis. Proceedings of the 51st Annual Meeting of the American Society of Hematology; December 5–8, 2009; New Orleans, LA. Abstract 3905. | ||
Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of INCB018424, an oral, selective JAK1/JAK2 inhibitor, in patients with advanced polycythemia vera (PV) and essential thrombocythemia (ET) refractory to hydroxyurea. Proceedings of the 51st Annual Meeting of the American Society of Hematology; December 5–8, 2009; New Orleans, LA. Abstract 311. | ||
Younes A, Fanale M, Mclaughlin P, et al. Phase-I study of the novel oral JAK-2 inhibitor SB1518 in patients with relapsed lymphoma: evidence of clinical and biologic activity. Proceedings of the 51st Annual Meeting of the American Society of Hematology; December 5–8, 2009; New Orleans, LA. Abstract 588. | ||
Younes A, Fanale M, Mclaughlin P, Copeland AR, Zhu J, De Castro Faria S. | ||
Komrokji RS, Wadleigh M, Seymour JF, et al. Results of a phase 2 study of pacritinib (SB1518), a novel oral JAK2 inhibitor, in patients with primary, post-polycythemia vera, and post-essential thrombocythemia myelofibrosis. Proceedings of the 53rd Annual Meeting of the American Society of Hematology; December 10–13, 2011; Chicago, IL. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. Long-term outcomes of ruxolitinib therapy in patients with myelofibrosis: 3-year update from COMFORT-I. Proceedings of the 55th Annual Meeting of the American Society of Hematology; December 7–10, 2013; New Orleans, LA. Abstract 2830. | ||
Mesa RA, Egyed M, Szoke A, et al. Results of the PERSIST-1 phase III study of pacritinib (PAC) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), or post-essential thrombocythemia-myelofibrosis (PET-MF). Proceedings of the 2015 Annual Meeting of the American Society of Clinical Oncology; May 29–June 2, 2015; Chicago, IL. | ||
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | ||
Lu X, Levine R, Tong W, et al. Expression of a homodimeric type 1 cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102(52):18962–18967. | ||
Rumi E, Pietra D, Pascutto C, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124(7):1062–1069. | ||
Reilly JT. FLT3 and its role in the pathogenesis of acute myeloid leukaemia. Leuk Lymphoma. 2003;44(1):1–7. | ||
Tickenbrock L, Muller-Tidow C, Berdel WE, Serve H. Emerging Flt3 kinase inhibitors in the treatment of leukaemia. Expert Opin Emerg Drugs. 2006;11(1):153–165. | ||
Steensma DP, McClure RF, Karp JE, et al. JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia. 2006;20(6):971–978. | ||
Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1039–1045. | ||
Yee TW, Hastie TJ. Reduced-rank vector generalized linear models. Stat Model. 2003;3:15–41. | ||
van der Loo MPJ. Distribution based outlier detection for univariate data, Discussion paper 10003. The Hague: Statistics Netherland; 2010. | ||
Rhyasen GW, Bolanos L, Fang J, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell. 2013;24(1):90–104. | ||
Puig de la Bellacasa R, Karachaliou N, Estrada-Tejedor R, Teixido J, Costa C, Borrell JI. ALK and ROS1 as a joint target for the treatment of lung cancer: a review. Transl Lung Cancer Res. 2013;2(2):72–86. | ||
Henderson MC, Gonzales IM, Arora S, et al. High-throughput RNAi screening identifies a role for TNK1 in growth and survival of pancreatic cancer cells. Mol Cancer Res. 2011;9(6):724–732. | ||
Polk A, Lu Y, Wang T, et al. Colony-stimulating Factor-1 receptor is required for nurse-like cell survival in chronic lymphocytic leukemia. Clin Cancer Res. Epub 2016 Jun 22. | ||
Cleary MM, Thompson R, Mahmood S, et al. Pacritinib, a dual FLT3/JAK2 inhibitor, reduces IRAK-1 signaling in acute myeloid leukemia. Proceedings of the 57th Annual Meeting of the American Society of Hematology; December 5–8, 2015; Orlando, FL. Abstract 570. | ||
Jakafi® (ruxolitinib) tablets, for oral use [prescribing information]. Wilmington, DE: Incyte Corporation; 2011. | ||
Gotlib J. JAK inhibition in the myeloproliferative neoplasms: lessons learned from the bench and bedside. Hematology Am Soc Hematol Educ Program. 2013;2013:529–537. | ||
Gowin K, Emanuel R, Geyer H, Mesa RA. The new landscape of therapy for myelofibrosis. Curr Hematol Malig Rep. 2013;8(4):325–332. | ||
Furqan M, Mukhi N, Lee B, Liu D. Dysregulation of JAK-STAT pathway in hematological malignancies and JAK inhibitors for clinical application. Biomark Res. 2013;1(1):5. | ||
Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23(8):1441–1445. | ||
Zhou T, Georgeon S, Moser R, Moore DJ, Caflisch A, Hantschel O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia. 2014; | ||
Looyenga BD, Hutchings D, Cherni I, Kingsley C, Weiss GJ, Mackeigan JP. | ||
Heine A, Held SA, Daecke SN, et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013; | ||
Schonberg K, Rudolph J, Vonnahme M, et al. JAK inhibition impairs NK cell function in myeloproliferative neoplasms. Cancer Res. 2015; | ||
Tefferi A. Ruxolitinib targets DCs: for better or worse? Blood. 2013; | ||
Ghoreschi K, Laurence A, O’Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be? Nat Immunol. 2009;10(4):356–360. | ||
Boldin MP, Taganov KD, Rao DS, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208(6):1189–1201. | ||
Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell R, Baltimore D. NF-ϰB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A. 2011; | ||
Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll-like receptors and cancer: MYD88 mutation and inflammation. Front Immunol. 2014;5:367. | ||
Agarwal A, Bumm TG, Corbin AS, et al. Absence of SKP2 expression attenuates BCR-ABL-induced myeloproliferative disease. Blood. 2008;112(5):1960–1970. | ||
McCleary MM, Thompson R, Mahmood S, et al. Pacritinib, a dual FLT3/JAK2 inhibitor, reduces Irak-1 signaling in acute myeloid leukemia. Proceedings of the 57th Annual Meeting of the American Society of Hematology; December 5–8, 2015; Orlando, FL. Abstract 570. | ||
Ma Y, Rix LR, Zhang Q, et al. Pacritinib (PAC) synergistically potentiates azacitidine (5AZA) cytotoxicity in chronic myelomonocytic leukemia (CMML). Proceedings of the 57th Annual Meeting of the American Society of Hematology; December 5–8, 2015; Orlando, FL. Abstract 1658. | ||
Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125(9):3365–3376. | ||
Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. | ||
Tap WD, Wainberg ZA, Anthony SP, et al. Structure-guided blockade of csf1r kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428–437. | ||
Paniagua RT, Chang A, Mariano MM, et al. c-Fms-mediated differentiation and priming of monocyte lineage cells play a central role in autoimmune arthritis. Arthritis Res Ther. 2010;12(1):R32. | ||
Blonska M, Agarwal NK, Vega F. Shaping of the tumor microenvironment: Stromal cells and vessels. Semin Cancer Biol. 2015;34:3–13. | ||
Borge M, Nannini PR, Morande PE, et al. CXCL12 is a costimulator for CD4+ T cell activation and proliferation in chronic lymphocytic leukemia patients. Cancer Immunol Immunother. 2013;62(1):113–124. | ||
Borge M, Remes Lenicov F, Nannini PR, et al. The expression of sphingosine-1 phosphate receptor-1 in chronic lymphocytic leukemia cells is impaired by tumor microenvironmental signals and enhanced by piceatannol and R406. J Immunol. 2014;193(6):3165–3174. | ||
Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma. 2002;43(3):461–466. | ||
Knapper S, Grech A, Cahalin P, et al. An evaluation of the tyrosine kinase inhibitor pacritinib in patients with relapsed FLT-3 mutated acute myeloid leukaemia (the UK NCRI AML 17 study). Poster presented at: 21st Congress of the European Hematology Association; June 9-12, 2016; Copenhagen, Denmark. Abstract P184. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.