")
Back to Journals » Journal of Pain Research » Volume 10
Complex management of a patient with refractory primary erythromelalgia lacking a SCN9A mutation
Authors Low SA, Robbins W, Tawfik VL
Received 7 December 2016
Accepted for publication 7 February 2017
Published 27 April 2017 Volume 2017:10 Pages 973—977
DOI https://doi.org/10.2147/JPR.S129661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Enrica Santarcangelo
Sarah A Low,1 Wendye Robbins,2,3 Vivianne L Tawfik2
1Department of Internal Medicine, Banner University Medical Center, University of Arizona College of Medicine, Tucson, AZ, 2Department of Anesthesiology, Perioperative & Pain Medicine, Stanford University, Palo Alto, 3Blade Therapeutics, South San Francisco, CA, USA
Abstract: A 41-year-old woman presented with burning and erythema in her extremities triggered by warmth and activity, which was relieved by applying ice. Extensive workup was consistent with adult-onset primary erythromelalgia (EM). Several pharmacological treatments were tried including local anesthetics, capsaicin, ziconotide, and dantrolene, all providing 24–48 hours of relief followed by symptom flare. Interventional therapies, including peripheral and sympathetic ganglion blocks, also failed. Thus far, clonidine and ketamine have been the only effective agents for our patient. Genetic testing was negative for an EM-associated mutation in the SCN9A gene, encoding the NaV1.7 sodium channel, suggesting a mutation in an alternate gene.
Keywords: erythromelalgia, chronic pain, genetic testing, sodium channels, ketamine
Introduction
Erythromelalgia (EM) is a rare neurovascular disorder characterized by swelling, erythema, and burning in the extremities in a stocking-glove distribution, which is relieved by exposure to cold but worsened by heat stimuli. Based on its etiology, EM is classified into two types: primary and secondary EM. Primary EM may be either hereditary with mutations in SCN9A resulting in defective NaV1.7 sodium channels or idiopathic with spontaneous gain-of-function mutations in SCN9A or other genes. Secondary EM is associated with hematological disorders involving thrombocytosis, certain immunological disorders, and some vasoactive medications.1 Despite the discovery of underlying NaV1.7 channel dysfunction in inherited EM,2 these patients often course through many different treatment modalities with inconsistent therapeutic responses.3 This report describes a case of refractory late-onset primary EM and raises key questions about its diagnosis, treatment, and the utility of genetic testing.
Consent for publication
We obtained written permission from the patient to publish this case report and the accompanying images.
Case description
A 41-year-old woman presented to our clinic with late-onset EM, diagnosed 9 years prior at another institution following prolonged casting for a left metatarsal stress fracture. Her clinical presentation included burning, erythema, and swelling throughout her left foot. Over the course of next 3–4 months, her symptoms progressed to involve all extremities in a stocking-glove distribution. Her pain grew more intense with the addition of “throbbing, stabbing, and jerking” sensations. The frequency of her symptoms also escalated over time. Initially, she suffered from monthly flares lasting up to 24–48 hours, but within 5 years, she was essentially confined to her home maintained at an ambient temperature of 15°C. Her past medical history was significant for Raynaud’s disease, focal alopecia, hirsutism, premature ovarian failure, and severe melanosis coli secondary to long-term laxative use for chronic constipation. Her family history was negative for EM.
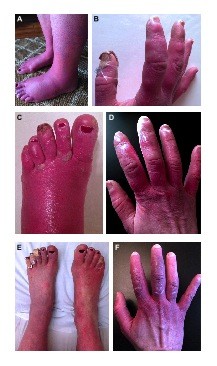
During her initial visit to our clinic in 2012, her symptoms were nearly constant and had spread to involve her nose and ears. Her extremities were swollen, erythematous, and mottled (Figure 1A and B), and she episodically developed purple patches on her digits with a characteristic evolution (Figure 1C and D). Over the course of approximately 4 weeks, hard immobile papules would appear, whiten, and then ulcerate. Her symptoms were exacerbated by warmth, stress, physical activity, certain foods, and alcohol. She also had allodynia and arthralgias that persisted between flares.
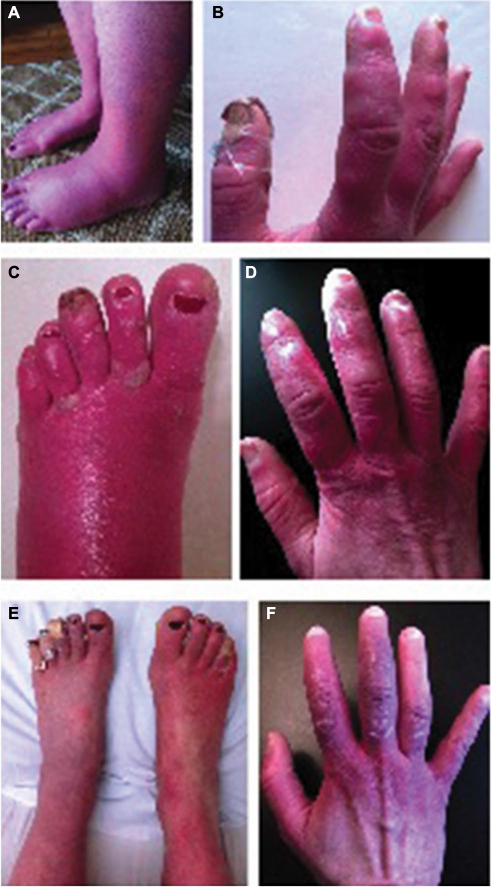 | Figure 1 Erythematous feet (A) and hands (B) at baseline with firm, nodular lesions in the beginning stages of development. An example of extremities (C and D) during a symptom flare with progression of lesions to blisters and ulceration. Demonstration of feet (E) and hands (F) after successful treatment with clonidine and ketamine, and lesions in the process of healing. |
Having failed several treatments at other institutions, she had stopped taking any medications when she presented to us but was chronically icing her limbs and receiving intermittent herbal body wraps that briefly mitigated her symptoms. Laboratory studies including complete blood count, comprehensive autoimmune panel (rheumatoid factor, erythrocyte sedimentation rate, C-reactive protein, anti-nuclear antibody, anti-dsDNA, anti-cyclic citrullinated peptide, anti-Ro, anti-La, anti-Sm, anti-ribonucleoprotein, anti-Scl70, anti-centromere), cold agglutinin screen, serum protein electrophoresis, and serum concentrations of IgG and heavy metals were all negative, ruling out secondary causes of EM. Autonomic reflex screen showed cardiovagal impairment consistent with limited neuropathy, as cardiovascular adrenergic and postganglionic sympathetic sudomotor function was preserved. There was evidence of length-dependent large-fiber peripheral neuropathy per nerve conduction studies, and transcutaneous oximetry (tcpO2) showed abnormal peripheral hemodynamic regulation with increased tcpO2 at the distal extremities, which decreased more proximally. Plain radiographs of the hands showed resorptive changes involving distal tufts of her distal phalanges, but there were normal degenerative changes in the feet where her symptoms were the most severe. Quantitative sensory testing revealed changes consistent with peripheral neuropathy, with the most significant changes noted with vibration and temperature sense. Thermography was positive, and her skin biopsy showed pernio, microvascular inflammation from repeated exposure to cold, which likely reflected her icing habits. Other special testing including hemodynamic responses to valsalva and tilt table testing as well as quantitative axon reflex sweat tests were negative.
Medical treatment over the past 4 years included trials of an array of oral, intravenous (IV), and topical medications including opioids, sodium and calcium channel blockers, antidepressants, antiepileptics, dantrolene, immunoglobulin infusions, compounded creams, and other adjuvants (Table 1 presents a summary of medical and interventional therapies). Medical management with these agents was either ineffective or amplified her symptoms. These flares involved 1 week of pain intensification followed by 2–4 weeks of swelling with fulminant development of ulcerating lesions on her digits. Sequential rounds of IV nitroglycerin and high-dose capsaicin patches were also tried but were discontinued secondary to desquamation. An initial improvement with IV dantrolene infusion was encouraging; however, the therapy was discontinued due to side effects. Transdermal clonidine improved her erythema and swelling; however, the associated vasodilation worsened her pain, which was controlled by the addition of IV ketamine. Clonidine and ketamine were the only agents that transiently dampened her symptoms without provocation of flares; however, she required monthly weeklong hospitalizations for these treatments, which provided only minimal relief (Figure 1E and F).
 | Table 1 Classes of medications and interventions tried with varying efficacy Abbreviations: NSAIDs, nonsteroidal anti-inflammatory drugs; GABA, gamma aminobutyric acid; IV, intravenous; IT, intrathecal; IVIG, intravenous immunoglobulin. |
Interventional therapies included sympathetic ganglion and perivascular blocks as well as intrathecal and epidural infusions of fentanyl, local anesthetics, clonidine, and ziconotide, which all resulted in brief symptom mitigation (up to 48 hours) followed by her characteristic flare lasting up to 4 weeks. Epidural catheter placement resulted several times in insertion-site erythema, pain, and sterile drainage in the absence of systemic signs of infection. In one instance, 48 hours after the initiation of an epidural infusion of 0.25% bupivacaine, she developed sudden onset complete lower extremity motor weakness and saddle anesthesia that resolved 3 hours after terminating the infusion.
Given the intractability of her symptoms, she was enrolled in a clinical trial testing of a topical small-molecule NaV1.7 inhibitor, XEN402, a novel compound that exhibits potent, voltage-dependent block of NaV1.7.4 She was prematurely discontinued from this trial due to dramatic desquamation that developed days after initial application of the inhibitor. With the goal of formulating our therapeutic strategy, we sought the results of her SCN9A exon sequencing, which was performed during the clinical trial. Surprisingly, it revealed a wild-type sequence for the SCN9A gene encoding the NaV1.7 α-subunit. This ruled out all known coding region genetic defects linked to inherited EM as well as any novel mutations.
Discussion
In this case report, we describe the condition of a patient severely affected by erythema, swelling, and pain in her hands and feet that progressed in spite of multiple therapeutic trials. Complex regional pain syndrome (CRPS) is a disease affecting the extremities that often develops after fracture or minor injury,5 as was the case with this patient. However, while she does meet the criteria for CRPS (including vasomotor, sudomotor, motor, and sensory signs),6 the triggering of her symptoms by even mild increases in ambient temperature or activity, the use of cooling behaviors to achieve symptom relief, and predominance of erythema and burning are highly suggestive of EM as the primary diagnosis.
In 2004, the SCN9A gene was implicated in the pathogenesis of inherited EM,2 and since then, an impressive amount of research has been performed to produce targeted inhibitors of the NaV1.7 channel.4,7 Interestingly, however, the heterogeneous clinical presentation of patients with EM explains the phenotype–genotype discrepancy of this disease,7,8 as even patients with the same mutation can exhibit a range of phenotypes.3,9 Nevertheless, genetic testing may still help develop targeted treatment strategies, as certain NaV1.7 mutations have been shown to be exquisitely responsive to specific agents such as mexiletine8,10 and carbamazepine.11,12
This case report highlights the challenge in managing symptoms of primary EM in patients for whom genetic testing rules out an SCN9A exonic mutation. Interestingly, one recent study of 48 patients meeting clinical criteria for EM found that only seven of the 48 patients carried protein-modifying mutations of NaV1.7, whereas 20 patients had rare protein-modifying mutations in genes encoding other sodium channel α- or β-subunits, transient receptor potential channels, or other pain-related targets.13 This suggests that screening our patient for mutations beyond the SCN9A gene may identify additional targets and clarify the genetic basis of her condition.
Treatment for our patient began with local anesthetics (LAs) in an effort to directly dampen aberrant sodium channel activity. Sodium channel-binding sites of LAs are highly dynamic, as LAs’ binding affinity is voltage-dependent and the charges within binding sites fluctuate as the channel cycles change between four different conformational states. Thus, several different LAs were tried given their varied state-dependent binding preferences, with lidocaine having greater affinity to the open state, for example, and flecainide to the closed state.14 Antiepileptics were then tried as their sodium channel antagonism is produced via a different mechanism allowing them to preferentially inhibit the high-frequency repetitive firing of hyperexcitable neurons.15 Even at high doses, however, antiepileptics, such as carbamazepine, oxcarbazepine, lamotrigine, and topiramate, were not effective. Consequently, our patient was enrolled in a clinical trial testing of topical application of a novel NaV1.7 small molecule inhibitor. Unfortunately, however, it was negligibly therapeutic for her, and she was discontinued from the trial due to severe desquamation.
We then extended our approach to target the vasculature using agents such as calcium channel blockers, TrpV1 antagonists, phosphodiesterase inhibitors, and nitrates. These vasoactive drugs notably increased our patient’s swelling, pain, and lesion/ulcer formation independent of the delivery method. Then, we hypothesized that dantrolene, a ryanodine receptor antagonist that blocks intracellular calcium release from the sarcoplasmic reticulum, may be beneficial. Dantrolene is effective in malignant hyperthermia, a pathological state involving build-up of available intracellular calcium leading to cellular hypermetabolism, and notably, increased autonomic activity. In our patient, IV dantrolene did not impart symptom relief. Instead, she experienced multiple side effects including eye pain, myalgias, slurred speech, and vomiting that continued for 2 weeks after cessation of the drug.
The characteristic symptoms of EM are both sensory and autonomic in nature, reflecting the known distribution of sodium channels in sensory and sympathetic ganglia.16 In our patient, clonidine and ketamine effectively mitigated her symptoms but only when administered systemically, suggesting that a central site of action was necessary for efficacy. As an adrenergic α2-receptor agonist, clonidine may address sympathetic overdrive in EM by blunting excess central outflow via medullary α2-receptors and peripherally counteracting aberrant vasoconstriction. Ketamine, an N-methyl-D-aspartate (NMDA)-receptor antagonist, has been effective in treating CRPS, a pain disorder marked by local sympathetic dysfunction.17 The mechanism by which ketamine blunts the apparent sympathetically mediated pain in these patients is unclear; however, inhibition of pathological synaptic changes has been suggested by a study which shows that NMDA-receptor antagonism impedes synaptic plasticity.18 The fact that EM symptoms typically affect the extremities in sequence suggests that synaptic plasticity may contribute to the progression of the disease.
We therefore suggest that successful management of patients with EM can be achieved when three major goals are simultaneously addressed: primary analgesic management, dampening sympathetic outflow, and reversing excessive vasoconstriction. Genetic testing may be informative if an SCN9A mutation is identified that may be responsive to specific agents. As our understanding of the pathophysiology and diverse genetic basis of primary EM continues to expand, the development of directed definitive treatments for this debilitating disease will continue to evolve. Until then, cases like this emphasize the need for multimodal approaches to achieving symptomatic relief for our patients.
Acknowledgment
Dr Tawfik is funded by the Foundation for Anesthesia Education and Research (FAER).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Drenth JP, Michiels JJ. Erythromelalgia and erythermalgia: diagnostic differentiation. Int J Dermatol. 1994;33(6):393–397. | ||
Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–174. | ||
Davis MD, O’Fallon WM, Rogers RS 3rd, Rooke TW. Natural history of erythromelalgia: presentation and outcome in 168 patients. Arch Dermatol. 2000;136(3):330–336. | ||
Emery EC, Luiz A, Wood JN. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets. 2016;20(8):1–9. | ||
de Mos M, de Bruijn AG, Huygen FJ, Dieleman JP, Stricker BH, Sturkenboom MC. The incidence of complex regional pain syndrome: a population-based study. Pain. 2007;129(1–2):12–20. | ||
Harden N, Bruehl S, Perez RS, et al. Validation of proposed diagnostic criteria (the ‘Budapest Criteria’) for Complex Regional Pain Syndrome. Pain. 2010;150(2):268–274. | ||
Cao L, McDonnell A, Nitzsche A, et al. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med. 2016;8(335):335ra56. | ||
Waxman S, Merkies IS, Gerrits MM, et al. Sodium channel genes in pain-related disorders: phenotype–genotype associations and recommendations for clinical use. Lancet Neurol. 2014;13(11):1152–1160. | ||
Estacion M, Han C, Choi JS, et al. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain. 2011;7:92. | ||
Choi JS, Zhang L, Dib-Hajj SD, et al. Mexiletine-responsive erythromelalgia due to a new Nav1.7 mutation showing use-dependent current fall-off. Exp Neurol. 2009;216(2):383–389. | ||
Fischer TZ, Gilmore ES, Estacion M, et al. A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol. 2009;65(6):733–741. | ||
Geha P, Yang Y, Estacion M, et al. Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol. 2016;73(6):659–667. | ||
Zhang Z, Schmelz M, Segerdahl M, et al. Exonic mutations in SCN9A (NaV1.7) are found in a minority of patients with erythromelalgia. Scand J Pain. 2014;5(4):217–225. | ||
Sheets MF, Fozzard HA, Lipkind GM, Hanck DA. Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc Med. 2010;20(1):16–21. | ||
Lipkind GM, Fozzard HA. Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol Pharmacol. 2010;78(4):631–638. | ||
Dib-Hajj S, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2012;14(1):49–62. | ||
Schwartzman RJ, Alexander GM, Grothusen JR, Paylor T, Reichenberger E, Perreault M. Outpatient intravenous ketamine for the treatment of complex regional pain syndrome: a double-blind placebo controlled study. Pain. 2009;147(1–3):107–115. | ||
Woolf CJ, Thompson SW. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44(3):293–299. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.