")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Compositional Alterations of Gut Microbiota in Patients with Diabetic Kidney Disease and Type 2 Diabetes Mellitus
Authors He X, Sun J, Liu C, Yu X, Li H, Zhang W, Li Y, Geng Y, Wang Z
Received 4 November 2021
Accepted for publication 25 February 2022
Published 6 March 2022 Volume 2022:15 Pages 755—765
DOI https://doi.org/10.2147/DMSO.S347805
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Xin He,1 Jiping Sun,2 Chao Liu,2 Xiaoyang Yu,2 Huixian Li,2 Wenjing Zhang,2 Yan Li,2 Yingzhou Geng,2 Zhigang Wang2
1Dialysis Department of Nephrology Hospital, First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, 710061, People’s Republic of China; 2Department of Nephrology, First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, 710061, People’s Republic of China
Correspondence: Zhigang Wang, First Affiliated Hospital of Xi’an Jiaotong University, West Yanta Road 277, Xi’an, 710061, People’s Republic of China, Email [email protected]
Purpose: Emerging evidence has revealed that gut microbiota plays a pivotal role in the pathogenesis of type 2 diabetes mellitus (T2DM) and diabetic kidney disease (DKD). However, few studies have used metagenomic sequencing to analyze the alterations of gut microbiota community in patients with early-stage DKD.
Methods: We carried out metagenomic sequencing in fecal samples of 10 DKD patients (DKD group) and 10 T2DM patients who appeared to be less prone to DKD (non-DKD group), aiming to compare the composition and function of gut microbiota between the DKD and non-DKD groups.
Results: The gut microbial community of the DKD group was significantly different from that of the non-DKD group, characterized by a marked increase in phylum Proteobacteria, genus Selenomonadales, Neosynechococcus, Shigella, Bilophila, Acidaminococcus, species, Escherichia coli, Bacteroides plebeius, Megasphaera elsdenii, Acidaminococcus unclassified, and Bilophila wadsworthia. The amounts of species Citrobacter farmeri and Syntrophaceticus schinkii were significantly and positively correlated with the urinary albumin creatinine ratio in the DKD group. Furthermore, functional analysis based on dbCAN and KEGG databases showed aberrant lipopolysaccharide (LPS) biosynthesis and carbohydrate metabolism in the gut microbiome of the DKD group.
Conclusion: Our findings provided evidence for alterations in the composition and function of gut microbiota in patients with DKD versus the non-DKD group. These data may contribute to a more comprehensive understanding of the pathological mechanisms of DKD.
Keywords: diabetic kidney disease, gut microbiota, metagenomics, type 2 diabetes mellitus
Introduction
With the rapidly increasing world population and proportion of elderly, type 2 diabetes mellitus (T2DM), a well-known metabolic disease, has become a major global public health problem.1 Diabetic kidney disease (DKD), characterized by proteinuria, hypertension, and progressive reductions in kidney function, is considered one of the most common diabetic complications and is the leading cause of end-stage renal disease (ESRD).2 Besides the traditional risk factors such as high blood glucose level, high blood pressure, and altered lipid profiles, previous epidemiological studies have suggested the emerging role of genetic factors in DKD. It is estimated that only 30−40% of T2DM patients with long-term hyperglycemia ultimately develop DKD.3 Also, DKD has been observed in some T2DM patients with good glycemic control, indicating that genetic susceptibility may play a critical role in the pathogenesis of DKD.4
Gut microbiota is increasingly recognized to be associated with various metabolic diseases, such as obesity,5 diabetes,6 and cardiovascular diseases.7 The human gastrointestinal tract harbors thousands of bacterial species, together comprising 10–100 trillion microbes, which encode approximately 150-fold more genes than the human genome.8,9 The gut microbiota has been shown to modulate both local and systemic immunity and metabolism by inducing chronic inflammation and altering microbial metabolism and microenvironment.10
Accumulating evidence has shown that changes in the composition and proportion of gut microbiota are closely related to an increased risk of developing T2DM and its associated complications.11 Improvement of the gut microbiota alleviates the symptoms of diabetes, glucose tolerance, and insulin sensitivity in patients with T2DM.12–14 The disturbance of the gut-kidney axis contributes to the pathogenesis of kidney disease through diverse mechanisms, including toxin secretion, epigenetic modification,15 inflammation,16 immune response,17 mitochondrial dysfunction,18 glycometabolism,19 and short-chain fatty acids (SCFAs) metabolism,20 all of which are key events during the progression of diabetes and DKD. However, no studies have compared the gut microbiota in T2DM patients with or without DKD. Whether gut microbiota alteration is a predisposing factor in DKD warrants exploration.
In this study, we performed metagenomic analysis to compare the composition and functional profiles of gut microbiota in T2DM patients with or without DKD, aiming to determine whether alterations in the existing gut dysbiosis may increase the susceptibility of T2DM patients to DKD. Our results may provide new insights into the role of gut microbiota in the pathogenesis of DKD.
Methods
Subjects
A total of 20 patients who were diagnosed with T2DM according to the WHO Diabetes Diagnostic Criteria (1999) were recruited from the First Affiliated Hospital of Xi’an Jiaotong University between March 2018 and January 2019. Among them, 10 patients were diagnosed with stage III/IV DKD, presenting normal renal function and a urinary albumin creatinine ratio (ACR) of >30 μg/mg (the DKD group). To better compare the role of gut microbiota in the pathogenesis of DKD, we selected 10 subjects who had suffered from T2DM for more than 10 years, had no renal damage, and showed an ACR of < 30 μg/mg as control subjects (the non-DKD group). Clinically, change of ACR is an early sign of DKD.21 All patients had an HbAlc level of 6−7% and normal blood pressure and lipid profiles.
The exclusion criteria for both groups were as follows: (1) patients with tumors, hypertension, obesity, coronary heart disease, liver cirrhosis, fatty liver, or thyroid disease; (2) patients using glucocorticoids, probiotics, antibiotics, and immunosuppressant within 1 month before sample collection; (3) patients with gastrointestinal diseases or surgery history; (4) patients with renal impairment due to other causes.
All patients were Han Chinese from Shaanxi Province in China. The two groups were matched for gender and age (Table 1). This study was approved by the Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University. All subjects provided written informed consent and were informed about the study.
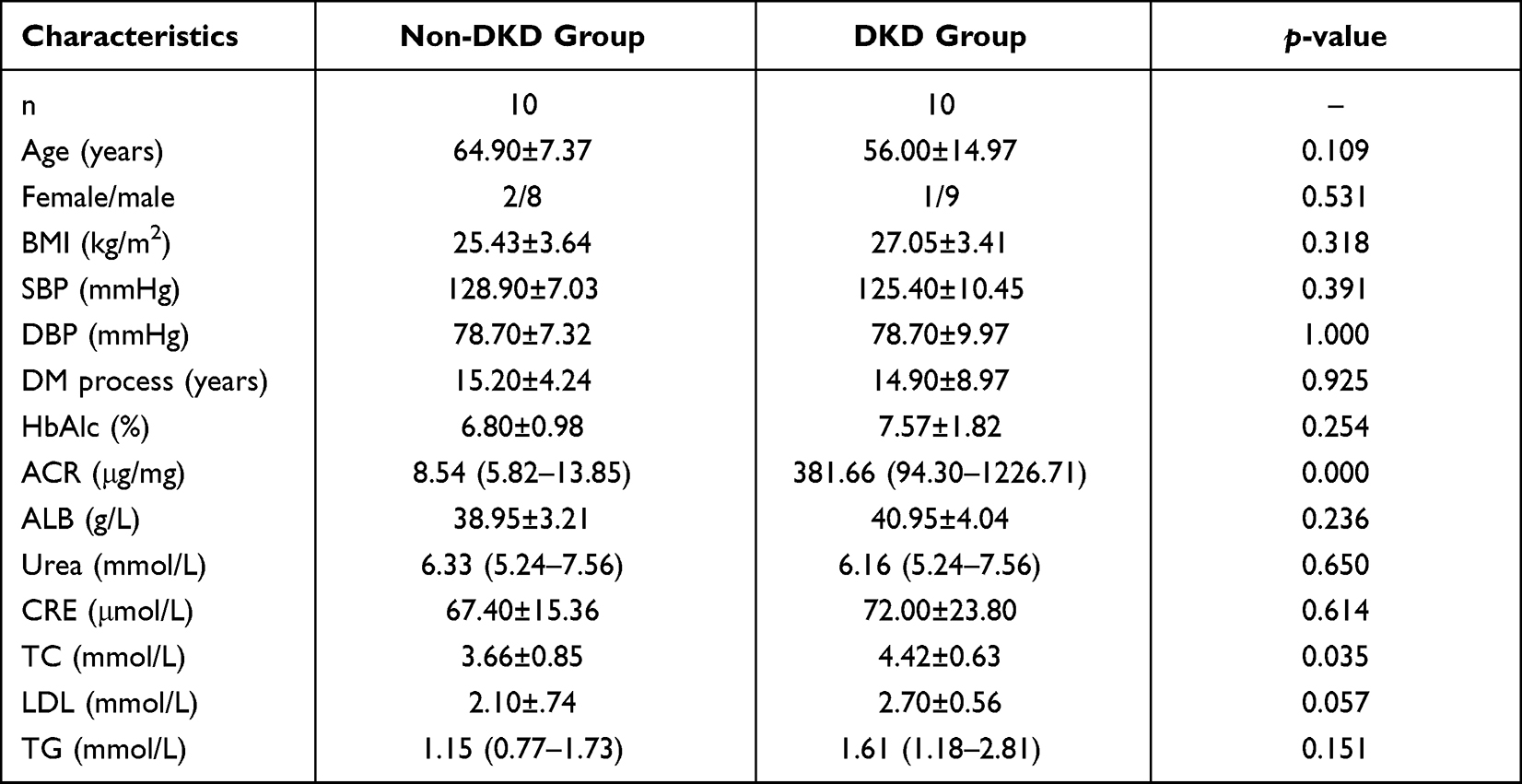 |
Table 1 Demographic and Clinical Characteristics of All Patients at Baseline |
Sample Collection and DNA Extraction
Fresh stool samples from each patient were collected in sterile collection containers, immediately transported on ice to the laboratory, and stored at −80°C until extraction. The bacterial DNAs were isolated from stool samples using the E.Z.N.A.® Stool DNA Kit (D4015; Omega Bio-Tek, Norcross, GA, USA). The integrity and concentration of DNAs were assessed using 1.2% agarose gel electrophoresis (LC-Bio Technology Co. Ltd., Hangzhou, China).
Bioinformatics and Statistical Analysis
The metagenomic libraries were generated using the TruSeq Nano DNA LT Library Preparation Kit (FC-121-4001). The library quality was evaluated by the [email protected] Fluorometer (Life Technologies, Eugene, OR, USA) and the Agilent Bioanalyzer 2100 system (Agilent Technologies Inc., Santa Clara, CA, USA). The sequencing was performed using the Illumina HiSeq4000 platform and PE150 mode.
The adapters were removed by Cutadapt (V1.9) and the host genome was discarded using Bowtie2 (V2.2.0). The quality of the sequencing reads was assessed using FastQC software (V0.10.1). The high-quality reads were assembled to contigs by SPAdes (V3.7.0) and evaluated using QUAST (V 3.2). MetaGeneMark software was used for the CDS (Coding Region) prediction on contigs (≥500 bp) from each sample. The reads shorter than 100 nt were excluded. Then, the prediction results were filtered using the CD-HIT software (V4.6.1) to obtain non-redundant genes and cluster predicted genes with 95% identity and 90% coverage. Then, the gene reads counts were calculated by Bowtie2 (V2.2.0) and the unigenes were obtained through the calculation of the corresponding abundance. Finally, the taxonomic assignment and functional annotation were performed by DIAMOND (V0.7.12) using the following databases: Non-Redundant (NR), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Carbohydrate-Active enzymes (CAZy) (e-value≤1e-5).
The KEGG or CAZy sequence hits with the highest scores were assigned as the annotations of unigenes. By combining the hierarchical structure of the KEGG pathway or CAZy database, the information at each level was obtained. The partial least square-discriminant analysis (PLS-DA) was performed using R software (V3.3.2). The LDA effect size (LEfSe) analysis was performed to identify the difference in the bacterial taxa between the DKD and non-DKD groups using LEfSe software (V1.0) with an effect size cutoff of 2.0. The differential abundance analysis between the two groups was performed using Welch’s t-test. The correlations of clinical characteristics with top 50 differential species were analyzed by Spearman’s rank correlation coefficient and visualized by the R package “heatmap”. The demographic data between the two groups were compared using the Student’s t-test. A p-value <0.05 was considered statistically significant.
Results
Demographic and Clinical Characteristics of All Subjects
To explore the differences in gut microbial composition between the DKD and non-DKD groups, we analyzed their fecal samples using metagenomic sequencing (n=10 per group). All participants (n=20) were of Han ethnicity, born in northeastern China, and had a similar dietary pattern. Their demographic and clinical characteristics are summarized in Table 1. No significant difference was observed in the clinic characteristics except for ACR and the level of total cholesterol (TC).
Taxonomic Characterization and Gut Microbial Composition
A total of 143.6 million high-quality reads were generated with an average (standard deviation) of 7.18 (1.13) million clean reads for each sample (Supplementary Table S1). The most abundant gut microbiota in each subject at phylum (top 10) and genus (top 20) levels is shown in Figure 1. The dominant phyla in both DKD and non-DKD groups are Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria (Figure 1A). At the genus level, Bacteroides, Prevotella, and Faecalibacterium are the most abundant genera in both groups (Figure 1B). Then, the PLS-DA was performed to analyze the samples at the phylum level. The plot shows the difference in the microbial profile between the DKD and non-DKD groups (Figure 2), indicating an altered gut microbial community structure in T2DM patients who were susceptible to early-stage DKD.
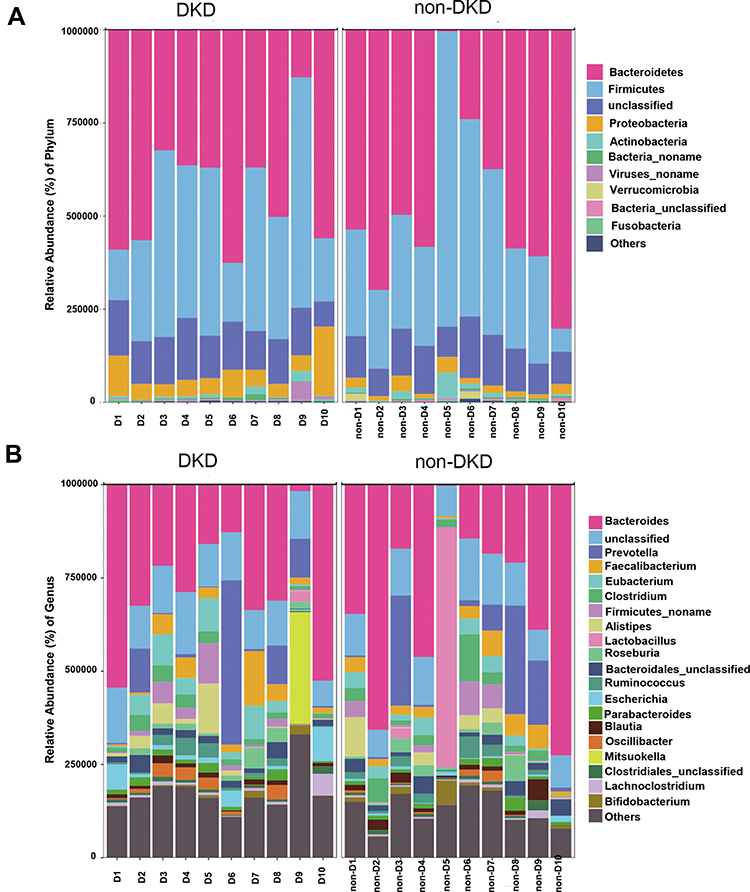 |
Figure 1 The relative abundance of gut microbiota in two groups. (A) The relative abundance of top 10 microbial taxa at the phylum level. (B) The relative abundance of top 20 microbial taxa at the genus level. Each bar represents a bacterial taxon and the length of the bar indicates the abundance level. |
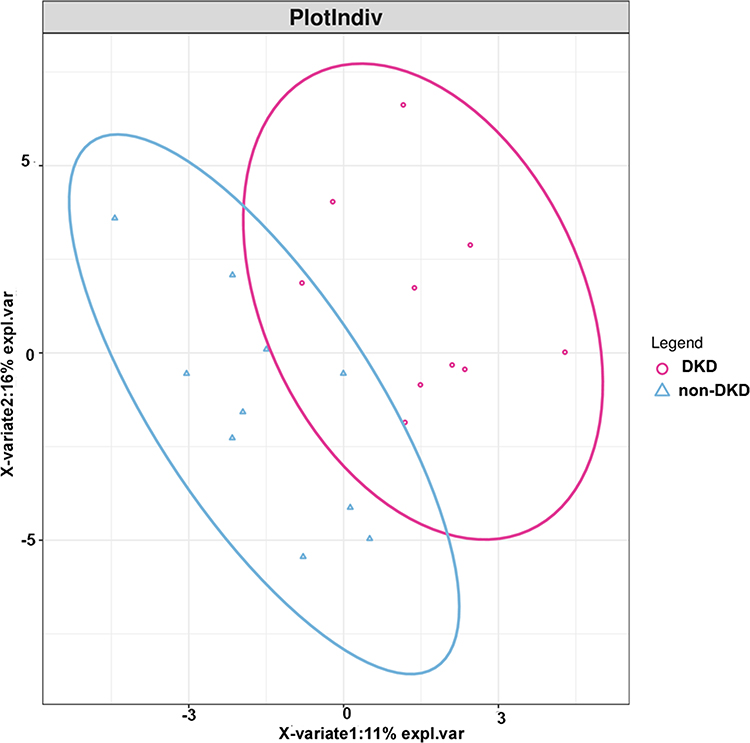 |
Figure 2 The score plot of the PLS-DA model between the DKD and non-DKD groups. Red cycle: DKD group; blue triangle: non-DKD group. |
Differences in Gut Microbial Communities Between Two Groups
To identify the microbial communities associated with DKD, the gut microbial composition of the DKD and non-DKD groups was compared using LEfSe analysis with a cutoff value of 2.0. As shown in Figure 3, there was a remarkable difference in the composition of gut microbiota between the DKD and non-DKD groups. The phylum Proteobacteria was increased in the DKD group. At the genus level, the DKD group possessed more abundant Selenomonadales, Neosynechococcus, Shigella, Bilophila, Acidaminococcus and Escherichia, while Phlyctochytrium was increased in the non-DKD group. Further, to analyze at the species level, the relative abundance of Escherichia coli, Bacteroides plebeius, Megasphaera elsdenii, Acidaminococcus unclassified, Ruminococcus torques, and Bilophila wadsworthia in the DKD group was higher than those in the non-DKD group, while the non-DKD group presented a higher abundance of Bacteroides coprocola, Bacteroides xylanisolvens, and Bacteroides sp D22, etc.
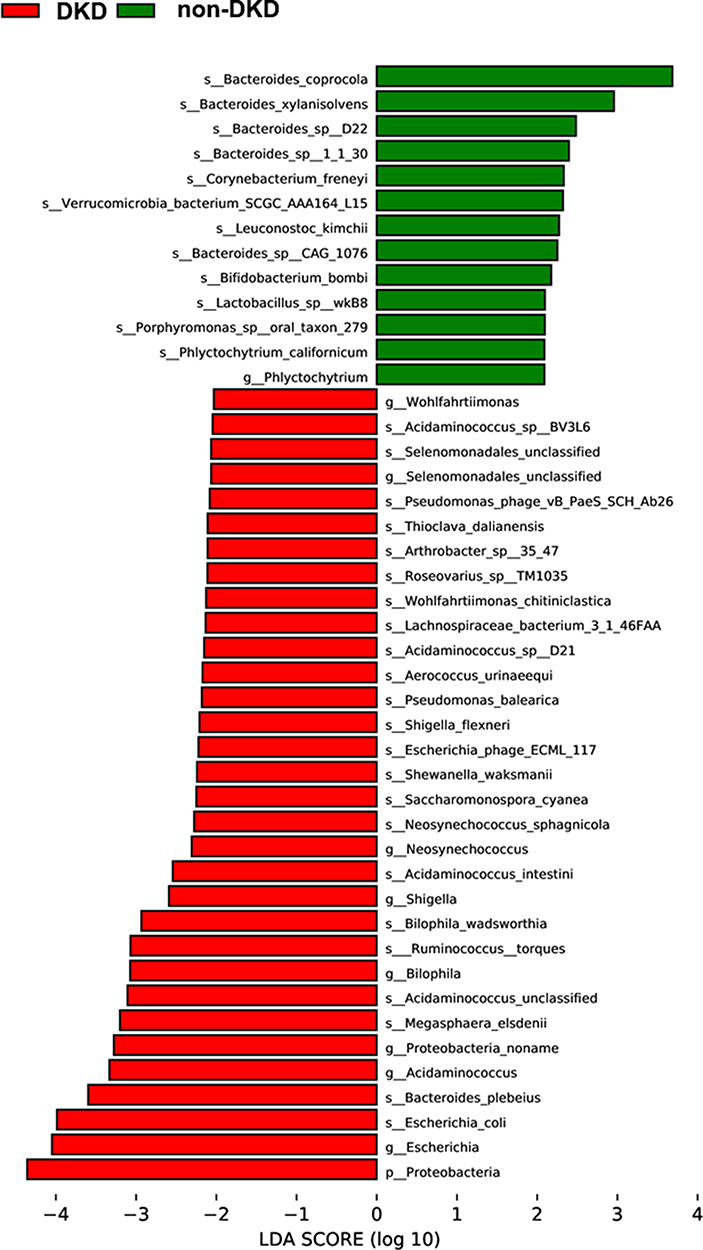 |
Figure 3 The LDA scores of differentially abundant microbiota at genus and species levels between the DKD and non-DKD groups. |
Correlations Between Gut Microbiota and Clinical Characteristics
Next, we explored whether the top 50 differential microbial species between the DKD and non-DKD groups correlated with the clinical characteristics of these patients (Figure 4). At the species level, Citrobacter farmeri (Spearman rho=0.53, P=0.0167) and Syntrophaceticus schinkii (Spearman rho=0.61, P=0.0052), which were abundantly present in the gut microbiota of the DKD group, were significantly and positively correlated with the ACR level. Enterococcus caccae (Spearman rho= −0.55, P=0.0125), which was enriched in the non-DKD group, was negatively correlated with the ACR level. Additionally, Lachnospiraceae bacterium 3 1 46FAA, which was abundant in the DKD group, showed a positive correlation with the levels of TC (Spearman rho=0.53, P=0.0182).
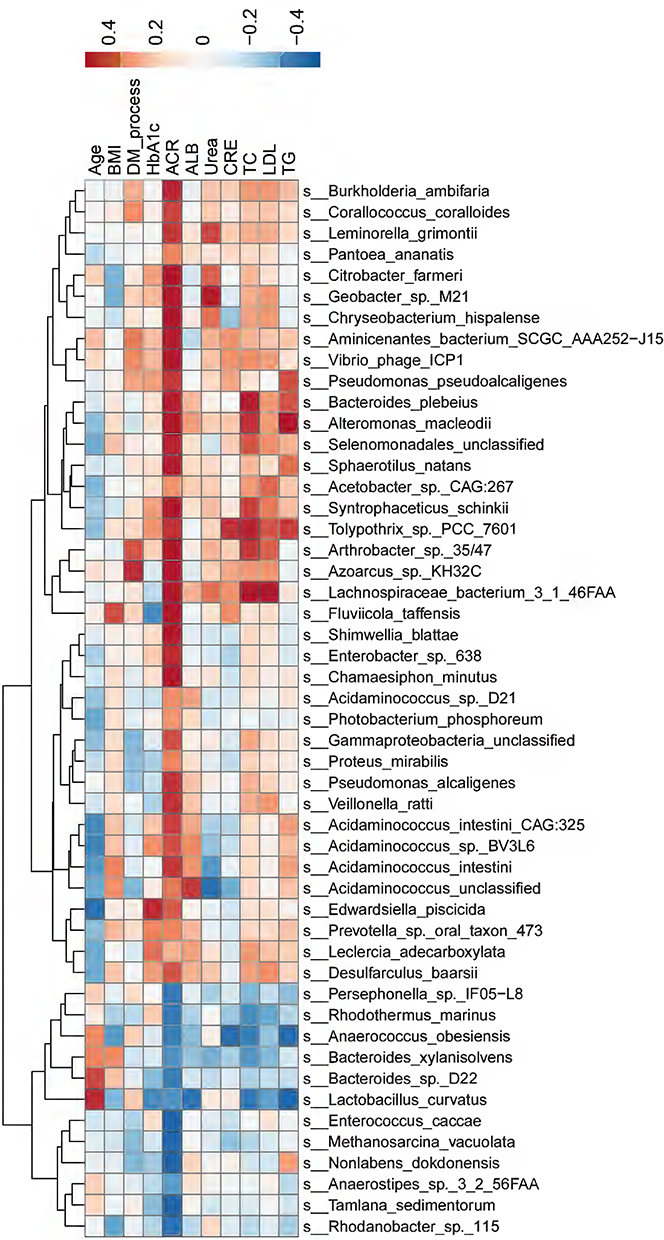 |
Figure 4 The correlations between gut microbiota and clinical characteristics. The color gradient indicates p-value; red: positive correlation; blue: negative correlation. |
Functional Pathways Analysis
To further investigate the functional profiles of gut microbiota in the DKD and non-DKD groups, the functional annotation based on KEGG and dbCAN databases was performed. The results showed that most genes were annotated with metabolism function (Figure S1). As shown in the differential gene enrichment bubble chart, there are five key pathways: glycosaminoglycan biosynthesis–heparan sulfate/heparin, glycosaminoglycan biosynthesis–chondroitin sulfate/dermatan sulfate, lipopolysaccharide (LPS) biosynthesis, phosphotransferase system (PTS), and histidine metabolism (Figure 5). It is well known that gut microbiota also plays a pivotal role in the metabolism of dietary carbohydrates of the host.22 The carbohydrate-active enzymes (CAZymes) produced by gut microbiota are associated with the synthesis, modification, and degradation of carbohydrates.23 Thus, we analyzed the genes related to carbohydrate metabolism. As illustrated in Figure 6, a total of 199 genes were annotated by the dbCAN database at level 2. Nineteen CAZymes were significantly different between the two groups, including GH50 (β-agarase), GT17 (β1, 4-N-acetylglucosaminyl transferase, GnTIII), etc, all of which were much less abundant in the fecal microbiome of DN patients (Table S2).
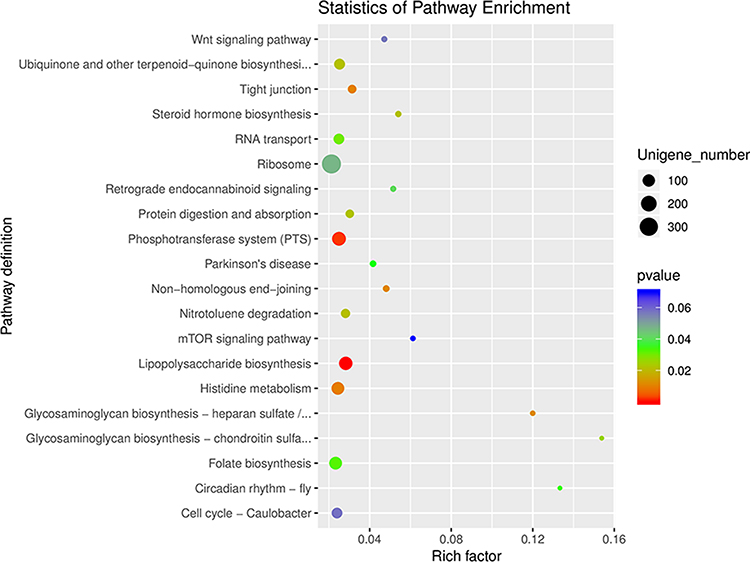 |
Figure 5 The KEGG enrichment analysis of gut microbiota in the DKD and non-DKD groups. The smaller the p-value, the higher the degree of enrichment. |
 |
Figure 6 The abundance of carbohydrate-active enzymes (CAZymes) in the gut microbiota of the DKD and non-DKD groups. |
Discussion
In this study, we performed a comprehensive metagenomics comparison of the taxonomic and functional profiles of gut microbiota in T2DM patients with or without DKD and its correlation with clinical characteristics. Our data revealed distinctions in the compositional and functional characteristics of gut microbiota between the DKD and non-DKD groups, suggesting that alterations in the intestinal microbiota may play a key role in the pathogenesis of DKD in T2DM patients who were susceptible to this disease.
Although the dominant microbial phyla in T2DM patients were similar, our data showed that the DKD group possessed a distinctive microbiota composition, characterized by a high abundance of phylum Proteobacteria. Recent evidence has shown that Proteobacteria in gut microbial dysbiosis is essential for the metabolic and immune disorders, inflammation, and nutritional status of the host.24 In clinical and experimental studies, gastrointestinal inflammation increases the risk of kidney disorders through the gut-kidney axis.25 Multiple studies have consistently demonstrated that Proteobacteria-dominated microbiome profiles are associated with elevated inflammation,26,27 which is a key contributor to the pathogenesis of DKD.28 Moreover, we found the non-DKD group presented a higher increase in the species Bacteroides coprocola, Bacteroides xylanisolvens, and Bacteroides sp D22, which were classified as Bacteroidetes. Bacteroidetes is well-known for its capability to ferment dietary fiber into SCFAs, an emerging therapeutic target for systemic inflammatory, immune, and metabolic diseases, especially in glucose tolerance, obesity, and insulin resistance.29 SCFAs have been shown to mediate cellular signaling pathways, including cell proliferation, apoptosis, and histone acetylation.30 Importantly, Li et al found that SCFAs protected diabetic mice against DKD by activating G protein-coupled receptors and reducing inflammation.31 Similarly, we found that Proteobacteria, were more abundant in the DKD group, resulting in a reduced abundance of SCFA-producing microbiota and SCFAs production, which might promote the progression of DKD.
ACR is a key indicator for the diagnosis of renal injury at the early stage of DKD. Tao et al showed that DKD patients with an eGFR of > 60 mL/min/1.73 m2 and an ACR of > 30 μg/mg could be distinguished from diabetic patients by g Escherichia-Shigella, which belongs to Proteobacteria.32 Consistently, we observed a significant correlation between ACR and increased numbers of Citrobacter farmeri and Syntrophaceticus schinkii in the DKD group, both of which were classified as Proteobacteria. A previous study also reported the abundance of Proteobacteria in the intestine of DKD patients,33 indicating that an elevated level of Proteobacteria is closely associated with an increased risk of DKD. Further studies are needed to explore the sole effect of Proteobacteria on the pathogenesis of DKD.
Our functional analysis demonstrated a differential abundance of functional pathways in the gut microbiota between the DKD and non-DKD groups. The PTS is an important bacterial signaling pathway related to carbohydrate uptake and is associated with the metabolism and longevity of the host.34 Imidazole propionate is a histidine derivative produced by gut microbiota and has been found to impair insulin signaling through mechanistic target of rapamycin complex 1 in T2DM patients.35 A high level of Proteobacteria in gut microbiota has been shown to induce the biosynthesis of LPS, resulting in a chronic low-grade inflammation via activating macrophages/monocytes and neutrophils.33 Furthermore, Toll-like receptors (TLRs), an important component of the innate immune system, play a key role in the pathogenesis of DKD. LPS is an endogenous ligand of TLRs and can promote the expression of inflammatory factors and activate the specific immune system by binding with TLRs.36 Altogether, these results suggest that the metabolic functions of gut microbiota are changed in DKD patients and the increased levels of Proteobacteria may contribute to the pathogenesis of DKD via a disturbed inflammatory and immune system.
The emerging correlation of gut microbiota with carbohydrate metabolism and glucose homeostasis in diabetes led us to investigate the genes associated with CAZymes. Compared to the non-DKD group, the DKD group showed a lower level of GnTIII, an N-acetylglucosaminyl transferase (OGT). OGT is a key enzyme involved in the post-translational modification of O-link N-acetylglucosamine (O-GlcNAc) and has been found to be closely related to the pathogenesis of DKD.37 During the process of O-GlcNAcylation, a single O-GlcNAc moiety was added to the serine and/or threonine residues of various proteins by OGT.38 Alejandro et al reported that the knockout of OGT in islet B cells resulted in reduced insulin secretion and increased blood glucose in mice.39 The host-microbe metabolic axis is a multi-directional interaction between the host and the gut microbiota.40 Our findings suggest that the decrease in OGT level, which is closely related to the glycemic level and kidney damage, may result in aberrant O-GlcNAc modification in DKD patients.
There are some limitations of this study that need to be taken into account. First, the sample size was small due to strict inclusion criteria, which required that all enrolled T2DM patients had normal renal function and a well-controlled blood glucose level. Second, our study is a cross-sectional study. The causal direction between the abundance of Proteobacteria in gut microbiota and the progression of DKD remains unknown. A longitudinal study with a large sample size is needed for causal inferences.
Conclusions
In summary, T2DM patients with DKD showed altered composition and function of gut microbiota compared to those without DKD. Increased abundance of Proteobacteria was observed in DKD patients, which might contribute to the pathogenesis of DKD via regulating LPS biosynthesis, SCFA production, and carbohydrate metabolism. Our result may provide new insights into the application of microbial intervention as a potential treatment for DKD patients.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of the First Affiliated Hospital of Xi’an Jiaotong University. All subjects provided written informed consent and were informed about the study.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81700644).
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Kakio Y, Uchida HA, Takeuchi H, et al. Diabetic nephropathy is associated with frailty in patients with chronic hemodialysis. Geriatr Gerontol Int. 2018;18:1597–1602. doi:10.1111/ggi.13534
2. Singh S, Sonkar SK, Sonkar GK, Mahdi AA. Diabetic kidney disease: a systematic review on the role of epigenetics as diagnostic and prognostic marker. Diabetes Metab Res Rev. 2019;35(5):e3155. doi:10.1002/dmrr.3155
3. Umanath K, Lewis JB. Update on diabetic nephropathy: core curriculum 2018. Am J Kidney Dis. 2018;71(6):884–895. doi:10.1053/j.ajkd.2017.10.026
4. Jeong KH, Kim JS, Woo JT, et al. Genome-wide association study identifies new susceptibility loci for diabetic nephropathy in Korean patients with type 2 diabetes mellitus. Clin Genet. 2019;96:35–42. doi:10.1111/cge.13538
5. Scheithauer TPM, Rampanelli E, Nieuwdorp M, et al. Gut microbiota as a trigger for metabolic inflammation in obesity and type 2 diabetes. Front Immunol. 2020;11:571731. doi:10.3389/fimmu.2020.571731
6. Gurung M, Li Z, You H, et al. Role of gut microbiota in type 2 diabetes pathophysiology. E Bio Med. 2020;51:102590. doi:10.1016/j.ebiom.2019.11.051
7. Witkowski M, Weeks TL, Hazen SL. Gut microbiota and cardiovascular disease. Circ Res. 2020;127(4):553–570. doi:10.1161/CIRCRESAHA.120.316242
8. Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi:10.1038/nature08821
9. Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13(4):260–270. doi:10.1038/nrg3182
10. Pickard JM, Zeng MY, Caruso R, Nunez G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279:70–89. doi:10.1111/imr.12567
11. Zhao L, Lou H, Peng Y, Chen S, Zhang Y, Li X. Comprehensive relationships between gut microbiome and faecal metabolome in individuals with type 2 diabetes and its complications. Endocrine. 2019;66(3):526–553. doi:10.1007/s12020-019-02103-8
12. Zhaz F, Liu Q, Cao J, et al. A sea cucumber (Holothuria leucospilota) polysaccharide improves the gut microbiome to alleviate the symptoms of type 2 diabetes mellitus in Goto-Kakizaki rats. Food Chem Toxicol. 2019;135:110886. doi:10.1016/j.fct.2019.110886
13. Beli E, Prabakaran S, Krishnan P, Evans-Molina C, Grant MB. Loss of diurnal oscillatory rhythms in gut microbiota correlates with changes in circulating metabolites in type 2 diabetic db/db mice. Nutrients. 2019;11(10):2310. doi:10.3390/nu11102310
14. Hui S, Liu Y, Chen M, et al. Capsaicin improves glucose tolerance and insulin sensitivity through modulation of the gut microbiota-bile Acid-FXR axis in type 2 Diabetic db/db mice. Mol Nutr Food Res. 2019;63(23):e1900608. doi:10.1002/mnfr.201900608
15. Felizardo RJF, de Almeida DC, Pereira RL, et al. Gut microbial metabolite butyrate protects against proteinuric kidney disease through epigenetic- and GPR109 a-mediated mechanisms. FASEB J. 2019;33:11190–11894. doi:10.1096/fj.201901080R
16. Li F, Wang M, Wang J, Li R, Zhang Y. Alterations to the gut microbiota and their correlation with inflammatory factors in chronic kidney disease. Front Cell Infect Microbiol. 2019;9:206. doi:10.3389/fcimb.2019.00206
17. Collison J. Gut microbiota linked to kidney disease in SLE. Nat Rev Rheumatol. 2019;15:188.
18. Mafra D, Borges NA, Lindholm B, Stenvinkel P. Mitochondrial dysfunction and gut microbiota imbalance: an intriguing relationship in chronic kidney disease. Mitochondrion. 2019;47:206–209. doi:10.1016/j.mito.2018.11.006
19. Ren M, Zhang H, Qi J, et al. An almond-based low carbohydrate diet improves depression and glycometabolism in patients with type 2 diabetes through modulating gut microbiota and GLP-1: a randomized controlled trial. Nutrients. 2020;12(10):3036. doi:10.3390/nu12103036
20. Wang Z, Zhao Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell. 2018;9(5):416–431. doi:10.1007/s13238-018-0549-0
21. Seidu S, Barrat J, Khunti K. Clinical update: the important role of dual kidney function testing (ACR and eGFR) in primary care: identification of risk and management in type 2 diabetes. Prim Care Diabetes. 2020;14:370–375. doi:10.1016/j.pcd.2020.02.006
22. Rowland I, Gibson G, Heinken A, et al. Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr. 2018;57(1):1–24. doi:10.1007/s00394-017-1445-8
23. Huang L, Zhang H, Wu P, et al. dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018;46:D516–D521. doi:10.1093/nar/gkx894
24. Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33:496–503. doi:10.1016/j.tibtech.2015.06.011
25. Stavropoulou E, Kantartzi K, Tsigalou C, et al. Focus on the gut-kidney axis in health and disease. Front Med. 2020;7:620102. doi:10.3389/fmed.2020.620102
26. Salguero MV, MAI A-O, Singh R, Siepmann T, Vasylyeva TL. Dysbiosis of gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp Ther Med. 2019;18:3461–3469. doi:10.3892/etm.2019.7943
27. Dicker AJ, Huang JTJ, Lonergan M, et al. The sputum microbiome, airway inflammation, and mortality in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2021;147:158–167. doi:10.1016/j.jaci.2020.02.040
28. Di Vincenzo A, Tana C, El Hadi H, Pagano C, Vettor R, Rossato M. Antioxidant, anti-inflammatory, and metabolic properties of tocopherols and tocotrienols: clinical implications for vitamin E supplementation in diabetic kidney disease. Int J Mol Sci. 2019;20(20):5101. doi:10.3390/ijms20205101
29. Kimura I, Ichimura A, Ohue-Kitano R, Igarashi M. Free fatty acid receptors in health and disease. Physiol Rev. 2020;100(1):171–210. doi:10.1152/physrev.00041.2018
30. Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119.
31. Li YJ, Chen X, Kwan TK, et al. Dietary fiber protects against diabetic nephropathy through short-chain fatty acid-mediated activation of G protein-coupled receptors GPR43 and GPR109A. J Am Soc Nephrol. 2020;31:1267–1281. doi:10.1681/ASN.2019101029
32. Tao S, Li L, Li L, et al. Understanding the gut-kidney axis among biopsy-proven diabetic nephropathy, type 2 diabetes mellitus and healthy controls: an analysis of the gut microbiota composition. Acta Diabetol. 2019;56:581–592. doi:10.1007/s00592-019-01316-7
33. Rizzatti G, Lopetuso LR, Gibiino G, Binda C, Gasbarrini A. Proteobacteria: a common factor in human diseases. Biomed Res Int. 2017;2017:9351507. doi:10.1155/2017/9351507
34. Pryor R, Norvaisas P, Marinos G, et al. Host-microbe-drug-nutrient screen identifies bacterial effectors of metformin therapy. Cell. 2019;178(1299–1312):e1229. doi:10.1016/j.cell.2019.08.003
35. Koh A, Molinaro A, Stahlman M, et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175(947–961):e917. doi:10.1016/j.cell.2018.09.055
36. Lin M, Tang SC. Toll-like receptors: sensing and reacting to diabetic injury in the kidney. Nephrol Dial Transplant. 2014;29(4):746–754. doi:10.1093/ndt/gft446
37. Gellai R, Hodrea J, Lenart L, et al. Role of O-linked N-acetylglucosamine modification in diabetic nephropathy. Am J Physiol Renal Physiol. 2016;311:F1172–F1181. doi:10.1152/ajprenal.00545.2015
38. Haltiwanger RS, Blomberg MA, Hart GW. Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-N-acetylglucosamine: polypeptide beta-N-acetylglucosaminyltransferase. J Biol Chem. 1992;267:9005–9013. doi:10.1016/S0021-9258(19)50380-5
39. Alejandro EU, Bozadjieva N, Kumusoglu D, et al. Disruption of O-linked N-acetylglucosamine signaling induces ER stress and beta cell failure. Cell Rep. 2015;13:2527–2538. doi:10.1016/j.celrep.2015.11.020
40. Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi:10.1126/science.1223813
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.