")
Back to Journals » International Journal of Nanomedicine » Volume 14
Comparisons of in vitro Fick’s first law, lipolysis, and in vivo rat models for oral absorption on BCS II drugs in SNEDDS
Authors Ye J, Wu H, Huang C , Lin W, Zhang C, Huang B, Lu B, Xu H, Li X, Long X
Received 2 February 2019
Accepted for publication 17 June 2019
Published 23 July 2019 Volume 2019:14 Pages 5623—5636
DOI https://doi.org/10.2147/IJN.S203911
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Jingyi Ye,1,* Huiyi Wu,1,* Chuanli Huang,1 Wanting Lin,2 Caifeng Zhang,1 Bei Huang,2 Banyi Lu,2 Hongyu Xu,2 Xiaoling Li,3 Xiaoying Long1,4
1Department of Pharmaceutics, School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, People’s Republic of China; 2Department of Pharmacy of Chinese Materia Medica, School of Traditional Chinese Medicine, Guangdong Pharmaceutical University, Guangzhou 510006, People’s Republic of China; 3Department of Pharmaceutics and Medicinal Chemistry, Thomas J. Long School of Pharmacy & Health Sciences, University of the Pacific, Stockton, CA 95211, USA; 4Department of Oral Delivery, Guangdong Engineering and Technology Research Center of Topical Precise Drug Delivery System, Guangdong Pharmaceutical University, Guangzhou 510006, People’s Republic of China
*These authors contributed equally to this work
Purpose: The objective of this study was to compare the in vitro Fick’s first law, in vitro lipolysis, and in vivo rat assays for oral absorption of Biopharmaceutical Classification Systems Class II (BCS II) drugs in self-nanoemulsifying drug delivery system (SNEDDS), and studied drugs and oils properties effects on the absorption.
Methods: The transport abilities of griseofulvin (GRI), phenytoin (PHE), indomethacin (IND), and ketoprofen (KET) in saturated water solutions and SNEDDS were investigated using the in vitro Madin-Darby canine kidney cell model. GRI and cinnarizine (CIN) in medium-chain triglycerides (MCT)-SNEDDS and long-chain triglycerides (LCT)-SNEDDS were administered in the in vivo SD rat and in vitro lipolysis models to compare the oral absorption and the distribution behaviors in GIT and build an in vitro-in vivo correlation (IVIVC).
Results: In the cell model, the solubility of GRI, PHE, IND, and KET increased 6–8 fold by SNEDDS, but their permeability were only 18%, 4%, 8%, and 33% of those of their saturated water solutions, respectively. However, in vivo absorption of GRI-SNEDDS was twice that of the GRI suspension and those of CIN-SNEDDS were 15–21 fold those of the CIN suspension. In the lipolysis model, the GRI% in aqueous and pellet phases of MCT were similar to that in LCT. In contrast, the CIN% in the aqueous and pellet phases were decreased but that of the lipid phase increased. In addition, an IVIVC was found between the CIN% in the lipid phase and in vivo relative oral bioavailability (Fr).
Conclusion: The in vitro cell model was still a suitable tool to study drug properties effects on biofilm transport and SNEDDS absorption mechanisms. The in vitro lipolysis model provided superior oral absorption simulation of SNEDDS and helped to build correlation with in vivo rats. The oral drug absorption was affected by drug and oil properties in SNEDDS.
Keywords: SNEDDS, MDCK cell model, in vivo rat model, in vitro lipolysis, in vitro-in vivo correlation
Introduction
Oral drug absorption in the gastrointestinal tract (GIT) depends on a complex interplay of multiple factors, such as gastrointestinal physiology, drug physicochemistry properties, and drug formulations. Drug solubility is considered critical factors in oral bioavailability of Biopharmaceutical Classification Systems Class II (BCS II) drugs. Self-nanoemulsifying drug delivery system (SNEDDS) is a promising lipid-based formulation and it enhances the drugs bioavailability by increasing their solubility, prolonging their retention time in the GIT, facilitating their lymphatic absorption, improving their penetration, reducing their pre-systemic metabolism, and inhibiting their P-gp efflux.1,2 Despite the proven excellence of SNEDDS, however, the oral absorption mechanisms of SNEDDS products have been clearly elucidated yet, unlike those of conventional formulations.3,4 The underlying reasons include the effect of drug properties, SNEDDS constituents, and the use of simulation and evaluation model and so on.
Several models have been used to investigate drug oral absorption mechanisms, such as in vitro cells, cellulose membrane permeation, in situ intestinal perfusion, and in vivo animal models.5,6 Based on BCS and classic Fick’s first law, passive drug diffusion is driven by the concentration of apical side as shown as Equation (1).7 Therefore, increasing the drug solubility (Cd) can improve J of BCS II drugs.
(1)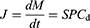
where J is the biomembrane transport flux, dM is the cumulative transport mass during dt, S is the diffusion area, P is the permeability coefficient, and Cd is the drug concentration in the donor chamber.
However, in the present study, solubility–permeability interactions of four BCS II drugs (griseofulvin [GRI], phenytoin [PHE], indomethacin [IND], and ketoprofen [KET]; Table 1) were found in SNEDDS (Figures 1 and 2). The solubility of four drugs was raised, but their permeability decreased. This trade-off relationship between the solubility and permeability of BCS II drugs in particle systems was previously reported and is related to drug state and particle size (free drugs or particles). Cinnarizine (CIN) solubility increased by ~600-fold in SNEDDS relative to the saturated water solution. However, a 4-hr transmembrane experiment with PermeapadTM (hydrophilic polymer membrane) revealed that only 0.00012% of the total CIN dose remained in the recipient pool.8 Moreover, estradiol and progesterone have >400-fold and >1,000-fold solubilization in nanoemulsions, respectively. When they were transported across a silicone rubber membrane, though, their Papp was only 0.9% of their saturated water solutions.9 Dahan et al confirmed the solubility–permeability interplay of progesterone within hydroxypropyl-β-cyclodextrin by using a Caco-2 cell model, a parallel artificial membrane permeation assay, and a rat jejunal perfusion model.10
 |
Table 1 Drug properties of model drugs |
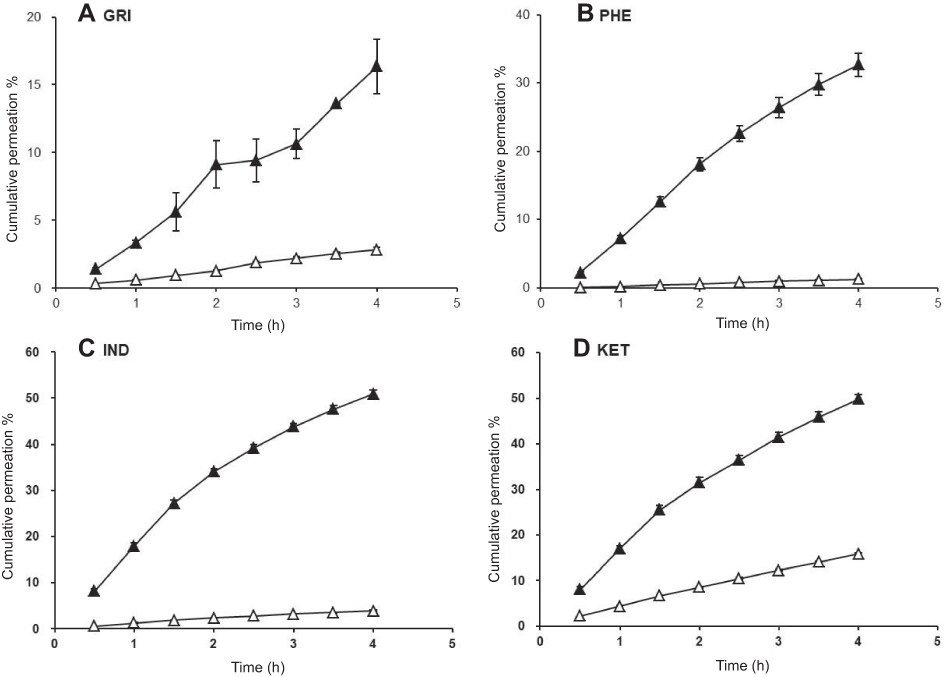 |
Figure 1 CP % of drug-loaded SNEDDSs (△) and saturated water solutions (▲). Abbreviations: CP%, cumulative permeation; GRI, griseofulvin; PHE, phenytoin; IND, indomethacin; KET, ketoprofen. |
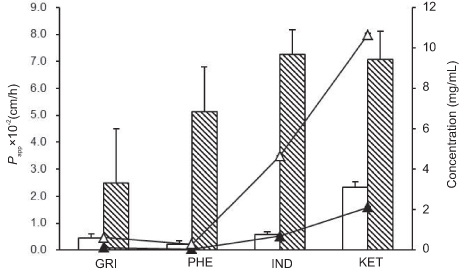 |
Figure 2 Papp of drug-loaded SNEDDSs (blank column) and saturated water solutions (twill column). Concentrations of drugs in SNEDDS (△) and saturated water solution (▲) at apical side. Abbreviations: GRI, griseofulvin; PHE, phenytoin; IND, indomethacin; KET, ketoprofen; Papp, apparent permeability coefficient. |
Recently, an in vitro lipolysis model was used to study the absorption mechanism of lipid-based formulations including SNEDDS and was based on oil-food digestion in the GIT.11,12 Oral SNEDDS facilitates the secretion of bile salt, phospholipids, and pancreatic lipases. The lipolytic products of pancreatic lipases are mixed with endogenous digestion products to form a complex particle system consisting of an upper lipid phase (triglycerides, diglycerides, and fatty acids), a middle aqueous phase (colloidal particles, vesicles, and emulsion droplets), and a bottom pellet phase (fatty acid soaps and drug precipitate) after ultracentrifugation.13 In contrast, the SNEDDS in the cell model is homogeneous and the drugs are encapsulated in oil cores.
The post-oral administration in vitro lipolysis model effectively simulates the true intestinal state of the drugs in SNEDDS. The distribution of the drugs in lipolysis is influenced by drug properties (solubility and lipophilicity) and oil species (medium-chain triglycerides [MCT] and long-chain triglycerides [LCT]).12,14 After digestion, the drugs may be absorbed in free form by passive diffusion or as particles such as mixed micelles, colloidal particles, vesicles or emulsion droplets via the transcytosis of enterocytes into the portal blood and the M cells into the lymph, or through chylomicrons as soluble lipid-phase drugs into the mesenteric lymph.15,16 In vitro lipolysis and in vivo rat models indicate that drug distributions in various phases are probably correlated with in vivo absorption.
In this paper, the permeability of four BCS II drugs (GRI, PHE, IND, and KET) in SNEDDS was compared by in vitro Madin-Darby canine kidney [MDCK] cell model. Drug properties (Table 1) and oil species (MCT/LCT) effects on oral absorptions of SNEDDS were studied by in vivo rat model. Furthermore, the GRI- and CIN-SNEDDSs digestion behaviors in GIT were investigated by in vitro lipolysis. The certain in vitro-in vivo correlation between the in vitro lipolysis and the in vivo rat model was built.
Materials and methods
Materials
GRI (98.3% purity) and IND (99% purity) were purchased from MP Biomedicals (Strasbourg, France). KET (purity >98%) was supplied by Tokyo Chemical Industry Corporation (Japan). A MDCK cell line and Eagle’s Minimum Essential Medium (EMEM) were purchased from ATCC (American Type Culture Collection, Rockville, MD, USA). The 24-well Transwell® plates (Diameter 6.5 mm) with 0.4-μm polyester membrane were acquired from Corning Inc., Corning, NY, USA. Hanks’ Balanced Salt Solution (HBSS solution) was obtained from Thermo Fisher Scientific, Waltham, MA, USA. CIN (purity >99%), PHE (purity >99%), Span® 80, sodium taurodeoxycholate hydrate (NaTDC), phosphatidylcholine (Lipoid E 80, PC), Trizma® maleate, 4-bromobenzene-boronic acid (4-BBBA), and pancreatin from porcine pancreas (50 TBU/mg, 8× USP) were supplied by Sigma-Aldrich Corp. (St. Louis, MO, USA). MCT (CNAC Pharma Co., Beijing, People’s Republic of China), soybean oil (LCT; Aladdin Industrial Corp., Ontario, CA, USA), n-Octanoic acid (Aladdin Industrial Corp.), oleic acid (Damo Chemical Reagent, Tianjin, People’s Republic of China), Cremophor® RH40 (BASF SE, Ludwigshafen, Germany), and Tween® 80 (Alfa Aesar®, Haverhill, MA, USA) were used as received. Pure water was obtained from a RephiLe direct water purification system (RephiLe, Boston, MA, USA). The reagents used in HPLC were all HPLC grade. All other chemicals were of analytical reagent grade.
Methods
In vitro permeability of GRI, PHE, IND, and KET in SNEDDS
MDCK cells were cultured in EMEM supplemented with 1% (w/v) penicillin-streptomycin solution, 1% (w/v) glutamate, and 10% (w/v) FBS at 37°C, 95% relative humidity, and a 5% CO2 atmosphere.17,18 The cells were seeded onto 24-well Transwell® plates at a density of 1.0×104 cell/cm2 after reaching 80% confluence. MDCK monolayers were formed after 4–6 days further incubation. The growth media were replaced every other day. When the transepithelial electrical resistance) was >90 Ω·cm2, the MDCK cells could be used in the transport studies.19
Prior to the experiment, MDCK monolayers were rinsed 3× with HBSS solution. Krebs-Ringer buffer (K-R buffer; pH 6.0) was added to the donor and recipient chambers in the incubator at 37°C for 30 mins to achieve equilibrium. The K-R buffer in the donor chamber was then replaced with SNEDDS or saturated solutions of GRI, PHE, IND, or KET. The constituents of the SNEDDS are listed in Table 2. Drug-loaded pre-emulsifying concentrates (PECs) were prepared by mixing the drugs with the excipients. PECs were then diluted with K-R buffer and gently blended at 37°C to prepare the SNEDDS. Transport experiments were run using either 0.2 mL saturated drug solutions or SNEDDS as test solutions on the apical side. One milliliter of plain K-R buffer was used as a recipient solution to fill the basal wells. Apical-to-basal transport experiments were performed on a shaker (Dubnoff Metabolic Precision®; Thermo Fisher Scientific, Waltham, MA, USA) at 50 rpm. Then, 0.8-mL recipient solution aliquots were withdrawn at 0.5-hr intervals over 4 hrs and compensated with equal volumes of fresh K-R buffer. The entire system was maintained at 37°C during the transport experiment and all tests were performed in triplicate. The GRI, PHE, IND, and KET levels were determined by HPLC. The apparent permeability coefficient (Papp) and the cumulative permeation (%) (CP%) were calculated according to the literature.20
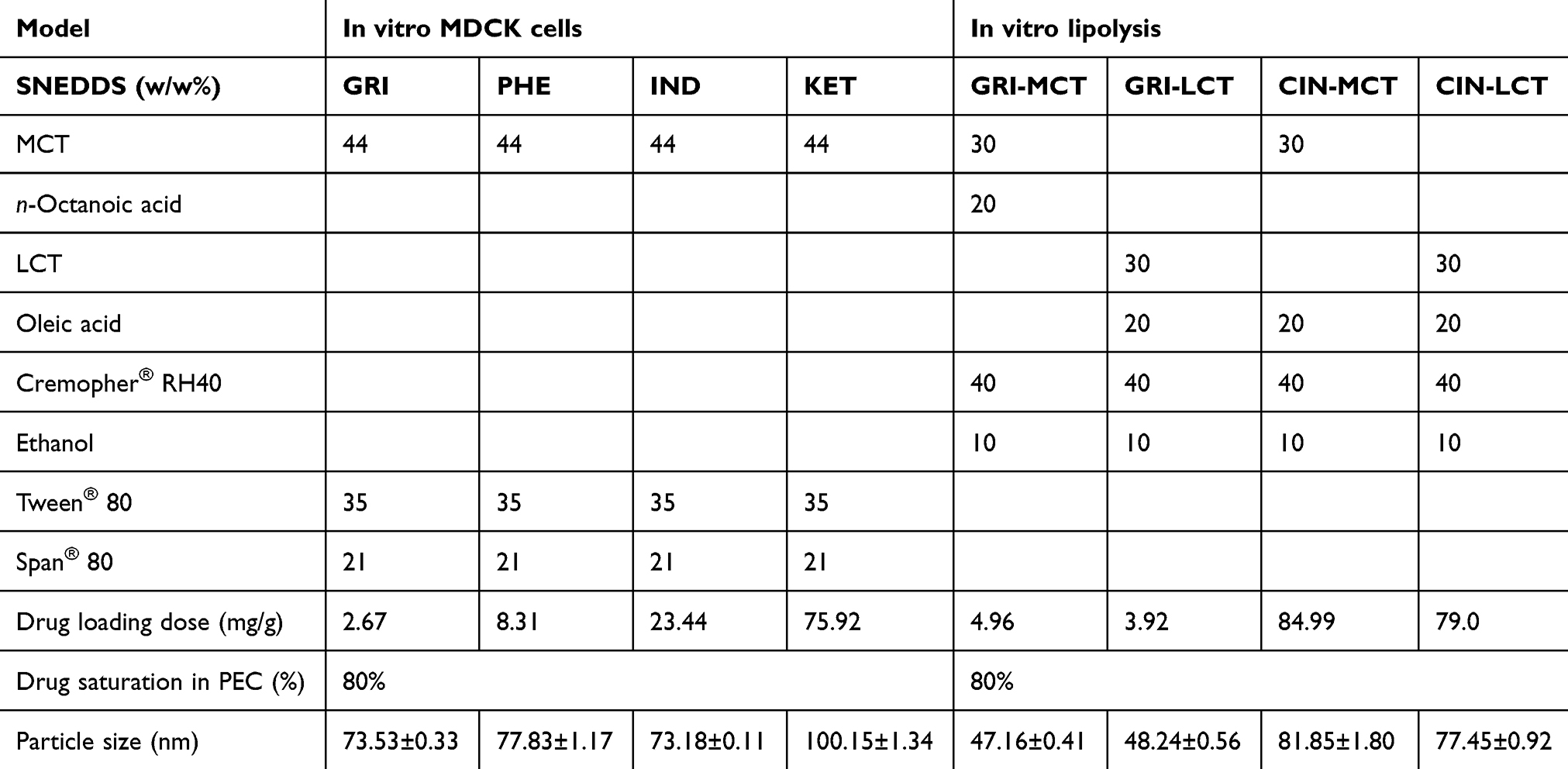 |
Table 2 Components of PECs and model drug dosage in PECs |
In vivo GRI/CIN-SNEDDSs absorption
Health male Sprague-Dawley rats (250±20 g) were purchased from the Experimental Animal Center of the Guangzhou University of Chinese Medicine (SCXK 2013-0034, Guangdong, People’s Republic of China) and acclimatized for 1 week. Prior to the experiment, the rats were fasted but had free access to water for 12 hrs. All the animal experiments were performed according to the China National Guide for the Care and Use of Laboratory Animals. The animal studies were conducted with the approval of the GuangDong Pharmaceutical University Undergraduate Laboratory Animal Center and the GuangDong Pharmaceutical University Experimental Animal Ethics Committee Inspection (no: SPF2017127). SNEDDS compositions are shown in Table 2. Drug-loaded PECs were prepared by mixing the excipients and GRI or CIN with stirring at 50°C. Rats were orally administered GRI-MCT/LCT-SNEDDS (4.5 mg/g) at 18 mg/kg or CIN-MCT/LCT-SNEDDS (80 mg/g) at 320 mg/kg. Equal doses of GRI or CIN were suspended in 0.5% (w/v) methylcellulose and orally administered as a suspension group (SUSP). Then, 0.4-mL blood samples were collected from the orbital veins of the GRI group rats at 0.167, 0.5, 1, 1.5, 2, 4, 6, 8, 10, and at 12 hrs. Plasma from the CIN group rats was collected at the same intervals before 12 hrs and then collected at 24 and 48 hrs. The treated samples were analyzed by HPLC as described in the subsection “GRI/CIN content during in vivo absorption”.
The maximum blood concentrations (Cmax) and their time of occurrence (Tmax) were obtained directly from the rat plasma concentration vs time profiles. The areas under the plasma concentration-time curves (AUC) of GRI and CIN were calculated using the linear trapezoidal rule. Relative oral bioavailability (Fr) was calculated according to Equation (2).
(2)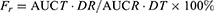
where T and R represent the test and reference sample, respectively, D is the drug dosage.
In vitro lipolysis of GRI/CIN-SNEDDSs
In vitro lipolysis was conducted based on Leab Sek et al.21 The constituents of the GRI/CIN-PECs are listed in Table 2. The PECs were dispersed in 45 mL of NaTDC/PC micelle solution (5 mM NaTDC +1.25 mM PC) to form SNEDDS. The pH was adjusted to 6.8 with 1 M NaOH solution and a PB-10 pH meter (Sartorius, Göttingen, Germany).22 Pancreatin digestion buffer was prepared with pancreatin (10,000 TBU/mL) and NaCl (150 mM). At the beginning of lipolysis, Trizma® maleate (50 mM), CaCl2·2H2O (5 mM), and 4 mL pancreatin digestion buffer were added to SNEDDS to a total volume of 50 mL. During lipolysis, the temperature was maintained at 37°C and the pH was fixed at 6.8 with 1 M NaOH. The volume of NaOH consumed and the titration time were recorded. At specific time points (0, 5, 10, 30, 60, 120, 180, and 240 mins), 6 mL of lipid-digested mixture was withdrawn and the lipase activity was immediately inhibited using 30 μL of 4-BBB (1 M). Then, they were ultracentrifuged (L-100XP ultracentrifuge, 100Ti rotor; Beckman Coulter, Brea, CA, USA) at 802,000×g and 25°C for 30 mins to separate the lipolysis products into the lipid, aqueous, and pellet phases. Each phase was analyzed by HPLC for drug content.
GRI, PHE, IND, and KET content in transport experiments
GRI, PHE, IND, and KET content was analyzed by HPLC according to United States Pharmacopoeia 35 (2012).23 The HPLC system was an Agilent 1100 Series HPLC (Agilent Technologies, Santa Clara, CA, USA). The columns used for the GRI, IND, and PHE/KET separation were the Luna CN (4.6 mm×150 mm, 5 μm), Luna C18 (4.6 mm×150 mm, 3 μm), and Gemini C18 (4.6 mm×50 mm, 5 μm), respectively, from Phenomenex (Torrance, CA, USA). The injection volumes were all 10 μL.
GRI/CIN content during in vivo absorptions
The GRI and CIN content was determined using slightly modified HPLC assays as described in previous studies.24,25 Briefly, blood samples were collected in heparin tubes and centrifuged to obtain the plasma. Twofold acetonitrile was added to precipitate the protein. The samples were centrifuged at 6,950× g and 0°C for 10 mins and the supernatants were analyzed in a Waters 1525 HPLC system (Waters Corp., Milford, MA, USA) equipped with a 2489 UV-VIS detector and a 2707 autosampler. CIN was separated with 48% (v/v) acetonitrile and 52% (v/v) of 20 mM NH4H2PO4 buffer (pH 4.5). The columns used to separate the GRI and CIN were the Gemini C18 (250 mm×4.6 mm, 5 μm; Phenomenex, Torrance, CA, USA) and the Luna C18 (250 mm ×4.6 mm, 5 μm; Phenomenex), respectively. The guard column was a Phenomenex C18 (30 mm×4 mm; Phenomenex). The column temperature, flow rate, and injection volume were 30°C, 1 mL/min, and 20 μL, respectively.
GRI/CIN contents in the in vitro lipolytic phases
The lipid and aqueous phase was collected in an Eppendorf tube and diluted 10× with acetonitrile, respectively. The pellet phase was dispersed in 1 mL pure water and vortexed, and then, 3 mL acetonitrile was added to dissolve the GRI or CIN. The Eppendorf tube was sealed and centrifuged at 7,040× g for 10 mins to remove the sediment. The supernatant was withdrawn, diluted with the mobile phase to an appropriate concentration, and analyzed by HPLC. The separation conditions were the same as those described in the subsection “GRI/CIN content during in vivo absorptions”.
Statistical analysis
All results were presented as mean±SD. Statistical differences were identified using one-way ANOVA and P<0.05 was considered statistically significant.
Results
In vitro permeability based on Fick’s first law
In the MDCK cells transport experiments, the CP% and Papp of the GRI, PHE, IND, and KET SNEDDS were significantly lower (P<0.05) than those of their saturated water solutions (Figures 1 and 2) even though the drug solubility was significantly improved by SNEDDS (P<0.05). The concentrations of GRI, PHE, IND, and KET in SNEDDS (Figure 2) in the donor chambers were 7.0-fold, 9.1-fold, 9.1-fold, and 6.7-fold those of their saturated water solutions, and ranged from 0.088‒0.617, 0.032‒0.292, 0.511‒4.648, and 1.592‒10.648 mg/mL, respectively. However, there were no apparent increases in CP% or Papp in the MDCK cells transport experiments when the solubility was enhanced by SNEDDS. After 4-hr transport, the CP% of GRI, PHE, IND, and KET-SNEDDS (Figure 1) were only 18%, 4%, 8%, and 33% of their respective saturated water solutions. The Papp (×10−2 cm/h) of SNEDDS and their saturated water solutions (Figure 2) were 0.45±0.15 and 2.48±2.03, 0.22±0.12 and 5.11±1.70, 0.56±0.13 and 7.24±0.92, and 2.34±0.20 and 7.06±1.06, respectively.
In vivo absorption of GRI/CIN-SNEDDS
The plasma concentration-time profiles and bioavailability parameters of GRI and CIN are presented in Figure 3 and Table 3. The oral absorption of GRI was enhanced by SNEDDS (Figure 3A, Table 3), and the AUC0–12h of the GRI-SNEDDS composed of MCT and LCT were ~2-fold those of the GRI-SUSP (P<0.05). The Cmax of MCT-SNEDDS was higher than GRI-SUSP (P<0.05), but the bioavailability parameters of GRI-MCT-SNEDDS and GRI-LCT-SNEDDS did not significantly differ.
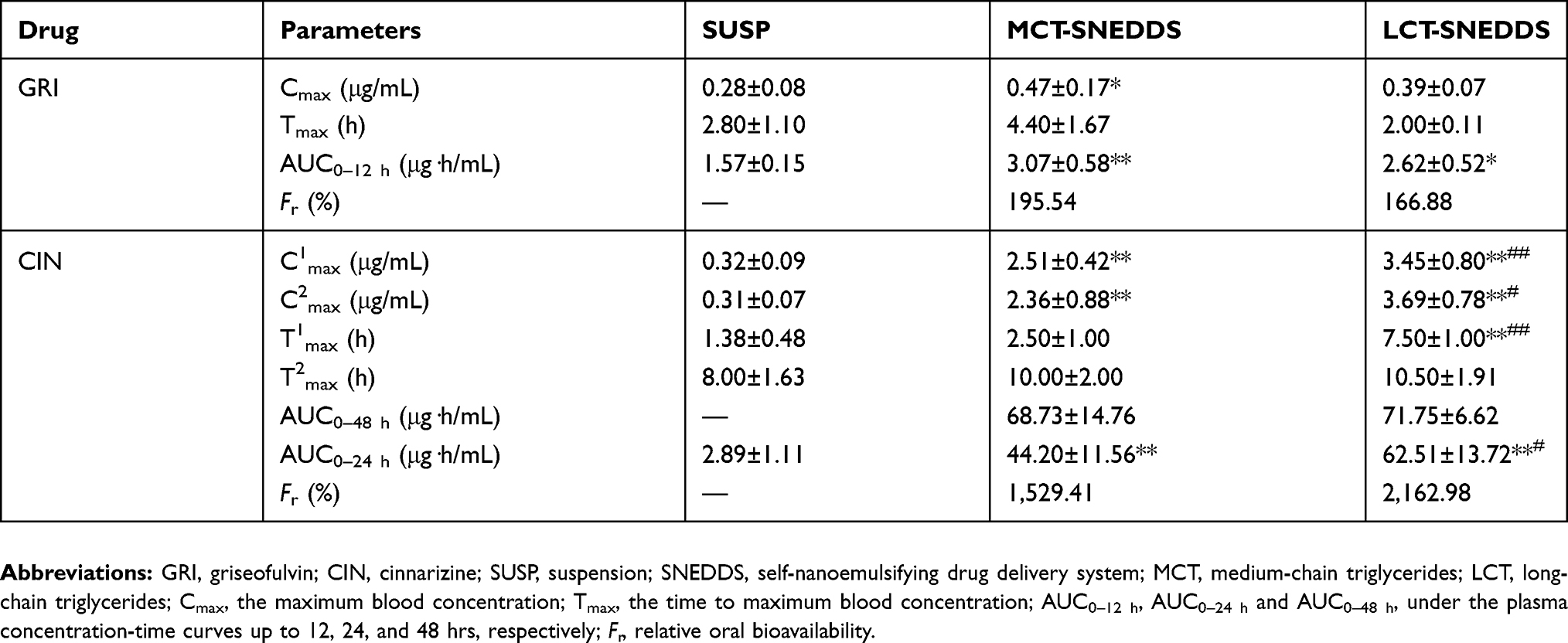 |
Table 3 Bioavailability parameters of GRI and CIN (mean±SD, n=4–5) |
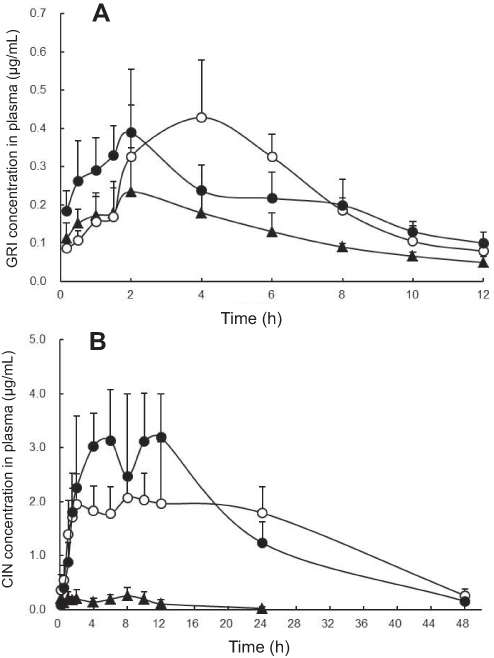 |
Figure 3 Plasma concentration-time profiles of GRI (A) and CIN (B) (n=5). MCT-SNEDDS (○), LCT-SNEDDS (●), SUSP (▲). Abbreviations: GRI, griseofulvin; CIN, cinnarizine; SUSP, suspension; SNEDDS, self-nanoemulsifying drug delivery system; MCT, medium-chain triglycerides; LCT, long-chain triglycerides. |
The absorption extent of CIN was substantially enhanced by SNEDDS (Figure 3B, Table 3). The AUC0–24h of CIN-SNEDDS were 15–22-fold larger than those of CIN-SUSP. The Cmax and AUC0–24h of LCT-SNEDDS were higher than that of MCT-SNEDDS (P<0.05), and T1max for LCT-SNEDDS was longer than that for MCT-SNEDDS (P<0.01). But, the AUC0–48h of MCT-SNEDDS and LCT-SNEDDS were not significantly differ. The high oral dose of CIN caused saturated absorption. For LCT-SNEDDS, the plateau regions of the CIN plasma concentration-time curves were maintained from the second to the twelfth hour while those for MCT-SNEDDS remained stable from the second to the twenty-fourth hour.
In vitro lipolysis of GRI/CIN-SNEDDS
The GRI and CIN distribution were described as GRI% and CIN% in the curves of Figure 4. Figure 4 shows that GRI and CIN partitioned mainly in the aqueous phase when there was no pancreatin in the lipolysis system, but 20–30% CIN distributed in pellet phase after ultracentrifugation. In contrast, separation phases formed and GRI and CIN were redistributed when pancreatin was added. The GRI% in the aqueous phase of LCT-SNEDDS was substantially higher than that in the MCT-SNEDDS (Figure 4A and B). The decreases in GRI in the aqueous phase were compensated by the increase in the pellet phase and the GRI% in the pellet phase of the LCT-SNEDDS and MCT-SNEDDS were no difference. The lipid phase was invisible during GRI-MCT-SNEDDS lipolysis, but the lipid phase was separated in the GRI-LCT-SNEDDS after 120 mins. In comparison, lipid phase was separated at the onset of lipolysis of CIN-SNEDDS (Figure 4C and D). The CIN% in the aqueous phase of LCT-SNEDDS were redistributed into lipid phase and significantly higher than that in lipid phase of MCT-SNEDDS at 240 mins (P<0.05). The CIN% in lipid phase of LCT-SNEDDS even to a level higher than that measured for the aqueous phase at 180 mins. The lowest CIN% in the lipid phase of the MCT-SNEDDS was recorded at 10 mins after the beginning of lipolysis which corresponded to 60 mins in the LCT-SNEDDS. In the pellet phase, CIN% was relatively lower in LCT-SNEDDS than that in MCT-SNEDDS.
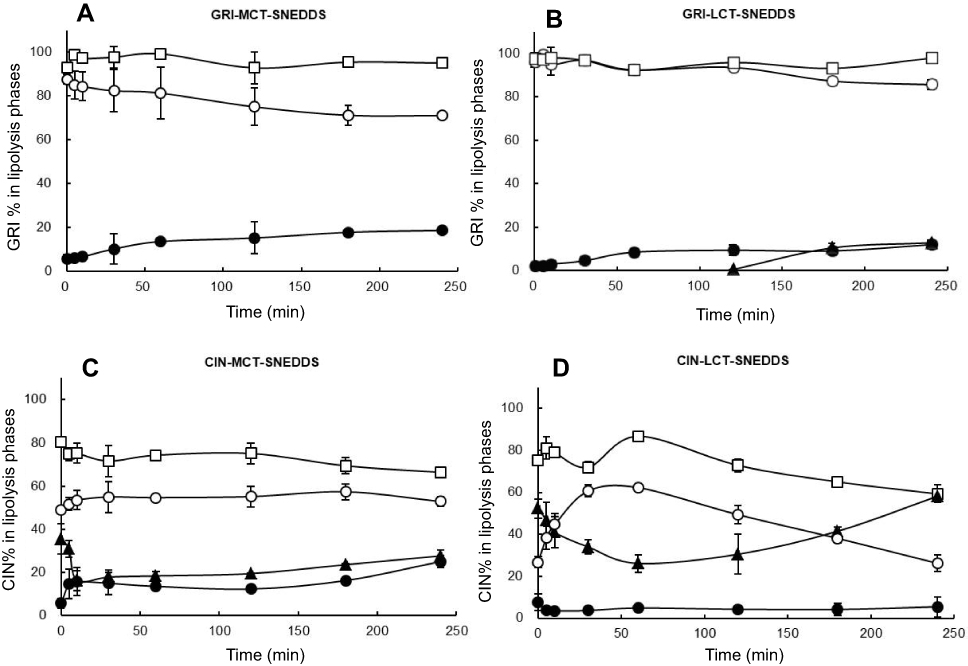 |
Figure 4 Dynamic distribution shown as GRI% (A, B) and CIN% (C, D) in the three phases of an in vitro lipolysis system; aqueous phase without pancreatin (□), lipid phase (▲), aqueous phase (○), pellet phase (●). Abbreviations: GRI, griseofulvin; CIN, cinnarizine; GRI%, griseofulvin distribution percentage content; CIN%, cinnarizine distribution percentage content; SNEDDS, self-nanoemulsifying drug delivery system; MCT, medium-chain triglycerides; LCT, long-chain triglycerides. |
Discussion
In vitro permeability based on Fick’s first law
Fast-growing MDCK cells (5–7 day to differentiation) have been used instead of Caco-2 cells to study cell growth regulation, drug metabolism, drug transport mechanisms, and drug permeability in lipid-based formulations.5,18,26–28 Moreover, several drugs have been compared in two different cell models and similar transport results were obtained for both of them.19 In the present study, MDCK cells were used to compare the oral absorption mechanisms of four drugs in SNEDDS.
Figure 2 shows that relative to the GRI saturated water solution, the concentration of GRI was sevenfold higher in SNEDDS, but its CP% was only 18% that of the saturated water solution (Figure 1) and its Papp was 0.45×10−2 cm/h. However, this result was by no means coincident. Similar solubility–permeability interplay outcomes were found in transport experiments involving three other drugs with different properties (Figures 1 and 2). As mentioned earlier, CIN, estradiol, and progesterone were reported on similar results.8,10,28,29
The drug states in formulations are related to the trade-off results. In saturated water solutions, drug molecules are dissolved and transported in free form. Based on Fick’s first law as shown in Equation (1), the solubility of a drug increases in nanoemulsion which leads to the higher concentration gradient and permeability. But, in fact, drug molecules are trapped inside oil cores and transported as particles. Since drugs within SNEDDS have diameters greater than those of macromolecules, their intestinal membrane permeability may be lower than that of free drugs (small molecules).30–35 As shown in Equation (3), every 0.8-Å increase in diameter (rca) reduces Papp by 50%.35
(3)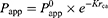
where rca is the molecular cross-sectional radius, Papp is the intrinsic pore permeability, and K is a hindrance intensity factor.
The diameters of the small molecules, including the drugs used in the research, were ~1 nm. However, the diameters of the SNEDDS prepared from the four drugs were in the range of 70‒100 nm (Table 2). Therefore, particle size increases much greater than solubility. Evidently, then, it is the increase in diameter which mainly accounts for the low permeability of the drugs inside the SNEDDS.
Though the results showed that in vitro cell model was unable to predict the oral absorption of SNEDDS correctly, but it was suitable to compare the drugs uptake in aqueous solutions or SNEDDS (Table 1 and Figure 2). The Papp of drug in free form was strongly correlated with drug lipophilicity (log P: IND>KET>PHE>GRI). But the Papp of drug within SNEDDS was consistent with the rank order of drug concentration at apical side (KET>IND>GRI>PHE), namely, free-form drugs concentration in aqueous solution determined the Papp of SNEDDS. In the previous study, GRI and dexamethasone have similar liposolubility (log P=1.8–2.1 vs log P=1.8) but with different water solubility (5 mg/mL vs 100 mg/mL), the bioavailability enhancement of dexamethasone was evidently higher than GRI in lipid formulations.12 It indicates that both of aqueous solubility and lipophilicity affect the biomembrane transport abilities of BCS II drugs.
Impact of drug properties and oil species used during in vivo absorption
The bioavailability results of the in vivo rat model demonstrated that SNEDDSs significantly increased the oral absorption of GRI and CIN compared to the drug suspensions (Table 3, Figure 3), were different from the CP% and Papp results gained by in vitro cell model.
There were significant differences between GRI and CIN in terms of their absorption because their properties markedly differ. Table 1 indicates that the GRI aqueous solubility was 35-fold higher than that of CIN, but the lipophilicity of CIN was nearly 1,000-fold higher than that of GRI. The lipophilicity also caused the difference of loading dose (GRI: 5 mg/g; CIN: 85 mg/g). Since the higher lipophilicity of CIN and the greater CIN concentration gradient in SNEDDS, the oral absorption of CIN was considerably more than it did GRI. This result was in agreement with the uptake mechanism of SNEDDS supported by MDCK cell model in this paper.
The formulation compositions also have a great influence on the drug absorption. Many studies reported that LCT formulations play a more important role in promoting drug oral absorption than MCT formulations.36–38 LC-SMEDDS (self-microemulsifying drug delivery system based long-chain lipids) significantly enhanced the oral bioavailability of danazol (log P=4.53), but the MC-SMEDDS (SMEDDS based on medium-chain lipids) resulted in little enhancement.36 The rank order of bioavailability of anethole trithione (log P=3.8) increased by SMEDDS was SCT<MCT<LCT and in accordance with the increase of solubilization data obtained by in vitro digestion.39 These conclusions were believed that there were significant differences in physiological response to medium and long-chain lipids.40 Despite the MCT-SNEDDS displayed the excellent dispersion properties and the higher drug loading dose, the capacity to form self-assembled mixed micelles and vesicles in the small intestine through interactions between lipid digestion products and amphiphilic endogenous molecules was considerably reduced compared to LCT-SNEDDS.41 In present studies, though the loading dose of GRI and CIN were both slightly higher in MCT-SNEDDS than it was in LCT-SNEDDS (Table 2), the oral bioavailability parameters of two drugs were significantly different. The Cmax between GRI-MCT-SNEDDS and GRI-LCT-SNEDDS were slightly different (Table 3), but their Tmax and AUC0–12 h were not statistical difference. In contrast, the C1max, T1max, and AUC0–24 h of CIN-SNEDDS between MCT and LCT were different significantly (P<0.05). The reasons were explained by the following absorption mechanisms of different drugs in SNEDDS composed of LCT or MCT.
Soybean oil as LCT (C >12) are hydrolyzed in the GIT and absorbed by enterocytes. The lipolysis products, free fatty acids, and monoglycerides are resynthesized to triglycerides. Hereafter, along with phospholipids, cholesterols, and drugs, they are incorporated and assembled into lipoproteins, processed into chylomicrons and carry drugs (log P>5) to the intestinal lymph. MCT (C <12) and homologous monoglycerides do not bind with lipoproteins. Consequently, they are absorbed into systemic circulation via the hepatic portal vein.42 CIN with log P>5 and the high solubility in LCT are easily absorbed into the lymphatic system.38,43,44 In addition, the oleic acid combines with CIN through ionic interaction resulted in the delay release of LCT-SNEDDS (Figure 5).45 The absorption mechanism not only explained the higher Cmax and AUC, and the longer Tmax in CIN-LCT-SNEDDS, but also illustrated its higher oral bioavailability of CIN than that of GRI.
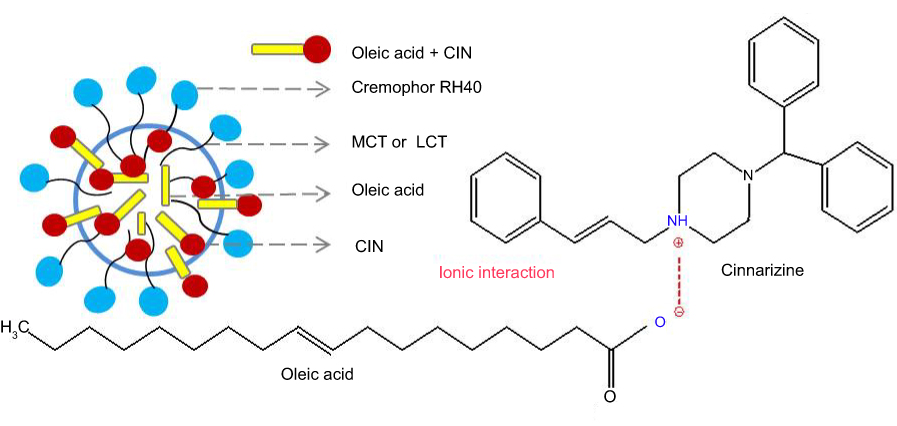 |
Figure 5 CIN solubilization in SNEDDS composed of oleic acid. Abbreviations: MCT, medium-chain triglycerides; LCT, long-chain triglycerides. |
Impact of drug properties and oil species on in vitro lipolysis
The effect of drugs properties and oils species on in vivo absorption is explained by in vitro model and the absorption routes expanded by SNEDDS are shown in Figure 6. In the in vitro cell model, the drugs in SNEDDS are absorbed by SNEDDS endocytosis and by free drug passive diffusion. However, there are seven potential uptake routes in the in vitro lipolysis model. When SNEDDSs are hydrolyzed by pancreatic lipase, the drugs they contained are released and redistributed. Free drug dispersed in the aqueous phase is either transported by passive diffusion (a) or by carrier-mediated uptake in the dissociated state (b).16 The upper lipid phase contained 2-monoglycerides, fatty acids, and residual LCT which stimulate lymphatic drug absorption (log P>5) by forming chylomicrons as mentioned before (c).2,15,46,47 In contrast, drugs with log P<5 are directly absorbed into the hepatic portal vein in the form of free drugs or vesicles (d). Drugs solubilized in the aqueous phase are transported to enterocytes by collisional transfer and binding then absorbed by vesicular-mediated uptake and endocytosis to the plasma (e) or the lymphatic system as 10‒100 nm particles because of the more permeable of lymphatic capillaries than blood capillaries (f).15,16 Precipitates of amorphous or metastable stereotypes are redissolved and transported by passive diffusion (g).45,48,49 Therefore, in vitro lipolysis more accurately interprets and predicts the oral absorption of drugs in SNEDDS than the in vitro cell model.
The GRI and CIN distribution results (Figure 4) were corresponding to previously study where CIN mainly distributed in lipid phase during lipolysis of LCT-suspension at 30 mins but GRI distributed in aqueous phase.50 Since MCT is rapidly and completely digested, the lipid phase was only observed during GRI-LCT-SNEDDS lipolysis (Figure 4A).51 In comparison, the gradual and incomplete hydrolysis of LCT and higher solubilizing capacity of the cubic liquid crystalline phase formed by LCT-SNEDDS increased GRI solubilization in the lipid and aqueous phases (Figure 4B).51,52 Since CIN had higher lipophilicity than GRI, the decrease of CIN% in the aqueous phase was compensated by CIN increased in the pellet and lipid phase (Figure 4C). The CIN% first decreased then increased in lipid phase during SNEDDS lipolysis should have been correlated with drug property an loading dose. At same excipient concentration but 20-fold higher loading doses than GRI, CIN rapidly released from the lipid cores of SNEDDS and precipitated due to poor water solubility. As lipolysis continued, the highly lipophilic and soluble amorphous CIN precipitate was gradually redistributed in the lipid phase.49 Figure 7 shows that the turning point of CIN% in lipid phase was corresponding to the turning point of fatty acid production (MCT-SNEDDS: 10 mins; LCT-SNEDDS: 60 mins). This indicated that the weak base CIN continuously redistributed from fatty acid to triglycerides or monoglycerides during lipolysis.
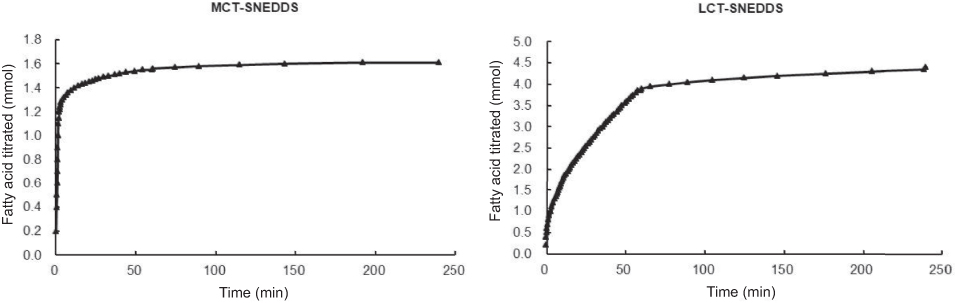 |
Figure 7 Fatty acid titrations of MCT-SNEDDS and LCT-SNEDDS. Abbreviations: CIN, cinnarizine; MCT, medium-chain triglycerides; LCT, long-chain triglycerides; SNEDDS, self-nanoemulsifying drug delivery system. |
In vitro-in vivo SNEDDS correlation
The in vitro lipolysis model effectively simulates SNEDDS behavior in the GIT and may be more strongly correlated than the in vitro cell model with in vivo absorption. Table 4 shows that rank order of GRI% in aqueous phase was LCT>MCT (P<0.05) during lipolysis, on the contrary with previous studies.12 In addition, the rank order of CIN% in aqueous phase was MCT-SNEDDS>LCT-SNEDDS (P<0.05) but the oral AUC0–24h was LCT-SNEDDS>MCT-SNEDDS (P<0.05) in the paper. Larsen et al compared SNEDDS with different loading level by in vitro lipolysis and in vivo dogs, and found that high aqueous phase distribution did not correspond to high oral absorption.11 The results indicated that drugs solubilizing in aqueous phase during lipolysis were not as important as previously suggested.
 |
Table 4 Correlation between drug% at 240 mins in in vitro lipolysis system and in vivo Fr |
Relative to that of GRI, CIN% was significantly decreased in the aqueous phase. In CIN-LCT-SNEDDS, the lipid-phase CIN% was twice that of the aqueous phase. In Table 4, the high CIN% in lipid phase was corresponding to the significantly higher Fr (LCT-SNEDDS>MCT-SNEDDS). Therefore, CIN in the lipid phase probably contributed comparatively more to oral uptake and were closely correlated with in vivo absorption in terms of drug lipophilicity and oil characteristics.
Despite the advantages of the in vitro lipolysis model in the investigation of the oral absorption mechanism, it still markedly differs from the real state of the drug in the GIT. The in vitro lipolysis model is limited and confined to a small space whereas the intestinal tract is relatively infinite and is an open space because of its large absorptive surface and continuous drug transport. However, a single in vitro lipolysis model cannot track drug transmembrane transport or metabolic transformations. In addition, drug absorption mechanisms in various states (Figure 6) remain to be elucidated and confirmed.
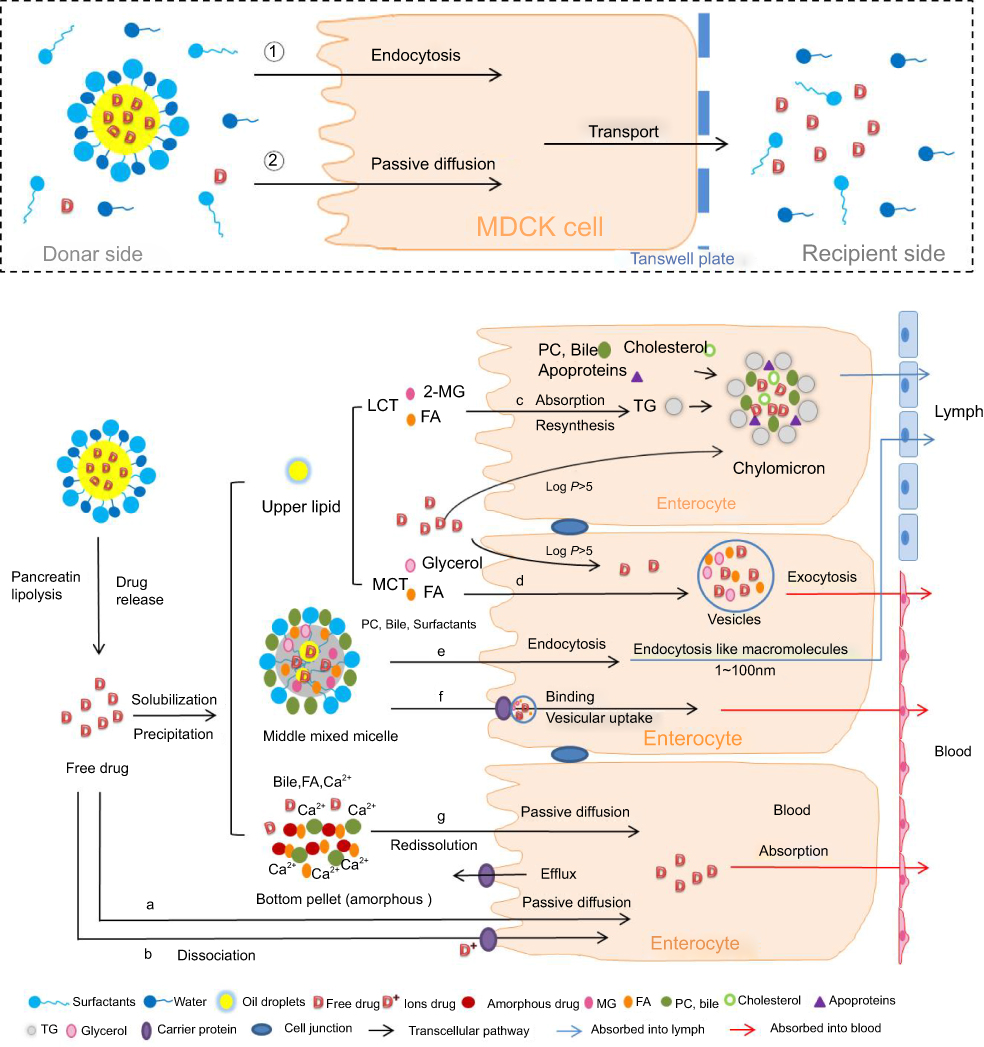 |
Figure 6 Absorption routes of SNEDDS with different oil species (MCT/LCT) and drug properties (log P>5/log P<5). Abbreviations: MDCK cell, Madin-Darby canine kidney cell; MCT, medium-chain triglycerides; LCT, long-chain triglycerides; 2-MG, 2-monoglycerides; FA, fatty acids; PC, phosphatidylcholine; TG, triglycerides; Ca2+, calcium ions. |
Only two model drugs significantly differing in lipophilicity and loading dose were investigated in the present study. Whether the distribution percentage of the drug in the lipid phase can serve as a reliable indicator of post-lipolysis oral absorption remains to be established. Other factors affecting SNEDDS formulation designed must be researched as well. These may include drug distribution changes in the lipid phase which occur when drugs with different properties and loading doses are used, the effects of oils with different saturation levels and ester bonds, the use of surfactants with different molecular weights and charges, and so on.
Conclusion
The in vivo SNEDDS absorption predicted by in vitro cell model might lead to incorrect results, but it still can be used for comparative study of drug transmembrane transport with different properties. The complex GIT behavior of drugs in SNEDDS was better simulated by an in vitro lipolysis model and thereby better simulated the in vivo absorption. The oral absorption of drugs in SNEDDS was closely related to the properties of the drugs and oil used in the SNEDDS. SNEDDS was more suitable for improving oral absorption of the high liposoluble drugs than the low one.
Abbreviation list
SNEDDS, self-nanoemulsifying drug delivery system; BCS II, Biopharmaceutical Classification Systems Class II; GRI, griseofulvin; PHE, phenytoin; IND, indomethacin; KET, ketoprofen; CIN, cinnarizine; GRI%, griseofulvin distribution percentage content; CIN%, cinnarizine distribution percentage content; GIT, gastrointestinal tract; MCT, medium-chain triglycerides; LCT, long-chain triglycerides; IVIVC, in vitro-in vivo correlation; Fr, relative oral bioavailability.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No. 81373361).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–420. doi:10.1023/A:1016212804288
2. Cherniakov I, Domb AJ, Hoffman A. Self-nano-emulsifying drug delivery systems: an update of the biopharmaceutical aspects. Expert Opin Drug Deliv. 2015;12(7):1121–1133. doi:10.1517/17425247.2015.999038
3. Fatouros DG, Karpf DM, Nielsen FS, Mullertz A. Clinical studies with oral lipid based formulations of poorly soluble compounds. Ther Clin Risk Manag. 2007;3(4):591–604.
4. Hauss DJ. Oral lipid-based formulations. Adv Drug Deliv Rev. 2007;59(7):667–676. doi:10.1016/j.addr.2007.05.006
5. Khatri P, Shao J. Transport of lipid nano-droplets through MDCK epithelial cell monolayer. Colloids Surf B. 2017;153:237–243. doi:10.1016/j.colsurfb.2017.02.024
6. Liu W, Pan H, Zhang C, et al. Developments in methods for measuring the intestinal absorption of nanoparticle-bound drugs. Int J Mol Sci. 2016;17:7. doi:10.3390/ijms17071171
7. Martin AN, Sinko PJ, Singh Y. Martin’s Physical Pharmacy and Pharmaceutical Sciences.
8. Bibi HA, Holm R, Bauer-Brandl A. Simultaneous lipolysis/permeation in vitro model, for the estimation of bioavailability of lipid based drug delivery systems. Eur J Pharm Biopharm. 2017;117:300–307. doi:10.1016/j.ejpb.2017.05.001
9. Land LM, Li P, Bummer PM. Mass transport properties of progesterone and estradiol in model microemulsion formulations. Pharm Res. 2006;23(10):2482–2490. doi:10.1007/s11095-006-9014-5
10. Dahan A, Miller JM, Hoffman A, Amidon GE, Amidon GL. The solubility-permeability interplay in using cyclodextrins as pharmaceutical solubilizers: mechanistic modeling and application to progesterone. J Pharm Sci. 2010;99(6):2739–2749. doi:10.1002/jps.22033
11. Larsen AT, Akesson P, Jureus A, et al. Bioavailability of cinnarizine in dogs: effect of SNEDDS loading level and correlation with cinnarizine solubilization during in vitro lipolysis. Pharm Res. 2013;30(12):3101–3113. doi:10.1007/s11095-013-1145-x
12. Dahan A, Hoffman A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm. 2007;67(1):96–105. doi:10.1016/j.ejpb.2007.01.017
13. Larsen AT, Sassene P, Mullertz A. In vitro lipolysis models as a tool for the characterization of oral lipid and surfactant based drug delivery systems. Int J Pharm. 2011;417(1–2):245–255. doi:10.1016/j.ijpharm.2011.03.002
14. Kaukonen AM, Boyd BJ, Porter CJ, Charman WN. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm Res. 2004;21(2):245–253. doi:10.1023/B:PHAM.0000016282.77887.1f
15. Trevaskis NL, Kaminskas LM, Porter CJ. From sewer to saviour - targeting the lymphatic system to promote drug exposure and activity. Nat Rev Drug Discov. 2015;14(11):781–803. doi:10.1038/nrd4608
16. Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. doi:10.1038/nrd2197
17. Zhang Q, He N, Zhang L, et al. The in vitro and in vivo study on self-nanoemulsifying drug delivery system (SNEDDS) based on insulin-phospholipid complex. J Biomed Nanotechnol. 2012;8(1):90–97. doi:10.1166/jbn.2012.1371
18. Rao SV, Agarwal P, Shao J. Self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of protein drugs: II. In vitro transport study. Int J Pharm. 2008;362(1–2):10–15. doi:10.1016/j.ijpharm.2008.05.016
19. Irvine JD, Takahashi L, Lockhart K, et al. MDCK (Madin-Darby canine kidney) cells: a tool for membrane permeability screening. J Pharm Sci. 1999;88(1):28–33. doi:10.1021/js9803205
20. Wu H, Long X, Fei Y, et al. Combined use of phospholipid complexes and self-emulsifying microemulsions for improving the oral absorption of a BCS class IV compound, baicalin. Acta Pharm Sin B. 2014;4(3):217–226. doi:10.1016/j.apsb.2014.03.002
21. Sek L, Porter CJ, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. J Pharm Pharmacol. 2002;54(1):29–41. doi:10.1211/0022357021771896
22. Armand M, Borel P, Pasquier B, et al. Physicochemical characteristics of emulsions during fat digestion in human stomach and duodenum. Am J Physiol. 1996;271(1 Pt 1):G172–183. doi:10.1152/ajpgi.1996.271.1.G172
23. USP. USP35-NF30 (united States Pharmacopoeia 35 - National Formulary 30) Ed. Rockville: United States Pharmacopeial; 2012.
24. Larsen AT, Ogbonna A, Abu-Rmaileh R, Abrahamsson B, Ostergaard J, Mullertz A. SNEDDS containing poorly water soluble cinnarizine; Development and in vitro characterization of dispersion, digestion and solubilization. Pharmaceutics. 2012;4(4):641–665. doi:10.3390/pharmaceutics4040641
25. Aggarwal N, Goindi S, Khurana R. Formulation, characterization and evaluation of an optimized microemulsion formulation of griseofulvin for topical application. Colloids Surf B, Biointerfaces. 2013;105:158–166. doi:10.1016/j.colsurfb.2013.01.004
26. Brandsch M, Ganapathy V, Leibach FH. H(+)-peptide cotransport in Madin-Darby canine kidney cells: expression and calmodulin-dependent regulation. Am J Physiol. 1995;268(3 Pt 2):F391–397. doi:10.1152/ajprenal.1995.268.3.F391
27. Ganapathy ME, Brandsch M, Prasad PD, Ganapathy V, Leibach FH. Differential recognition of beta -lactam antibiotics by intestinal and renal peptide transporters, PEPT 1 and PEPT 2. J Biol Chem. 1995;270(43):25672–25677. doi:10.1074/jbc.270.43.25672
28. Taub ME, Kristensen L, Frokjaer S. Optimized conditions for MDCK permeability and turbidimetric solubility studies using compounds representative of BCS classes I-IV. Eur J Pharm Sci. 2002;15(4):331–340. doi:10.1016/S0928-0987(02)00015-5
29. Miller JM, Beig A, Krieg BJ, et al. The solubility-permeability interplay: mechanistic modeling and predictive application of the impact of micellar solubilization on intestinal permeation. Mol Pharm. 2011;8(5):1848–1856. doi:10.1021/mp200181v
30. Wang S, Chen K, Li L, Guo X. Binding between proteins and cationic spherical polyelectrolyte brushes: effect of pH, ionic strength, and stoichiometry. Biomacromolecules. 2013;14(3):818–827. doi:10.1021/bm301865g
31. Dembczynski R, Jankowski T. Determination of pore diameter and molecular weight cut-off of hydrogel-membrane liquid-core capsules for immunoisolation. J Biomater Sci Polym Ed. 2001;12(9):1051–1058. doi:10.1163/156856201753252552
32. Stadalius MA, Ghrist BF, Snyder LR. Predicting bandwidth in the high-performance liquid chromatographic separation of large biomolecules. II. A general model for the four common high-performance liquid chromatography methods. J Chromatogr. 1987;387:21–40. doi:10.1016/S0021-9673(01)94511-X
33. Reddy ST, Berk DA, Jain RK, Swartz MA. A sensitive in vivo model for quantifying interstitial convective transport of injected macromolecules and nanoparticles. J Appl Physiol (1985). 2006;101(4):1162–1169. doi:10.1152/japplphysiol.00389.2006
34. Drisko GL, Cao L, Kimling MC, Harrisson S, Luca V, Caruso RA. Pore size and volume effects on the incorporation of polymer into macro- and mesoporous zirconium titanium oxide membranes. ACS Appl Mater Interfaces. 2009;1(12):2893–2901. doi:10.1021/am9006098
35. Lane ME, O’Driscoll CM, Corrigan OI. The relationship between rat intestinal permeability and hydrophilic probe size. Pharm Res. 1996;13(10):1554–1558. doi:10.1023/A:1016091915733
36. Porter CJH, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–1412. doi:10.1023/B:PHAM.0000036914.22132.cc
37. Iwanaga K, Kawabata Y, Miyazaki M, Kakemi M. Quantitative analysis of the effect of triglyceride alkyl-chain length on the partitioning of highly lipophilic compounds to the mesenteric lymph in intestinal cells. Arch Pharm Res. 2014;37(7):937–946. doi:10.1007/s12272-013-0249-5
38. Imada C, Takahashi T, Kuramoto M, et al. Improvement of oral bioavailability of N-251, a novel antimalarial drug, by increasing lymphatic transport with long-chain fatty acid-based self-nanoemulsifying drug delivery system. Pharm Res. 2015;32(8):2595–2608. doi:10.1007/s11095-015-1646-x
39. Han SF, Yao TT, Zhang XX, et al. Lipid-based formulations to enhance oral bioavailability of the poorly water-soluble drug anethol trithione: effects of lipid composition and formulation. Int J Pharm. 2009;379(1):18–24. doi:10.1016/j.ijpharm.2009.06.001
40. Kossena GA, Charman WN, Wilson CG, et al. Low dose lipid formulations: effects on gastric emptying and biliary secretion. Pharm Res. 2007;24(11):2084–2096. doi:10.1007/s11095-007-9363-8
41. Phan S, Salentinig S, Prestidge CA, Boyd BJ. Self-assembled structures formed during lipid digestion: characterization and implications for oral lipid-based drug delivery systems. Drug Deliv Transl Res. 2014;4(3):275–294. doi:10.1007/s13346-013-0168-5
42. O’Driscoll CM. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002;15(5):405–415. doi:10.1016/S0928-0987(02)00051-9
43. Trevaskis NL, Charman WN, Porter CJH. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv Drug Deliv Rev. 2008;60(6):702–716. doi:10.1016/j.addr.2007.09.007
44. Khoo SM, Shackleford DM, Porter CJ, Edwards GA, Charman WN. Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm Res. 2003;20(9):1460–1465. doi:10.1023/A:1025718513246
45. Khan J, Rades T, Boyd BJ. Lipid-based formulations can enable the model poorly water-soluble weakly basic drug cinnarizine to precipitate in an amorphous-salt form during in vitro digestion. Mol Pharm. 2016;13(11):3783–3793. doi:10.1021/acs.molpharmaceut.6b00594
46. Holm R, Porter CJ, Mullertz A, Kristensen HG, Charman WN. Structured triglyceride vehicles for oral delivery of halofantrine: examination of intestinal lymphatic transport and bioavailability in conscious rats. Pharm Res. 2002;19(9):1354–1361. doi:10.1023/A:1020311127328
47. Zgair A, Wong JC, Lee JB, et al. Dietary fats and pharmaceutical lipid excipients increase systemic exposure to orally administered cannabis and cannabis-based medicines. Am J Transl Res. 2016;8(8):3448–3459.
48. Alskar LC, Keemink J, Johannesson J, Porter CJH, Bergstrom CAS. Impact of drug physicochemical properties on lipolysis-triggered drug supersaturation and precipitation from lipid-based formulations. Mol Pharm. 2018;15(10):4733–4744. doi:10.1021/acs.molpharmaceut.8b00699
49. Tanaka Y, Kawakami A, Nanimatsu A, et al. In vivo evaluation of supersaturation/precipitation/re-dissolution behavior of cinnarizine, a lipophilic weak base, in the gastrointestinal tract: the key process of oral absorption. Eur J Pharm Sci. 2017;96:464–471. doi:10.1016/j.ejps.2016.10.023
50. Kaukonen AM, Boyd BJ, Charman WN, Porter CJ. Drug solubilization behavior during in vitro digestion of suspension formulations of poorly water-soluble drugs in triglyceride lipids. Pharm Res. 2004;21(2):254–260. doi:10.1023/B:PHAM.0000016283.87709.a9
51. Benito-Gallo P, Franceschetto A, Wong JC, et al. Chain length affects pancreatic lipase activity and the extent and pH-time profile of triglyceride lipolysis. Eur J Pharm Biopharm. 2015;93:353–362. doi:10.1016/j.ejpb.2015.04.027
52. Kossena GA, Charman WN, Boyd BJ, Dunstan DE, Porter CJ. Probing drug solubilization patterns in the gastrointestinal tract after administration of lipid-based delivery systems: a phase diagram approach. J Pharm Sci. 2004;93(2):332–348. doi:10.1002/jps.10554
53. Fischer LJ, Riegelman S. Absorption and activity of some derivatives of Griseofulvin. J Pharm Sci. 2010;56(4):469–476. doi:10.1002/jps.2600560410
54. Widanapathirana L, Tale S, Reineke TM. Dissolution and solubility enhancement of the highly lipophilic drug phenytoin via interaction with poly(N-isopropylacrylamide-co- vinylpyrrolidone) excipients. Mol Pharm. 2015;12(7):2537–2543. doi:10.1021/acs.molpharmaceut.5b00202
55. Aloisio C, Longhi MR, De Oliveira AG. Development and characterization of a biocompatible soybean oil-based microemulsion for the delivery of poorly water-soluble drugs. J Pharm Sci. 2015;104(10):3535–3543. doi:10.1002/jps.24555
56. Bhatt B, Kumar V. Regenerated cellulose capsules for controlled drug delivery: part IV. In-vitro evaluation of novel self-pore forming regenerated cellulose capsules. Eur J Pharm Sci. 2017;97:227–236. doi:10.1016/j.ejps.2016.11.027
57. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2017 Nov 8. doi:10.1093/nar/gkx1037 46 387-D1082
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.