")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Complement system and age-related macular degeneration: drugs and challenges
Received 22 February 2019
Accepted for publication 1 July 2019
Published 19 July 2019 Volume 2019:13 Pages 2413—2425
DOI https://doi.org/10.2147/DDDT.S206355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Jiali Wu,1 Xiaodong Sun1–3
1Department of Ophthalmology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China; 2Shanghai Key Laboratory of Fundus Disease, Shanghai, People’s Republic of China; 3Shanghai Engineering Center for Visual Science and Photomedicine, Shanghai, People’s Republic of China
Abstract: Age-related macular degeneration (AMD) is directly attributable to vision loss, posing significant pressure on public health. AMD is recognized to be a multi-factorial disease and among them, complement system is under heated discussion in recent years. In this review, we start with an overview of complement pathways involved in AMD and their therapies correspondingly. Finally, we discuss the development of the therapeutics existed now. Also, we enclose a list of drugs undergoing clinical trials.
Keywords: age-related macular degeneration, inflammation, complement system, immunotherapy
Introduction
Age-related macular degeneration (AMD) is a progressive disease and the leading cause of irreversible vision impairment among people over 50 years old in Western populations.1 Asia, especially People’s Republic of China, is undergoing similar scenario now. Although Asia has a relatively low prevalence right now, it is estimated to see a lot more AMD patients considering the large population.2 Up to 26.65 million people were suffering from AMD in 2015. It is projected to reach 31.23 million in 2020 and 55.19 million in 2050 in People’s Republic of China.3 There also sees a surge in AMD prevalence, bringing out severe financial burden and time constraints on patients.
AMD is classified into two stages: early and late stage. Clinical examinations can see drusen, the yellow deposits under the retinal pigment epithelium in the central region of the retina in early AMD.4 It can also be differentiated in the other way: exudative (wet) AMD and geographic (dry) AMD (Figure 1). Repeated injection of anti-vascular endothelial growth factor (VEGF) has become a well-recognized therapy for patients with wet AMD. However, there is no effective treatment yet for dry AMD to slowdown the progression5 while it occupies approximately 90% of the AMD patients.
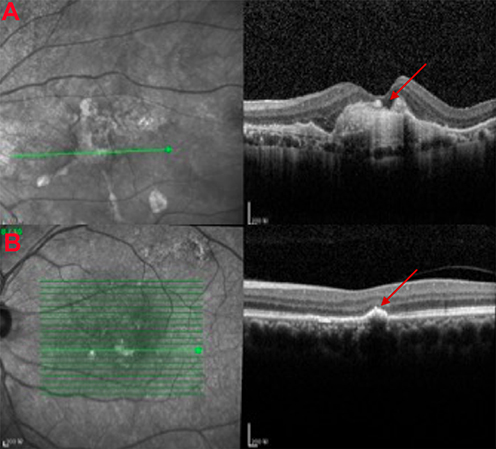 |
Figure 1 Clinical manifestations of age-related macular degeneration. (A) Images of optical coherence tomogram (OCT) (above) of choroidal neovascularization (CNV). Continuity of the retinal pigment epithelial is destroyed with local thickening and protuberance (red arrow). (B) Images of OCT of drusen (below) (red arrow). |
AMD is presumed to be a multi-factorial disease.6 The age group that contributed most was 60–64 years in 2015 and actually, aging is the strongest demographic risk factor for AMD. Except for aging, smoking is also a common environmental factor in Asians.7 Among its complex pathogenesis, immune dysfunction is one of the recurring topics currently.8 Among the immune dysfunction, the complement pathway is the most widely discussed and well established as emerging evidence suggests its causative role in the development of AMD. Several complement members such as complement component 5 (C5) and C3 have been identified in drusen,9,10 proving that complement dysregulation is an important element in drusen biogenesis.11 Furthermore, early in 2005, genetic evidence has been discovered between AMD and variants of complement pathway-associated genes. A common variant in complement factor H increased a 7.4- fold risk in developing AMD in individuals homozygous for this risk allele.12 Recent years also saw identification of multiple genes involving in early-onset and familial occurrence of AMD. Nearly all of them are located in complement genes, emphasizing the role of complement system.13,14 From this perspective, complement inhibition is a potential target to treat AMD. In this review, we will discuss diverse immune cell types involved in AMD and their therapies accordingly.
The complement pathways
The complement system (Figure 2) works as the major part of the innate immune system, defending the foreign pathogens and modified self-tissues. It makes up of three pathways: classical pathway (CP), lectin pathway (LP) and alternative pathway (AP). Each has its own trigger15 while they seem to merge sometime to activate the same protein: complement component 3 (C3).16 The CP usually responds to antigen-antibody complexes. It is initiated through the interaction between C1q and antibodies. After the activation of C1 complex, it cleaves the complement C2 and C4 into C2a, C2b, C4a and C4b. C4b and C2a bind to form C4b2a, which is a C3 convertase. It further binds to C3b to form C5 convertase and so on. The LP is usually activated by binding mannose residues, either via mannose-binding lectinor ficolin.17 They resemble C1q and bind to the homologues of C1r and C1s called Mannan-binding lectin-associated serine proteases (MASPs). MASP-1, MASP-2 and MASP-3 are discovered in turn. MASP-2 is similar to C1s in catalytic specificity and able to cleave C2 and C4.18 The AP is implicated closely with the pathogenesis of AMD in many studies.19 It is regarded as the amplification mechanism for the other two pathways. It is usually initiated by C3 convertase to cleave complement C3 into C3a and C3b. Both C3 and C5 convertases play a vital role in forming the final product membrane attack complex (MAC) and cell lysis.20
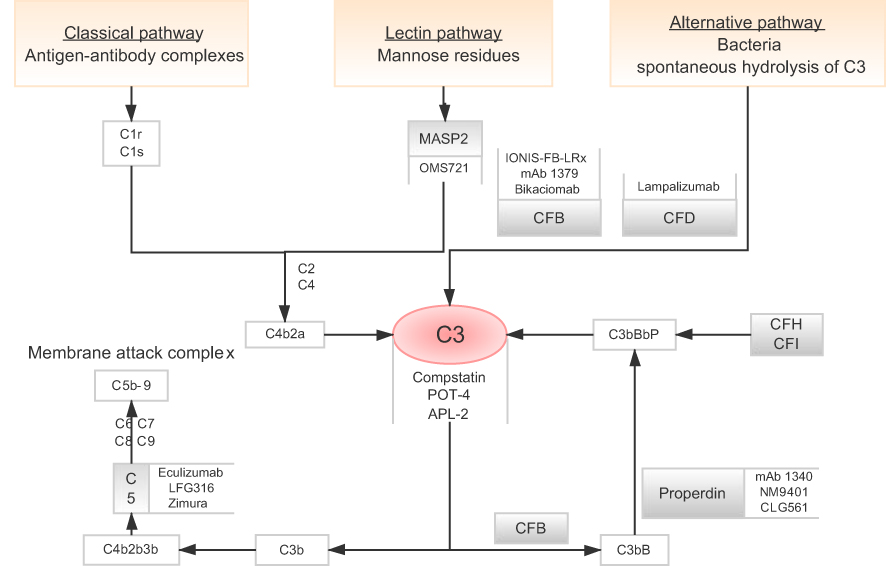 |
Figure 2 Flow diagram shows three separate complement pathways: lectin pathway, alternative pathway and classical pathway (orange box). All three pathways converge at complement protein C3 (red circle). Many drugs targeting specific complement components are being studied (gray line).Abbreviations: CFB, complement factor B; CFD, complement factor D. |
The suspects involving complement system in AMD dated back to the reports that revealed drusen, the early marker of AMD contained a large number of complement proteins in the mid-1990s.11,21–23 Since that, discussion on complement cascade started in the field of AMD therapy.
Drugs targeting specific complement components in AMD
Complement component 3 (C3)
C3 is a dominant figure in the complement cascade, presumably controlling the entire complement system. The cleavage of C3 by C3 convertases starts the convergence of three activation pathways to the terminal phase via an amplification loop, producing more C3b and C3a and further contributes to the cell destruction.24 Researchers found that C3 knockdown mice did not develop choroidal neovascularization (CNV) after laser photocoagulation, elucidating the essential role C3 plays in AMD development.25 Hence, C3 becomes an attractive target in the complement system. However, its powerful effect makes it impossible for us to ignore the risk of inhibiting its function completely. The complement system functions as the first defense line of innate immune response, protecting human organism from infections.26 The balance between its therapeutic benefits and risks brought out by general inhibition should be recognized clearly, especially considering AMD as a chronic disease which needs long-time treatment.27 Some researchers proposed that probably we can find a so-called adequate extent of inhibition instead of totally complete inhibition.28 For example, safety concerns on increased susceptibility to infections may be less pronounced when inhibitors are administered locally.24
Compstatin (POT-4, AL-78898A)
Compstatin is a 13-residue, cyclic peptide that selectively binds to C3b and C3c.29 POT-4 (AL-78898A), a compstatin analog, was the first complement inhibitor to be tested in macular degeneration which has reignited a vibrant interest in this field.30 An early experiment on eight monkeys showed some encouraging results. Diffusion of drusen in the macula was noticed after 6 months of injection in four monkeys with 50 μg dose at 1-week interval and partial disappearance of drusen was further appeared by 9 months in all four monkeys, while higher level of drug dose (1 mg) exhibited unfavorable results.31 This might be caused by the unique characteristic of compstatin. After injection, it precipitates and forms a kind of gel in the vitreous which takes about 6 months to dissolve gradually. So it is possible that overdose might lead to opacity. AL-78898A via intravitreal injection significantly reduces the light-induced elevation of complement C3a on cynomolgus monkeys.32 Afterward, it was developed for application and showed some clinical efficacy in its Phase I trial (NCT00473928). However, both Phase II trials (NCT01157065, NCT01603043) failed to duplicate the success of Phase I.33,34 Much-reduced dose used in the Phase II trials than that in Phase I was considered to be part of the reasons.29 However, AL-78898A still remains valuable given the early success it has achieved. New trials with improved design and dosage should be carried out to reassess the feasibility.
APL-2
APL-2 is a synthetic cyclic peptide conjugated to a polyethylene glycol polymer35 that binds specifically to C3 and C3b, blocking all the three complement activation. In its Phase II trial FILLY enrolling 246 patients, APL-2 presented impressive reduction in the rate of GA lesion growth: 29% reduction in the GA lesion growth compared to control group after 12 months via intravitreal injection monthly.36 When analyzed post hoc, the monthly dosing group witnessed a significant 47% reduction when the every-other-month dosing group saw 33% in the second 6 months of the study,37 suggesting a better outcome with a higher injection frequency. However, it is noteworthy that APL-2 therapy group has a higher risk for developing wet AMD. In the monthly dosing group, the risk was 18% compared to 8% in every-other-month dosing group and only 1% in the control group.38 Whether there is any relationship between APL-2 and wet AMD remains uncertain, but this is a point we should figure out. Still, Apellis Pharmaceuticals (APLS) saw potential in its statistically significant effect in the second 6 months of the study; thus, they planned to move forward with Phase III studies as soon as possible. The Phase III trial (NCT03525600) with 600 enrolled participants is assigned to four arms. Experimental groups receive 15 mg APL-2 monthly or every other month for 24 months via intravitreal injection.39 So far, the recruitment is still going on.36
Complement component 5 (C5)
C5 is an effector molecule that acts downstream of C3,16 thus it may be a safer target posing less influence on the opsonization functions of complement system. Selective inhibition of C5a via intravitreal injection is reported to be effective and safe in reducing vascular leakage and CNV area significantly in laser-induced CNV in mice.40 But some research also claimed the contrary. Christopher B. Toomey reported41 the potential of C5a blockade as both monotherapy and combination therapy with anti-VEGF agents in wet AMD; however, he found it still insufficient to stop the progression in early/intermediate dry AMD in mice models.
Eculizumab
Eculizumab (Soliris) is the first FDA-approved anti-C5 humanized monoclonal antibody (mAb) for the systemic treatment of paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome that came to the market in 2007. Eculizumab blocks C5a and C5b, thus abrogating its downstream pathways that contribute to AMD. The COMPLETE study (NCT00935883) applied it in the treatment of geographic atrophy (GA) via intravenous infusion to assess its safety and efficacy in AMD.42 The trial enrolled 30 patients with a follow-up of 52 weeks. Results showed no elimination in enlargement rate of GA in the eculizumab group from the placebo group. Blood samples collected showed that C5 activity did decrease to <1% of normal levels by week 2 after treatment, proving the ability of eculizumab in inhibiting C5; however, this inhibition has made no contribution to treatment effects so far. The authors provided three possible explanations: 1) complement activation does not work in the growth of GA at all. Even if many researchers assume that complement activation plays a role in AMD, it is still a question today whether it only has limited influence on a certain duration of the disease. GA is classified as an advanced form of AMD, in which complement inhibitors can probably no longer make any change. 2) Inappropriate dose or delivery way of eculizumab. Giving drug systemically probably cannot reach the adequate level in the retina. The large size of eculizumab (148 kDa) makes it difficult to penetrate to the major site of complement deposition of AMD43 while the lack of tools to monitor the complement activities in the eye makes it also difficult to detect. Searching for an analog of eculizumab with a smaller molecule size can help to rule out this possibility. About the delivery way, administration intravitreally can achieve more direct results.44 But relatively, it can be only applied to the single eye at single time and a series of adverse effects like endophthalmitis could happen due to intravitreal injection. 3) C5 inhibition prevents only terminal complement activation with no effect on the earlier part of the complement pathway. This controversy has been existed up to now. Blocking C3 rather than C5 is a more direct but less sound way without doubt. All these might explain the failure of eculizumab in thwarting GA progression. To be mentioned, when eculizumab is applied to PNH, similar depressing clinical response is also reported. Research supposed that insufficient dosing and/or pharmacodynamic breakthrough might be the contributor.
Combining eculizumab with an agent controlling upstream activation or another C5 inhibitor can achieve better efficacy than eculizumab monotherapy.45 In this way, clinical trials can be carried out to combine eculizumab with C3 inhibitors in a bid to ramp up the efficacy. Researchers should also pay high attention to the risk of inhibiting general complement pathway. In addition, as C3 works in the upstream of C5, we should also consider if such combination can make any complimentary improvement to C3 mono-inhibition.
Despite of this failure, many researchers still expressed their interest in it as they believed what really matters behind GA is MAC. MAC, as a complement effector, is formed by C5b, C6 and some other soluble proteins. As the final product of the activated complement cascade, the importance of MAC has been discussed extensively. The MAC level increases in both normal aging and diseases, including AMD. Puran S. Bora25 suggested that the CNV will not occur at all without the formation of MAC. Kenneth J. Katschke, Jr.46 suggested that inhibiting both C5a and C5b provides better protection on the retina over inhibiting C5a alone. Some researchers also indicated that MAC assembly has to be initiated near C5 convertase.47 In view of these, MAC is closely bound up with C5 and actually plays a role in GA. What is more, MAC accumulation occurs along the microvascular injury, which is the early sign of AMD.48 Targeting MAC may be a protective therapy before advanced GA actually happens. CD59, a glycosylphosphatidylinositol-anchored membrane inhibitor was found inhibitive to MAC in a fairly strict setting, but no evidence has reported about its inhibition in vivo thus far.49 AAVCAGsCD59 is an ocular gene therapy product that can increase the expression of sCD59, a soluble form of CD59 via intravitreal injection and it is now in the Phase I clinical trial (NCT03144999). As a gene therapy, the study expects to apply a single injection for whole life and evaluates its safety. We are looking forward to its results. Despite of the growing focus on MAC now, the clinical efficacy of eculizumab and other anti-C5 antibodies is still necessary to be elucidated, as C5b is closely involved in the formation of MAC.
LFG316
LFG316 is a fully human IgG1 targeting C5 and inhibits CP or AP. In its early experiments, results indicated that LFG316 could bind to C5 and prevent its cleavage.50 However, it did not demonstrate any success in its 12-month Phase II trial, announced on 6 February 2016, at the Angiogenesis, Exudation and Degeneration conference in Miami, USA.51
LFG316 has an acceptable safety profile … for further development, but monthly treatment with LFG316 monotherapy was not efficacious, either in GA lesion reduction or in visual acuity [gains]
Parisa Zamiri, MD, PhD said.52
Avacincaptad pegol
Zimura (avacincaptad pegol, ARC1905) is a selective C5 inhibitor. It was at first planned to be administered in combination with anti-VEGF agents to provide synergistic merits to anti-VEGF monotherapy.53 Supplementing anti-VEGF therapy with a complement inhibitor such as Zimura is a way believed to have the potential to further enhance the efficacy of anti-VEGF monotherapy in wet AMD. The Phase II study (NCT03362190) estimated to enroll 60 patients and finished on 15 November 2018. It aims to establish the safety and tolerability of Zimura with Lucentis® 0.5 mg in wet AMD but no results have been posted on ClinicalTrials.gov yet.54 The Phase IIb trial (NCT02686658) with estimated enrollment of 200 participants in an attempt to evaluate the safety and efficacy of intravitreous administration of Zimura monotherapy in GA patients has finished the data collection now.55
Complement factor D (CFD)
Factor D works as the rate-limiting enzyme, activating the AP and amplificating the complement response.56 It is reckoned as a prime candidate to inhibit the AP given that it occupies the rather upstream position. Also, it has the lowest plasma concentration among the complement proteins.57 Anti-complement factor D blocks factor D cleavage of factor B to factor Bb, thus inhibiting the AP while the other two pathways still remain intact.58 Researchers tried to apply the anti-human FD antibody in rabbits via intravitreal delivery.59 Since FB is the substrate of FD, researchers investigated the FD inhibition through FB breakdown product Ba. Both Ba generation in plasma and anterior segment were not affected by intravitreal anti-FD antibody. This result indicated that ocular anti-human FD antibody is generated rather locally with barely any systematic influence.
Lampalizumab (FCFD4514S)
The anti-complement FD Fab lampalizumab is the first therapeutic antibody that demonstrated the farthest therapeutic effect toward clinical use. M. van Lookeren Campagne60 elucidated its potent and selective blockade of AP activation through binding to the C-terminal portion of FD. Preclinical tests based on cynomolgus monkeys suggested the potential of intravitreous injection (IVT) administered anti-complement FD as a treatment of AMD. Serum AP activity was reduced between 2 and 6 hrs post-injection for the 20 mg IVT dose group compared to the range of 5 mins to 3 hrs in 0.2and 20 mg intravenous groups. The minimum serum AFD levels should be ≥2 µg/mL to make some difference.44
In its Phase Ia study (NCT00973011), researchers found its maximum tolerated dose was 10 mg.61 The maximum serum concentration following a single 10 mg IVT dose was only 257 ng/mL, which was eightfold lower than 2 µg/mL, suggesting 10 mg IVT dose may not be enough to make any influence on the systemic FD activity.44 In its Phase II study called MAHALO (NCT01602120) with 129 participants, a significant 20.4% reduction rate in GA lesion area was reported with 10 mg lampalizumab monthly after 18 months.62 It has two Phase III clinical trials called CHROMA (NCT02247531) with 906 participants and SPECTRI (NCT02247479) with 975 participants, both applying 10 mg dose every 4 or 6 weeks. Failure was declared in SPECTRI to meet its primary endpoint of reducing mean change in GA lesion area in patients treated with lampalizumab compared with sham treatment during 48 weeks of treatment.63 Differences in adjusted mean change in GA lesion area (lampalizumab minus sham) were 0.05 mm2 for lampalizumab every 6 weeks in CHROMA, and 0.09 mm2 for lampalizumab every 6 weeks in SPECTRI.64 But in MAHALO, it was −0.6 mm2 for lampalizumab monthly after 18 months, which is totally opposite.65 As lampalizumab has been given such great expectations before, this failure was so devastating that some researchers even hesitate again whether complement cascade is an appropriate therapeutic target. The reason behind the failure has not been elucidated clearly but according to Frank G. Holz, they inclined to believe MAHALO as a sweet accident.64 This completely subverted the original assumption.
Anti-MASP-2
OMS721
MASP-2 functions in translating binding of the lectin complement pathway-recognition complexes into complement activation. C3b generation is inhibited and LP activation is prevented through solely blocking MASP-2 as it can autoactivate and generate a C3 convertase by its own.66 OMS721 is a humanized IgG molecule against MASP-2. To date, there are no clinical trials identified on ClinicalTrials.gov about OMS721 in GA. But some researchers indicated that anti-MASP-2 antibodies intraperitoneally in advance reduce 50% of the CNV in the laser-induced wet AMD afterward.67 To be clear, the feasibility and efficacy of OMS721 will remain intriguing until its debut in clinical trials. But as József Dobó pointed out, when only MASP-2 was inhibited by anti-MASP-2 agent, the level of C3 deposition only dropped to 61% of the non-inhibited value. While only MASP-1 was inhibited, the level decreased to 43.5%. If both inhibited, the level decreased sharply to 3.3%.66 MASP-1 is usually overqualified as an auxiliary enzyme when put together with MASP-2, but it seems to make some amelioration in this regard. What level should C3 inhibition achieve is still far from conclusive. It is also possible that OMS721 alone will not wield encouraging influence just like many other anti-complement factors. Combining OMS721 with another anti-MASP-1 agent is also a way worth a try.
Anti-properdin
Properdin is the only positive complement regulator that acts by stabilizing C3 and C5 convertases and initiating the AP. It was reported to be detected in 50% of the patients with wet AMD.68 Many evidence support that anti-properdin shows counterintuitive effects on C3 inhibition.69
mAb 1340 and NM9401
Both of the two antibodies bind properdin with high avidity and prevent the interaction with C3, causing the cessation of AP in vitro.70 According to Diana Pauly, mAb 1340 was up to 15 times more efficient in inhibiting the complement pathway compared with C5 antibodies in vitro.71 Such binding seems to be more specific as some mentioned that therapeutic targeting of properdin may produce less side effects than inhibiting other components of the AP.72 To some extent, we can deduce that inhibiting the complement pathway through inhibiting properdin wields less influence on the function of rest complement pathway, thus retaining necessary cascade function. Theoretically, we can speculate that they will achieve similar effects in vivo. But temporarily, both anti-properdin remain preclinical.
CLG561
CLG561 was reported fail to slow the enlargement of GA in the Phase II study at the Angiogenesis, Exudation and Degeneration 2016 meeting in February.73 The Phase I clinical trial of CLG561 (NCT01835015) enrolled 50 participants with the follow-up up to 84 days in 2014. Generally, improvement of best corrected visual acuity (BCVA) was seen in different dose groups, however, the IOP also increased. The most common adverse effect is conjunctival hemorrhage, effecting 9 of 31 participants.74 The Phase II clinical trial of CLG561 (NCT02515942) enrolled 114 participants with the follow-up of 16 weeks. The purpose of this trial is to evaluate the efficacy of 12 intravitreal injections of CLG561 with the interval of every 28 days as well as the efficacy of CLG561 in combination with LFG316. This trial has finished on 1 December 2017, however, results showed no statistically difference between CLG561 group and CLG561+LFG316 group in BCVA.75
Anti-CFB
Complement factor B (CFB) is a serine protease that is active in the early stages of the alternative complement cascade.76 Although there seems no significant increase of FB by itself in AMD, it can bind to properdin-bound C3b and further be cleaved by FD. Inhibition of FB influences other factors competing to bind to PC3b, such as C3a and C5a.76 C3a is a potent anaphylatoxin and it is reported to be associated with the formation of basal deposits beneath retinal pigment epithelial.77 Furthermore, genetic studies have revealed that haplotype (H1) variant of FB is a risk factor for AMD, suggesting the relationship between FB and AMD.78 Therefore, anti-CFB might work.
Experiments on mice or cynomolgus monkeys have proved that subcutaneous injection instead of IVT can also markedly reduce the ocular FB levels. In this way, the risk of inflammation can decrease.79
IONIS-FB-LRx (ISIS 696844)
IONIS-FB-LRx affect AP through directly reducing the production of FB. It has undergone the Phase I trial (ACTRN12616000335493) with the sample size of 30 participants in 2017 to evaluate the safety and tolerability of two different doses (10 and 20 mg) administered subcutaneously. According to Daniel Ricklin, IONIS-FB-LRx showed dose-dependent reduction of FB level up to 50%.45 The Phase II clinical trial of IONIS-FB-Lrx (NCT03446144) has carried out to assess the safety and efficacy of it with an estimated enrollment of 120 patients over a 69-week treatment period; however, it is withdrawn now due to the business objective change.80
T0A106 (mAb 1379) and Bikaciomab (NM9308)
MAb 1379 also inhibits AP at the level of factor B, keeping the rest complement system intact. Up to 4 μg of mAb 1379 is needed to fully inhibit AP in 10 μL of sera from various of species in vitro. For example, a 1 mg dose per intraperitoneal injection is able to completely take effect for 48 hrs at maximum in mice.81
Bikaciomab is a murine IgG, reducing immune effector functions.
To date, there are no clinical trials about these two identified on ClinicalTrials.gov on GA yet.
IBI302
Most drugs nowadays are confined to a single target and there seems room for their efficacy. Some began to consider about the feasibility of combination therapy such as combining two different complement inhibitors or combining complement inhibitor with anti-VEGF agents as I have mentioned before. Anti-VEGF agents are the most widely used treatment for AMD nowadays. IBI302 (Innovent) is a fairly novel bispecific decoy receptor fusion protein which inhibits both VEGF and complement cascade at the same time. The affinity characteristics and pharmacokinetics of it have been evaluated in vitro and in rhesus monkeys. With a single IVT injection, retina and choroid can still detect IBI302 at 504 hrs after dose while the highest level is seen in vitreous humor, suggesting its well distribution and long remaining time. The binding affinity of IBI302 for complement proteins is up to 9.96 nM for C3b and 7.75 nM for C4b. Besides, its binding affinity to VEGF isoforms is also high, similar to aflibercept, a common anti-VEGF agent. The molecular size is approximately 156 kD, which is larger than eculizumab, suggesting the need to consider about the penetration problem. No concrete rate of penetration has been reported, but researchers are positive about its effective tissue targeting ability due to rather low systemic IBI302 level.82 Study about IBI302 is limited up till now, but we can infer that IBI302 is a potential candidate for AMD based on its favorite PK profiles and affinity ability revealed in existing information. IBI302 is undergoing the Phase I clinical trial now and we are really looking forward to the results.
Challenges and discussion
Complement activation takes part in many neurodegenerative diseases. With the growing awareness that complement system also plays an integral role in AMD, there sees a surge in novel immune-modulatory strategies in recent decades.
However, to date, approved treatment options still remained so scarce considering so many candidates undergoing clinical trials (Table 1). Despite the fact that most of the agents seemed promising in theory and even in vitro, their efficacy was still far from satisfactory when being evaluated in clinical trials.
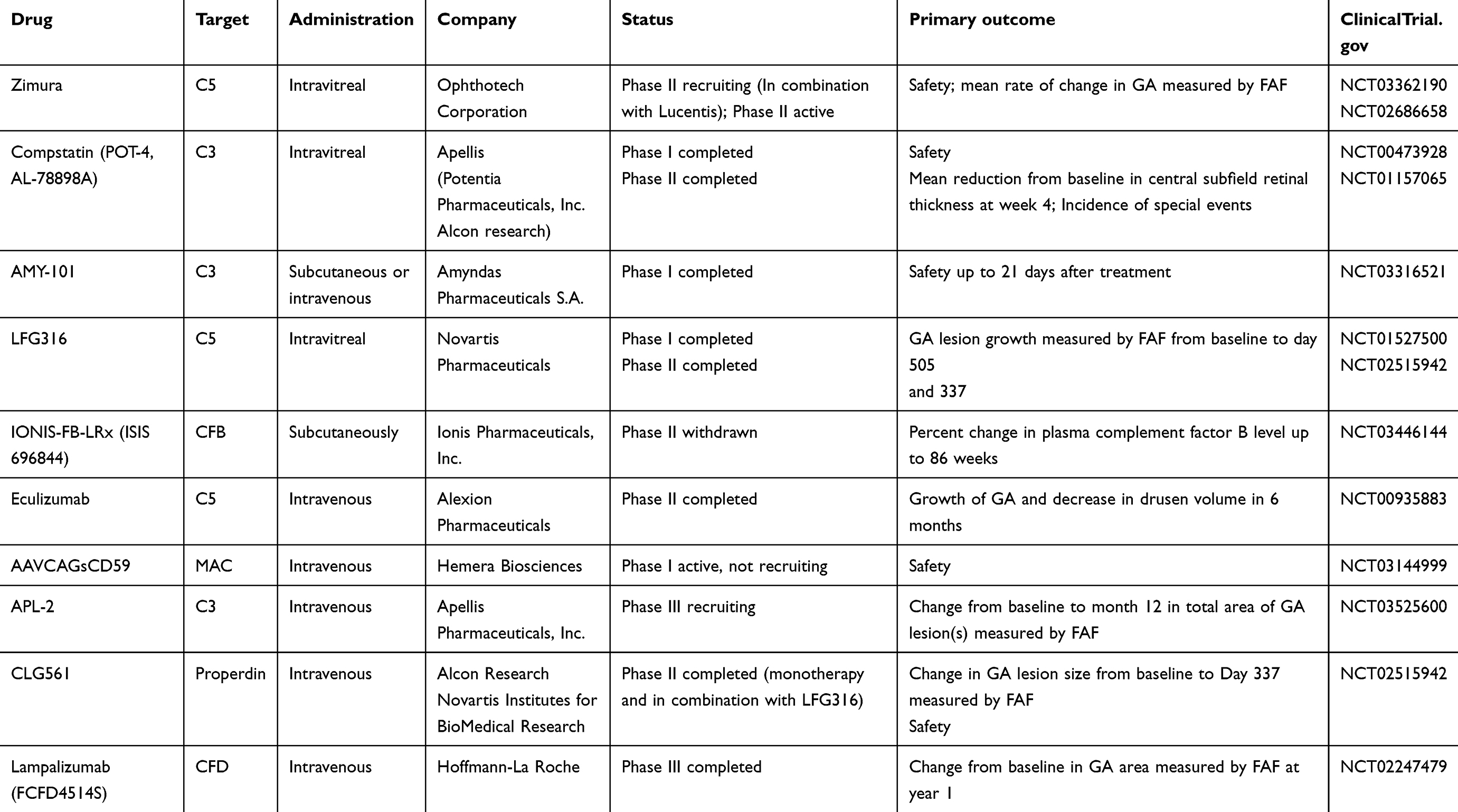 |
Table 1 Complement therapeutics in clinical trials |
Below categories are several possible reasons for the failure in treatment options.
Action stage
GA is the advanced stage of AMD. Nearly all the complement inhibitors today focus on treating GA that has already taken place but barely succeeded in reversing it. Such limited effect seems to suggest that the intervention should be initiated at an earlier action stage. What about applying them before GA really happens? Will they help postponing or even prevent the progression from the early stage of AMD? This is a new point of research now. As we have not found out the exact initiator of AMD, it is intricate to choose the appropriate target level and pathway.
Precise and early diagnosis
Innovation should be fostered to achieve a more sensitive evaluation, not only when diagnosing but also when assessing the treatment effect. Fundus examination is a rather recognized means in many trials ongoing recently, however, it does not necessarily correlate with progression of visual loss.83 The drawbacks of fundus autofluorescence also include susceptibility to media opacities and difficulty in imaging the fovea due to macular pigment that absorbs blue light.84 Moreover, although some researchers have raised the idea that combining autofluorescence imaging with other technologies like confocal scanning laser ophthalmoscopy or near-infrared images to help qualifying GA areas, most clinical trials now have not applied yet. Thus, quantitative analysis is still a challenge right now. One of the clinical trials of eculizumab applied the reduction of drusen volume based on spectral-domain optical coherence tomography (SD-OCT) as the endpoint while also ended with no significant outcome. Authors argued that probably inhibition of drusen growth rather than reduction of drusen volume works as a better parameter.85 Researchers also compared the atrophy area measured by FAF and SD-OCT. It turns out that any area measured by FAF was larger than that measured by SD-OCT, suggesting the unreliability of these existed means to some extent.86 BCVA, another common functional endpoint, is also far from enough. It often underrepresents the functional deficits.87 Actually, the process of visual loss is rather slow, therefore, patients can preserve the foveal function as lesions appear outside the fovea first. Based on the existed study, at least during the period of 12–24 months, eyes with foveal-sparing GA can show no obvious visual decrease.88 All these suggest that considering right now we have limited understanding about how complement therapy works out thoroughly, new and more measurement parameters should be applied together to achieve a more well-grounded assessment.
Rare variants
As we mentioned above, several rare variants recently identified contributing to AMD risk are located in the complement genes. It is rational that patients carrying certain rare variant in a complement gene might benefit more from the same complement inhibition treatment than others, especially if they carry the variants that severely affect complement activation. Some researchers believe that recognizing the predisposing genetic factors in AMD is helpful to identify the high-risk populations89 in the early period. For example, Arg1210Cys, a rare variant of CFH gene was found about 18 times more in AMD patients than in control individuals and these carriers had the disease onset 6 years earlier.90 In this way, uncovering the rare variants behind AMD is meaningful. But there is still much controversy about this. Nicole T. M. Saksens did not recognize the hypothesis that rare variant can make any difference in treatment efficacy.13 Also, in the well-known Phase III trial of lampalizumab, researchers debunked the claim proposed in its early MAHALO study that CFI-profile status worked as a genetic biomarker for progression of GA64 or CFI risk-allele group showed better response to lampalizumab. Larger cohorts should be carried out to identify more rare variants and find out the genetic association with AMD. This is helpful to unravel the pathogenic mechanisms.
Ocular pharmacokinetics and drug delivery issues
About the complement inhibitors themselves, ocular pharmacokinetics and drug delivery issues of the candidates are also important points to consider. The eye is a highly protected organ with the blood-ocular barrier preventing large molecules from crossing it and penetrating into the eye. Usually this so-called “large” is 76.5±1.5 kDa for human eyes. Therefore, some researchers blame the failure of eculizumab on its molecule size, which is 148 kDa. A smaller molecule size is desirable for its permeation if it is given intravenously while it may also cause easier clear by systemic circulation rapidly, decreasing the time of necessary therapeutic concentration.91 The size should be weighed between high penetration and high absorption. IVT is another common way of administration, which can broach the barrier directly.92 But it can also bring out adverse effect like increased IOP and subconjunctival hemorrhage due to such an invasive maneuver. In addition, IVT enables the drug concentration to peak immediately while has no improvement in maintaining the effective concentration. Both administration routes are used frequently now. By extension, different dose should be given according to these two different routes. Adequate dose is definitely the major premise for success. Studies on other novel delivery techniques pursuing long-lasting and stable effect are also urgent and fairly popular right now in AMD, especially in wet AMD. For example, nanocarriers have been a hot topic owing to their biocompatibility in delivering anti-VEGF agents in wet AMD.93 This affords a salutary lesson for GA. With the help of a better delivery system and improvement of drug design, the outcomes of the trials might be more encouraging.
Animal models
Accurate animal models help a lot in developing novel therapies, however, we all know that no animal model available now can faithfully recapitulate the complex anatomic features of human eye.94 Researchers usually use monkeys, rabbits and mice models. But they all have some defects. For example, mice do not have macular at all; the vascular endothelial cells of rabbits distributed unevenly and unstably. Results of animal experiments are not comparable to human trials. This might be part of the reason why some complement inhibitor achieved positive outcomes in animal experiments but failed in following clinical trials.
Actual role of complement modulation in AMD
Beyond these different reasons above, however, lies a fundamental doubt. That is the role of complement modulation in AMD. Researchers have reached the consensus that AMD is a multi-factorial disease and complement modulation is part of it, but what is its actual share in AMD pathogenesis? How it interacts with other pathological mechanisms like oxidative damage and lipid metabolism? Which subgroup of AMD it plays a role in? At this time, they are still not well understood. Therefore, it is urgent to clarify whether complement system plays a crucial role. Existed clinical trials on complement inhibitors have covered both dry and wet types, mostly in the field of GA. Some complement inhibitors have been evaluated in both types, such as LFG316.
Conclusion
Complement cascade plays a pivotal role in AMD pathogenesis, which has been revealed in so many studies in recent years. Thus, it is probably a fairly potential strategic target for therapy. But to date, there is no encouraging success in clinical trials to support this assumption. Further studies are urgent in order to provide a better understanding of the pathogenesis and identify valid therapeutic target.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Key Program) (81730026, 81425006), Frontier Project of Shanghai Hospital Development Center (SHDC12016105), Science and Technology Commission of Shanghai Municipality (17411953000, 16140900800, 0303N17001).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Miller JW. Age-related macular degeneration revisited–piecing the puzzle: the LXIX Edward Jackson memorial lecture. Am J Ophthalmol. 2013;155(1):1–35.e13. doi:10.1016/j.ajo.2012.10.018
2. Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–e111. doi:10.1016/S2214-109X(13)70145-1
3. Song P, Du Y, Chan KY, Theodoratou E, Rudan I. The national and subnational prevalence and burden of age-related macular degeneration in China. J Glob Health. 2017;7(2):020703. doi:10.7189/jogh.07.020703
4. Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. 2012;379:1728–1738. doi:10.1016/S0140-6736(12)60282-7
5. Nebbioso M, Lambiase A, Cerini A, Limoli GP, La Cava M, Greco A. Therapeutic approaches with intravitreal injections in geographic atrophy secondary to age-related macular degeneration: current drugs and potential molecules. Int J Mol Sci. 2019;20:7. doi:10.3390/ijms20071693
6. Modenese A, Gobba F. Macular degeneration and ocupational risk factors: a systematic review. Int Arch Occup Environ Health. 2019;92(1):1–11. doi:10.1007/s00420-018-1355-y.
7. Wong CW, Yanagi Y, Lee W-K, et al. Age-related macular degeneration and polypoidal choroidal vasculopathy in Asians. Prog Retin Eye Res. 2016;53:107–139. doi:10.1016/j.preteyeres.2016.04.002
8. Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013;13(6):438–451. doi:10.1038/nri3459
9. Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14(7):835–846.
10. McHarg S, Clark SJ, Day AJ, Bishop PN. Age-related macular degeneration and the role of the complement system. Mol Immunol. 2015;67:43–50. doi:10.1016/j.molimm.2015.02.032
11. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. PubMed: 11846519. doi:10.1006/exer.2001.1094
12. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi:10.1126/science.1109557
13. Saksens NTM, Geerlings MJ, Bakker B, et al. Rare genetic variants associated with development of age-related macular degeneration. JAMA Ophthalmol. 2016;134(3):287–293. doi:10.1001/jamaophthalmol.2015.5592
14. Geerlings MJ, de Jong EK, Den Hollander AI. The complement system in age-related macular degeneration: a review of rare genetic variants and implications for personalized treatment. Mol Immunol. 2017;84:65–76. doi:10.1016/j.molimm.2016.11.016
15. Khandhadia S, Cipriani V, Yates JRW, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2012;217(2):127–146. doi:10.1016/j.imbio.2011.07.019
16. Dolgin E. Age-related macular degeneration foils drugmakers. Nat Biotechnol. 2017;35(11):1000–1001. doi:10.1038/nbt1117-1000
17. Osthoff M, Dean MM, Baird PN, et al. Association study of mannose-binding lectin levels and genetic variants in lectin pathway proteins with susceptibility to age-related macular degeneration: a case-control study. PLoS One. 2015;10(7):e0134107. doi:10.1371/journal.pone.0134107
18. Wallis R, Mitchell DA, Schmid R, Schwaeble WJ, Keeble AH. Paths reunited: initiation of the classical and lectin pathways of complement activation. Immunobiology. 2009;215(1):1–11. doi:10.1016/j.imbio.2009.08.006
19. Loyet KM, DeForge LE, Katschke KJ, et al. Activation of the alternative complement pathway in vitreous is controlled by genetics in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2012;53(10):6628–6637. doi:10.1167/iovs.12-9587
20. Lorthiois E, Anderson K, Vulpetti A, et al. Discovery of highly potent and selective small-molecule reversible factor D inhibitors demonstrating alternative complement pathway inhibition in vivo. J Med Chem. 2017;60(13):5717–5735. doi:10.1021/acs.jmedchem.7b00425
21. Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. PubMed: 12208254. doi:10.1016/S0002-9394(02)01624-0
22. Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78:243–256. PubMed: 14729357. doi:10.1016/j.exer.2003.10.011
23. Hageman GS, Mullins RF. Molecular composition of drusen as related to substructural phenotype. Mol Vis. 1999;5:28. PubMed: 10562652.
24. Dobó J, Kocsis A, Gál P. Be on target: strategies of targeting alternative and lectin pathway components in complement-mediated diseases. Front Immunol. 2018;9:1851. doi:10.3389/fimmu.2018.01851
25. Bora PS, Sohn J-H, Cruz JMC, et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J Immunol. 2005;174(1):491–497. doi:10.4049/jimmunol.174.1.491
26. Zipfel PF. Complement and immune defense: from innate immunity to human diseases. Immunol Lett. 2009;126(1–2):1–7. doi:10.1016/j.imlet.2009.07.005
27. Weber BH, Charbel Issa P, Pauly D, Herrmann P, Grassmann F, Holz FG. The role of the complement system in age-related macular degeneration. Dtsch Arztebl Int. 2014;111:133–138.
28. Gorham RD, Forest DL, Tamamis P, et al. Novel compstatin family peptides inhibit complement activation by drusen-like deposits in human retinal pigmented epithelial cell cultures. Exp Eye Res. 2013;116:96–108. doi:10.1016/j.exer.2013.07.023
29. Mastellos DC, Yancopoulou D, Kokkinos P, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45(4):423–440. doi:10.1111/eci.12419
30. Leung E, Landa G. Update on current and future novel therapies for dry age-related macular degeneration. Expert Rev Clin Pharmacol. 2013;6(5):565–579. doi:10.1586/17512433.2013.829645
31. Chi Z-L, Yoshida T, Lambris JD, Iwata T. Chapter 9. Suppression of drusen formation by compstatin, a peptide inhibitor of complement C3 activation, on cynomolgus monkey with early-onset macular degeneration. Adv Exp Med Biol. 2010;703:127–135. doi:10.1007/978-1-4419-5635-4_9
32. Collier RJ, Smith S, Hoang H, et al. AL-78898A inhibits complement deposition in a primate light damage model. Invest Ophthalmol Vis Sci. 2012;53(14):5362.
33. ClinicalTrials.gov. Alcon research evaluation of AL-78898A in exudative age-related macular degeneration (RACE). 2013. NLM identifier: NCT01157065. Available from: https://clinicaltrials.gov/ct2/show/NCT01157065?term=01157065&rank=1.
34. ClinicalTrials.gov. Alcon research a multicenter, proof-of-concept study of intravitreal AL-78898A in patients with Geograhic Atrophy (GA) associated with Age-Related Macular Degeneration (AMD). 2013. NLM identifier: NCT01603043. Available from: https://clinicaltrials.gov/ct2/show/NCT01603043?term=01603043&rank=1.
35. What APL-2 means for geographic atrophy, Michelle Dalton, ELS. July 22, 2018. Available from: http://www.modernretina.com/asrs/what-apl-2-means-geographic-atrophy.
36. Apellis Pharmaceuticals. Apellis Pharmaceuticals announces that APL-2 met its primary endpoint in a phase 2 study in patients with geographic atrophy, an advanced form of age-related macular degeneration. August 24, 2017. Available from: http://apellis.com/pdfs/Press%20Release%20FILLY%2012%20Month%20Results%20FINAL%20FINAL%20170823.pdf.
37. Apellis’ geographic atrophy treatment meets phase 2 endpoint. Available from: https://www.healio.com/ophthalmology/retina-vitreous/news/online/%7bb24b7e1c-61fb-4b8b-baaa-13590a87dcec%7d/apellis-geographic-atrophy-treatment-meets-phase-2-endpoint.
38. APL-2 slows growth of GA in phase II safety and efficacy trial by Steve Lenier. February 26, 2018. Available from: http://www.modernretina.com/amd/apl-2-slows-growth-ga-phase-ii-safety-and-efficacy-trial.
39. ClinicalTrials.gov. Study to compare the efficacy and safety of intravitreal APL-2 therapy with sham injections in patients with Geographic Atrophy (GA) secondary to age-related macular degeneration. 2018. NLM identifier: NCT03525600. Available from: https://clinicaltrials.gov/ct2/show/NCT03525600?term=03525600&rank=1.
40. Brockmann C, Brockmann T, Dege S, et al. Intravitreal inhibition of complement C5a reduces choroidal neovascularization in mice. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2015;253(10):1695–1704. doi:10.1007/s00417-015-3041-z
41. Toomey CB, Landowski M, Klingeborn M, et al. Effect of Anti-C5a therapy in a murine model of early/intermediate dry age-related macular degeneration. Invest Ophthalmol Vis Sci. 2018;59(2):662–673. doi:10.1167/iovs.17-23134
42. Yehoshua Z, Filho CADAG, Nunes RP, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014;121(3):693–701. doi:10.1016/j.ophtha.2013.09.044
43. Mullins RF, Warwick AN, Sohn EH, Lotery AJ. From compliment to insult: genetics of the complement system in physiology and disease in the human retina. Hum Mol Genet. 2017;26(R1):R51–R57. doi:10.1093/hmg/ddx181
44. Loyet KM, Good J, Davancaze T, et al. Complement inhibition in cynomolgus monkeys by anti-factor D antigen-binding fragment for the treatment of an advanced form of dry age-related macular degeneration. J Pharmacol Exp Ther. 2014;351(3):527–537. In this important preclinical study of the anti-FD antibody lampalizumab, the researchers elucidate important aspects of local and systemic administration of a complement inhibitor considered for AMD treatment. doi:10.1124/jpet.114.215921
45. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. 2018;14(1):26–47. doi:10.1038/nrneph.2017.156
46. Katschke KJ, Xi H, Cox C, et al. Classical and alternative complement activation on photoreceptor outer segments drives monocyte-dependent retinal atrophy. Sci Rep. 2018;8(1):7348. doi:10.1038/s41598-018-25557-8
47. de Jorge EG, Yebenes H, Serna M, Tortajada A, Llorca O, de Córdoba SR. How novel structures inform understanding of complement function. Semin Immunopathol. 2018;40(1):3–14. doi:10.1007/s00281-017-0643-z
48. Chirco KR, Tucker BA, Stone EM, Mullins RF. Selective accumulation of the complement membrane attack complex in aging choriocapillaris. Exp Eye Res. 2015;146:393–397. doi:10.1016/j.exer.2015.09.003
49. Cashman SM, Ramo K, Kumar-Singh R. A non membrane-targeted human soluble CD59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS One. 2011;6(4):e19078. doi:10.1371/journal.pone.0019078
50. Roguska M, Splawski I, Diefenbach-Streiber B, et al. Generation and characterization of LFG316, a fully-human anti-C5 antibody for the treatment of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2014;55(13):3433.
51. LFG316 Found Ineffective. Available from: https://www.mdfoundation.com.au/content/lfg316-ineffective-dry-age-related-macular-degeneration.
52. Anti-complement C5 monotherapy ineffective in reducing geographic atrophy lesion size. 2016. Available from: https://www.healio.com/ophthalmology/retina-vitreous/news/online/%7Bb24258c2-0a16-425c-9476-a591485d3cf6%7D/anti-complement-c5-monotherapy-ineffective-in-reducing-geographic-atrophy-lesion-size.
53. Ophthotech. Ophthotech provides update on Zimura complement programs for treatment of eye diseases. September 19, 2017. Available from: https://www.streetinsider.com/FDA/Ophthotech+Corp.+(OPHT)+Reports+Completion+of+Patient+Recruitment+in+the+Phase+2a+Clinical+Trial+of+Zimura+in+Combination+with+Anti-VEGF+Therapy/14123271.html.
54. ClinicalTrials.gov. ZIMURA in combination with LUCENTIS in patients with Neovascular Age Related Macular Degeneration (NVAMD). 2017. NLM identifier: NCT03362190.
55. ClinicalTrials.gov. Zimura in subjects with geographic atrophy secondary to dry age-related macular degeneration. 2016. NLM identifier: NCT02686658.
56. Katschke KJ, Wu P, Ganesan R, et al. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J Biol Chem. 2012;287(16):12886–12892. doi:10.1074/jbc.M112.345082
57. Volanakis JE, Barnum SR, Giddens M, et al. Renal filtration and catabolism of complement protein D. N Engl J Med. 1985;312:395–399. doi:10.1056/NEJM198502143120702
58. Jack LS, Sadiq MA, Do DV, Nguyen QD. Emixustat and lampalizumab: potential therapeutic options for geographic atrophy. Retinal Pharmacotherapeutics. n.d.;302–309. doi:10.1159/000438954
59. Crowley MA, Delgado O, Adrian Will-Orrego NM, et al. Induction of ocular complement activation by inflammatory stimuli and intraocular inhibition of complement factor D in animal models. Invest Ophthalmol Vis Sci. 2018;59(2):940–951. doi:10.1167/iovs.17-22605
60. van Lookeren Campagne M, Katschke KJ
61. Do DV, Pieramici DJ, van Lookeren Campagne M, et al. A phase ia dose-escalation study of the anti-factor D monoclonal antibody fragment FCFD4514S in patients with geographic atrophy. Retina. 2014;34:313–320. doi:10.1097/IAE.0b013e3182979ddd
62. Holz, F. G. The MAHALO Phase 2 Study: Safety, Tolerability and Evidence of Activity of Lampalizumab (Anti-factor D) in Patients with Geographic Atrophy(GA) Secondary to Age-Related Macular Degeneration(AMD). in Euretina Meeting 28. Sept, 2013.
63. Roche. Roche provides update on first lampalizumab phase III study for geographic atrophy, an advanced form of age-related macular degeneration. 2017. Available from: https://www.roche.com/media/store/releases/med-cor-2017-09-08b.htm.
64. Holz FG, Sadda SR, Busbee B, et al. Efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: chroma and spectri phase 3 randomized clinical trials. JAMA Ophthalmol. 2018;136(6):666–677. doi:10.1001/jamaophthalmol.2018.1544
65. Yaspan BL, Williams DF, Holz FG, Regillo CD, Li Z, Dressen A. Targeting factor D of the alternative complement pathway reduces geographic atrophy progression secondary to age-related macular degeneration. Sci Transl Med. 2017;9(395):eaaf1443. doi:10.1126/scitranslmed.aaf1443
66. Dobó J, Pál G, Cervenak L, Gál P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunol Rev. 2016;274(1):98–111. doi:10.1111/imr.12460
67. Volz C, Pauly D. Antibody therapies and their challenges in the treatment of age-related macular degeneration. Eur J Pharm Biopharm. 2015;95:158–172. doi:10.1016/j.ejpb.2015.02.020
68. Wolf-Schnurrbusch UE, Stuck AK, Hess R, Wolf S, Enzmann V. Complement Factor P in choroidal neovascular membranes of patients with age-related macular degeneration. Retina. 2009;29(7):966–973. doi:10.1097/IAE.0b013e3181a2f40f
69. Chen JY, Cortes C, Ferreira VP. Properdin: A multifaceted molecule involved in inflammation and diseases. Mol Immunol. 2018;102:58–72. doi:10.1016/j.molimm.2018.05.018
70. Gupta-Bansal R, Parent JB, Brunden KR. Inhibition of complement alternative pathway function with anti-properdin monoclonal antibodies. Mol Immunol. 2000;37:191–201.
71. Pauly D, Nagel BM, Reinders J, et al. A novel antibody against human properdin inhibits the alternative complement system and specifically detects properdin from blood samples. PLoS One. 2014;9:e96371. doi:10.1371/journal.pone.0096371
72. Lesher A, Nilsson B, Song W-C. Properdin in complement activation and tissue injury. Mol Immunol. 2013;56(3):191–198. doi:10.1016/j.molimm.2013.06.002
73. Pipeline for dry AMD features diverse compounds. Modern medicine feature articles, modern medicine feature articles, ophthalmology. April 15, 2016. Available from: http://www.ophthalmologytimes.com/modern-medicine-feature-articles/pipeline-dry-amd-features-diverse-compounds.
74. ClinicalTrials.gov. Pharmacokinetics of CLG561 in patients with advanced age-related macular degeneration. 2013. NLM identifier: NCT01835015.
75. ClinicalTrials.gov. CLG561 proof-of-concept study as a monotherapy and in combination with LFG316 in subjects with Geographic Atrophy (GA). 2015. NLM identifier: NCT02515942.
76. Bansal R. Method of inhibiting complement activation with factor Bb specific antibodies WO2009/029669A1. 2009.
77. Fernandez-Godino R, Pierce EA. C3a triggers formation of sub-retinal pigment epithelium deposits via the ubiquitin proteasome pathway. Sci Rep. 2018;8:9679. doi:10.1038/s41598-018-28143-0
78. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38(4):458–462. doi:10.1038/ng1750
79. Grossman TR, Carrer M, Shen L, et al. Reduction in ocular complement factor B protein in mice and monkeys by systemic administration of factor B antisense oligonucleotide. Mol Vis. 2017;23:561–571.
80. ClinicalTrials.gov. Safety and efficacy of IONIS-FB-Lrx in up to 120 patients 55 and older with geographic atrophy (GA) secondary to age-related macular degeneration (AMD). NLM identifier: NCT03446144. Available from: https://clinicaltrials.gov/ct2/show/NCT03446144?term=IONIS-FB-Lrx&rank=2.
81. Thurman JM, Kraus DM, Girardi G, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody-induced pregnancy loss in mice. Mol Immunol. 2005;42(1):87–97. doi:10.1016/j.molimm.2004.07.043
82. Ren X, Li J, Xu X, Wang C, Cheng Y. IBI302, a promising candidate for AMD treatment, targeting both the VEGF and complement system with high binding affinity in vitro and effective targeting of the ocular tissue in healthy rhesus monkeys. Exp Eye Res. 2016;145:352–358. doi:10.1016/j.exer.2016.02.004
83. Sayegh RG, Sacu S, Dunavölgyi R, et al. Geographic atrophy and foveaal-sparing changes related to visual acuity in patients with dry age-related macular degeneration over time. Am J Ophthalmol. 2017;179:118–28.una. doi:10.1016/j.ajo.2017.03.031
84. Garrity ST, Sarraf D, Freund KB, Sadda SR. Multimodal imaging of nonneovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. 2018;59(4):AMD48–AMD64. doi:10.1167/iovs.18-24158
85. de Amorim Garcia Filho C, Yehoshua Z, Gregori G, et al. Change in drusen volume as a novel clinical trial endpoint for the study of complement inhibition in age-related macular degeneration. Ophthalmic Surg Lasers Imaging Retina. 2014;45:18–31. doi:10.3928/23258160-20131217-01
86. Simader C, Sayegh RG, Montuoro A, et al. A longitudinal comparison of spectral-domain optical coherence tomography and fundus autofluorescence in geographic atrophy. Am J Ophthalmol. 2014;158:557–566.e1. doi:10.1016/j.ajo.2014.05.026
87. Fleckenstein M, Mitchell P, Freund KB, et al. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology. 2018;125(3):369–390. doi:10.1016/j.ophtha.2017.08.038
88. Lindner M, Nadal J, Mauschitz MM, et al. Combined fundus autofluorescence and near infrared reflectance as prognostic biomarkers for visual acuity in foveal-sparing geographic atrophy. Invest Ophthalmol Vis Sci. 2017;58(6):BIO61. doi:10.1167/iovs.16-21210
89. Liu MM, Chan CC, Tuo J. Genetic mechanisms and age-related macular degeneration: common variants, rare variants, copy number variations, epigenetics, and mitochondrial genetics. Hum Genomics. 2012;6(1):13. doi:10.1186/1479-7364-6-13
90. Raychaudhuri S, Iartchouk O, Chin K, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet. 2011;43:1232–1236. doi:10.1038/ng.976
91. Urtti A. Challenges and obstacles of ocular pharmacokinetics and drug delivery. Adv Drug Deliv Rev. 2006;58(11):1131–1135. doi:10.1016/j.addr.2006.07.027
92. Moisseiev E, Loewenstein A. Drug delivery to the posterior segment of the eye. Dev Ophthalmol. 2017;87–101. doi:10.1159/000455276
93. Joseph RR, Tan DWN, Ramon MRM, et al. Characterization of liposomal carriers for the trans-scleral transport of Ranibizumab. Sci Rep. 2017;7:16803. doi:10.1038/s41598-017-16791-7
94. Pennesi ME, Neuringer M, Courtney RJ. Animal models of age related macular degeneration. Mol Aspects Med. 2012;33(4):487–509. doi:10.1016/j.mam.2012.06.003
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.