")
Back to Journals » Infection and Drug Resistance » Volume 15
Comparison of Molecular Characteristics Between Methicillin-Resistant and -Susceptible Staphylococcus aureus Clinical Isolates by Whole-Genome Sequencing
Authors Zhu H , Luo H, Zhong Q, Cao X, Gu S , Peng S , Xiao Y, Chen Y, Hang Y, Fang X, Zou S, Yu F, Hu L
Received 8 February 2022
Accepted for publication 12 May 2022
Published 9 June 2022 Volume 2022:15 Pages 2949—2958
DOI https://doi.org/10.2147/IDR.S359654
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Hongying Zhu,1,* Hong Luo,1,* Qiaoshi Zhong,1 Xingwei Cao,1 Shumin Gu,1 Suqin Peng,1 Yanping Xiao,1 Yanhui Chen,1 Yaping Hang,1 Xueyao Fang,1 Shan Zou,1 Fangyou Yu,2 Longhua Hu1
1Department of Clinical Laboratory Medicine, Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 2Department of Clinical Laboratory Medicine, Shanghai Pulmonary Hospital, Tongji UniversitySchool of Medicine, Shanghai, 200082, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Longhua Hu; Fangyou Yu, Email [email protected]; [email protected]
Introduction: The transmission of methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible Staphylococcus aureus (MSSA) are great public health concern worldwide. To better understand S. aureus evolution and dissemination, we compared the molecular features of MSSA and MRSA isolates.
Methods: In this study, 74 MSSA and 102 MRSA non-duplicate isolates were recovered from clinical samples between 2016 and 2020. Molecular epidemiology, antimicrobial resistance determinants, and virulence gene profiles were carried out by whole-genome sequencing (WGS).
Results: Twenty distinct sequence types were identified in MRSA isolates, with the most common being ST59, ST630, and ST338. The major genotypes of MSSA were ST188 and ST7. The toxin genes clfA, sek, and seq were significantly associated with MRSA, while splA/B, clfB, map, sdrC/D, and sem-sen-seo-seu were detected more frequently in MSSA isolates than MRSA (P < 0.05). The tst positive isolates were more commonly identified in CC1 and CC72, whereas lukE/D was mainly found in the CC7, CC15, CC88, and completely absent in CC59 clones.
Conclusion: Our results compared the genetic diversity between MRSA and MSSA strains, suggesting efforts to fight infections caused by MSSA need to be intensified due to MSSA isolates carrying wide range of virulence factors. Comparative epidemiological studies of large populations of MSSA and MRSA will be necessary in the future to understand how MSSA and MRSA populations may co-evolve and interact in the future.
Keywords: methicillin-susceptible Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, virulence, resistance, molecular characteristics, whole-genome sequencing
Introduction
Staphylococcus aureus (S. aureus) is one of the most infamous pathogens, causing an incalculable number of uncomplicated infections and probably tens of millions of more severe, rapidly progressive infections globally every year. A study from the University of Washington reported that S. aureus ranked in the two top priority pathogens for deaths associated with resistance. And during 2019, methicillin-resistant S. aureus (MRSA) accounting for more than 100, 000 deaths.1 This invasive bacterial continues to evolve, and recognition of recent trends in molecular characteristics and future horizons are highly meaningful.
MRSA has been reported to evolve via two primary mechanisms: spread of existing resistant clones and acquisition of staphylococcal cassette chromosome mec (SCCmec) by a methicillin-susceptible S. aureus (MSSA) strain. To date, 13 SCCmec types and three mec genes have been discovered in S. aureus.2,3 The epidemiological influence of MRSA strains is considered to originate from combination of several hypervirulence loci and methicillin resistance genes, which enable these strains to colonize and invade healthy individuals and transmit rapidly through the population.4 Molecular epidemiological studies have shown that a few epidemic MRSA lineages are given rise to the majority of infections worldwide.5,6 Hence, it is crucial to understand the molecular characteristics of MRSA isolates to achieve infection control more effectively. Noteworthy, significant progress has been achieved in the research of antimicrobial activity and its mechanism of action.7–13
Most studies focus on MRSA, although infections with MSSA might be even more clinically important, given their higher incidence of invasive infections when compared with MRSA.14 However, genetic diversity between MRSA and MSSA isolates may exist but remain unelucidated. Karauzum et al reported that MSSA ST8 is more virulent than MRSA ST8.15 Antimicrobial resistance genes and virulence factors located in staphylococci are often carried on large mobile DNA elements that can be transferred horizontally. Horizontal gene transfer of these mobile elements may ultimately enhance pathogenicity, epidemics, and resistance to antibiotics.16 Some previous studies have found an increasing transfer of mobile elements, leads to an increase in S. aureus pathogenicity and resistance.17,18
Whole-genome sequencing (WGS) provides a cost-effective approach to investigating and confirming or refuting outbreaks of S. aureus.19,20 Such sequencing appears to be well suited for comprehensive analysis of the genetic relatedness among isolates, detect uncommon antibiotic resistance genes, and reveal complex patterns of virulence genes. Therefore, this study aimed to unravel the molecular characteristics between MRSA and MSSA isolated from patients admitted to a tertiary hospital in southern China over a 5-year period utilizing WGS.
Materials and Methods
Collection of Clinical Isolates and S. aureus Confirmation
From January 2016 to December 2020, 102 non-duplicate MRSA isolates were recovered from a university hospital in central China. In addition, 74 MSSA isolates were randomly collected during this period. S. aureus isolates were cultured on blood chocolate agar plates at 37 °C (±1 °C) for 16–18 h, and confirmed by colony morphology, Gram-staining, cell morphology, catalase, and coagulase tests according to routine laboratory procedures. S. aureus isolates were identified using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF). Briefly, single colony was suspended in distilled water, and mixture was spotted onto a MALDI target plate. Then, the bacterial pellet was resuspended by adding 1 μL of an α-cyano-4- hydroxycinnamic acid matrix solution and allowed to dry. Before identification, quality control and calibration were carried out using a standard calibration mixture of an Escherichia coli (ATCC 8739) extract. Spectra were recorded using a MALDI-TOF mass spectrometer in positive linear mode. The pattern-matching algorithm in the software performed comparative analysis with reference spectra and processed spectra of bacteria. Further antimicrobial susceptibility tests of all isolates were implemented using the standard methods provided by VITEK 2 Compact system (bioMérieux, Marcy-l’Étoile, France).
Whole Genome Sequencing and Analysis of Isolates
DNA was extracted using a Bacteria GenDNA Kit (TIANGEN, China) following the manufacturer’s protocols, and added 20 mg/mL lysostaphin solution. The NEBNext R UltraTM II DNA Library PrepKit for Illumina R was utilized to construct sequencing libraries and loaded onto NovaSeq S4 flow cell, in accordance with standard Illumina instructions (Illumina, Inc., United States). The raw sequenced reads quality was assessed using FastQC v.0.11.5. The WGS information were used for typing characterization, clonality, resistome, and virulence determinant analysis. Multilocus sequence typing (MLST) was performed using the MLST database website (https://cge.cbs.dtu.dk/services/MLST/). Spa typing was performed by submitting sequences to the spaTyper database website (https://cge.cbs.dtu.dk/services/spatyper/). BioNumerics software was applied to cluster related sequence types (STs), defined as clonal complexes (CCs). The SCCmec of the 102 MRSA isolates was determined using the database website SCCmecFinder. Resistome analysis was performed using the ResFinder database website (https://cge.cbs.dtu.dk/services/ResFinder/).
Statistical Analysis
All data were analyzed using the SPSS Data Editor, version 25 (IBM Corp, Armonk, NY, USA). Categorical variables were presented as counts and percentages (%) and compared using the chi-square test. P >0.05 was considered statistically insignificant.
Results
Antimicrobial Susceptibility Testing and Concordance Between Phenotypic and Genotypic
All the strains were susceptible to linezolid and vancomycin (Table 1). Among the 102 MRSA isolates, the majority were resistant to penicillin (100.0%), erythromycin (72.5%), and clindamycin (57.8%); The resistance rates to other antibiotics tested were 34.3% to tetracycline, 16.7% to ciprofloxacin, 11.8% to gentamicin, and 5.9% to rifampicin. The correlation between detected resistance genes and penicillin, erythromycin and trimethoprim-sulfamethoxazole resistance were 92.9%, 96.0% and 93.6%, respectively. The correlation between other drugs and sequences data was 100%. The most prevalent erm gene at erythromycin-resistant S. aureus isolates were ermB (54.5%, 54/99), followed by ermC (45.5%, 45/99), ermA (7.1%, 7/99) and mph (1.0%, 1/99).
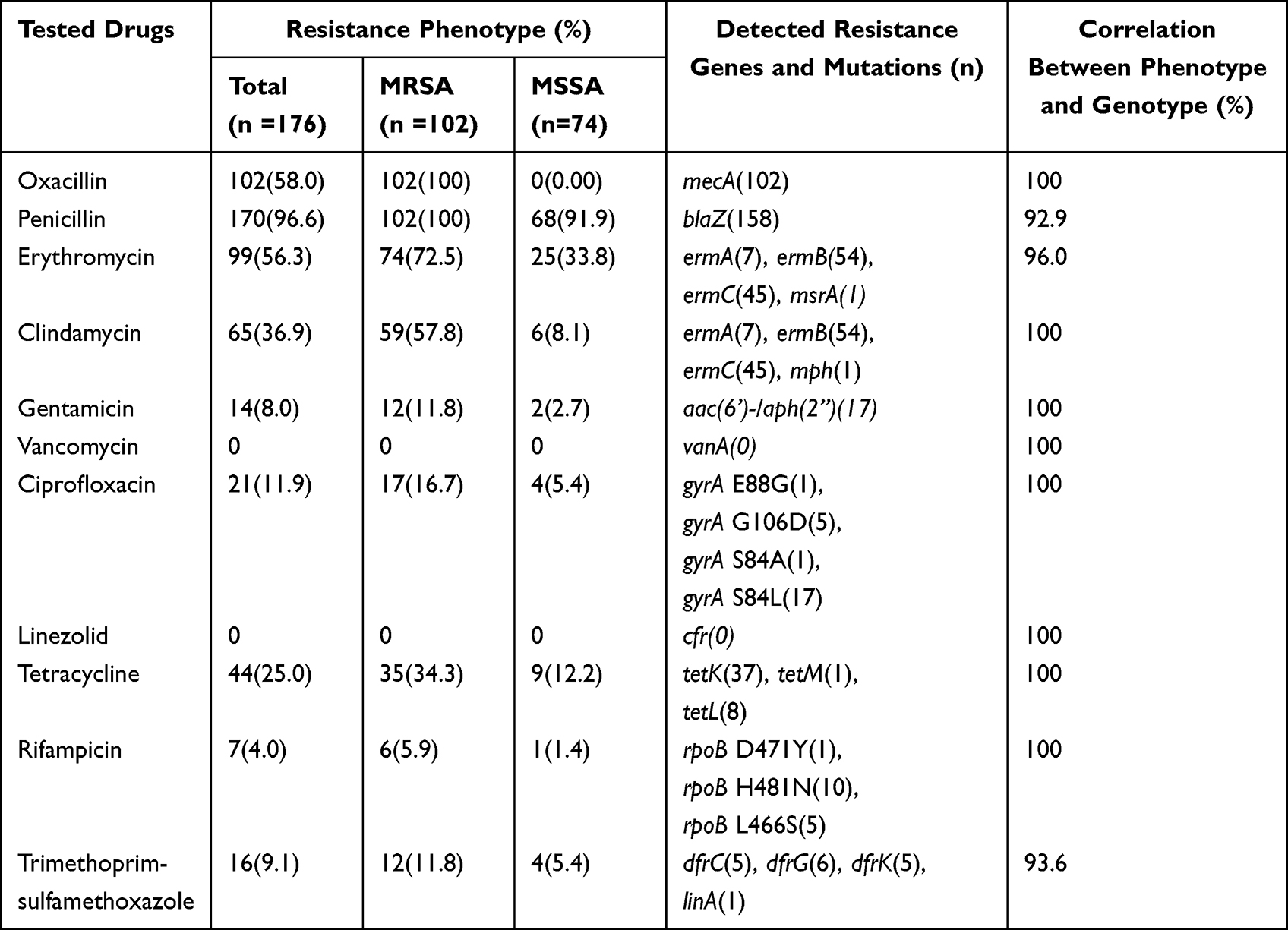 |
Table 1 Correlation of Resistance Phenotype and Predicted Genotype for 176 Staphylococcus aureus Isolates |
Virulence Gene Profiling of MRSA and MSSA
We compared the toxin gene frequency among MSSA and MRSA (Table 2). Almost all S. aureus (>95%) isolates carried certain toxin genes (icaA, hly/hla, hysA, and sspA/B), with a low prevalence (<10%) of other genes (tst, sed, seh, ser).
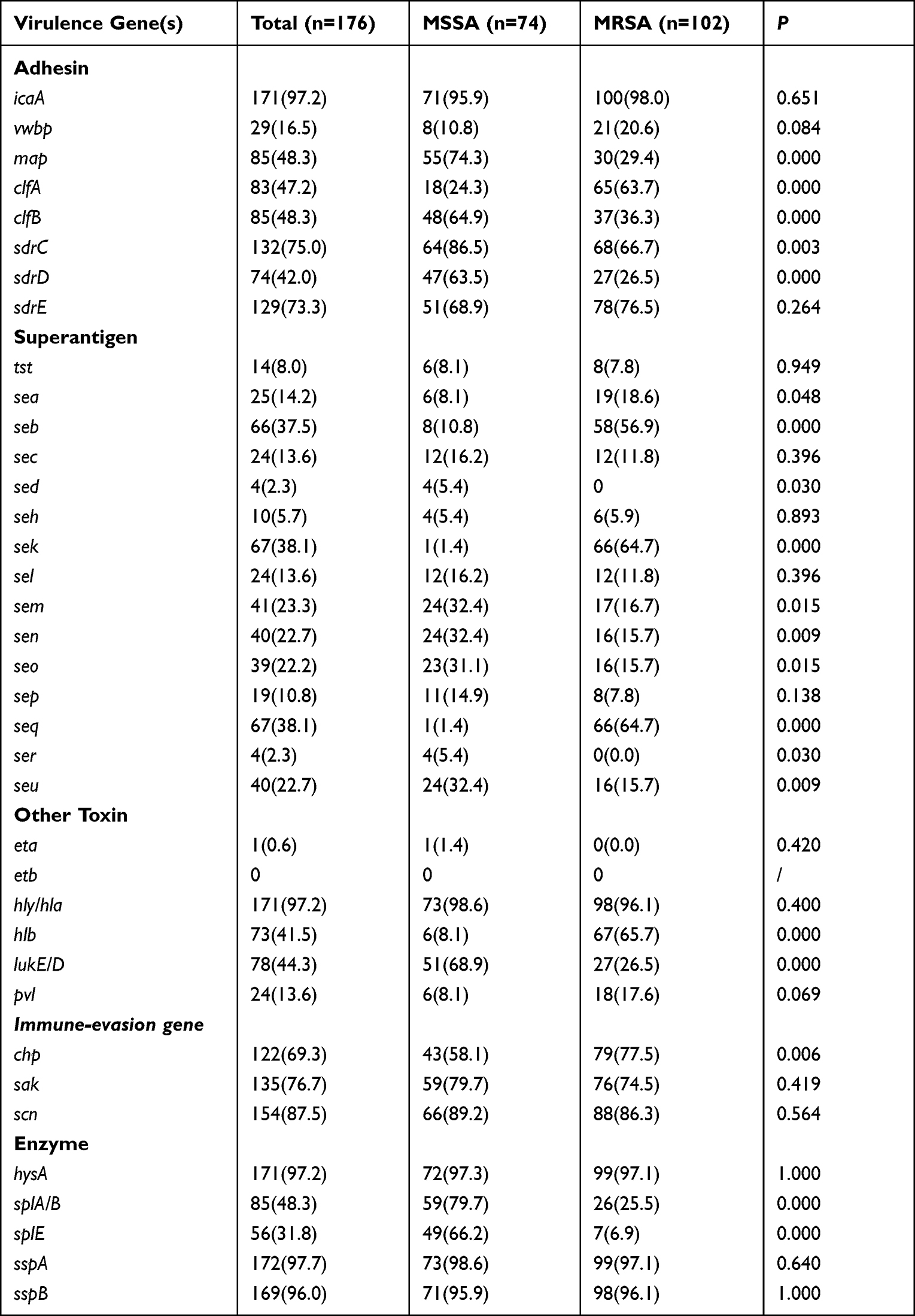 |
Table 2 Virulence Genes in Staphylococcus aureus Isolates as Determined by Whole-Genome Sequencing – Comparison Between MSSA and MRSA |
The tst gene was found in 7.8% of MRSA isolates, whereas 8.1% of MSSA strains contained the tst gene. In addition, clfB, map, sdrC/D, sem-sen-seo-seu, and splE genes were more common in MSSA than in MRSA. In our study, pvl gene was found in 24 isolates, with a prevalence of 17.6% (18/102) in MRSA and 8.1% (6/74) in MSSA. No etb genes were found among the 176 isolates, and eta gene was only found in one MSSA isolate.
Typing Characteristics Between MRSA and MSSA
The genetic differences of S. aureus isolates tested were assessed by MLST and spa typing, and 102 MRSA isolates were additionally tested for SCCmec typing. Among the 176 S. aureus isolates, 31 unique STs belonging to 16 CCs were observed, except for the untypable isolates (Supplementary Figure). Among the MRSA isolates, ST59 was most prevalent (41.2%, 42/102), followed by ST630 (9.8%, 10/102), ST338 (5.9%, 6/102), and ST88 (5.9%, 6/102) (Table 3). In the MSSA group, the major STs were ST188 (20.3%, 15/74) and ST7 (10.8%, 8/74), followed by ST6 (6.8%, 5/74) and ST15 (6.8%, 5/74) (Table 4).
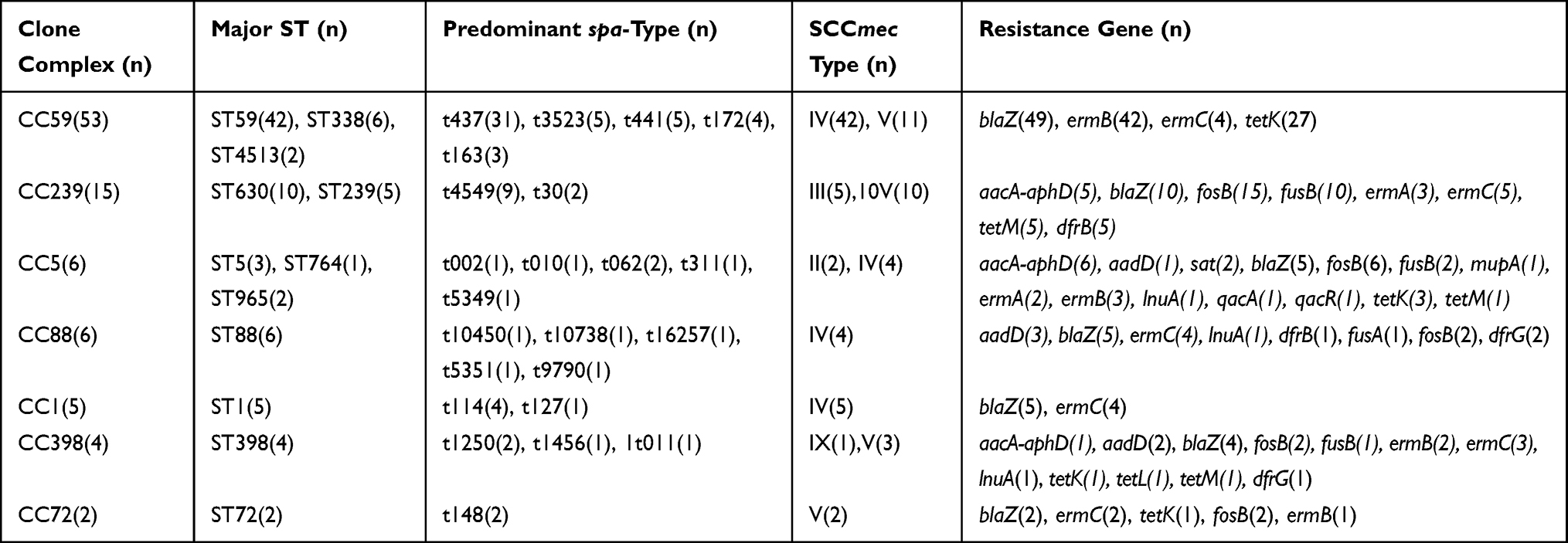 |
Table 3 Molecular Characteristics and Antibiotic Resistance Genes Profiles of 102 MRSA Isolates |
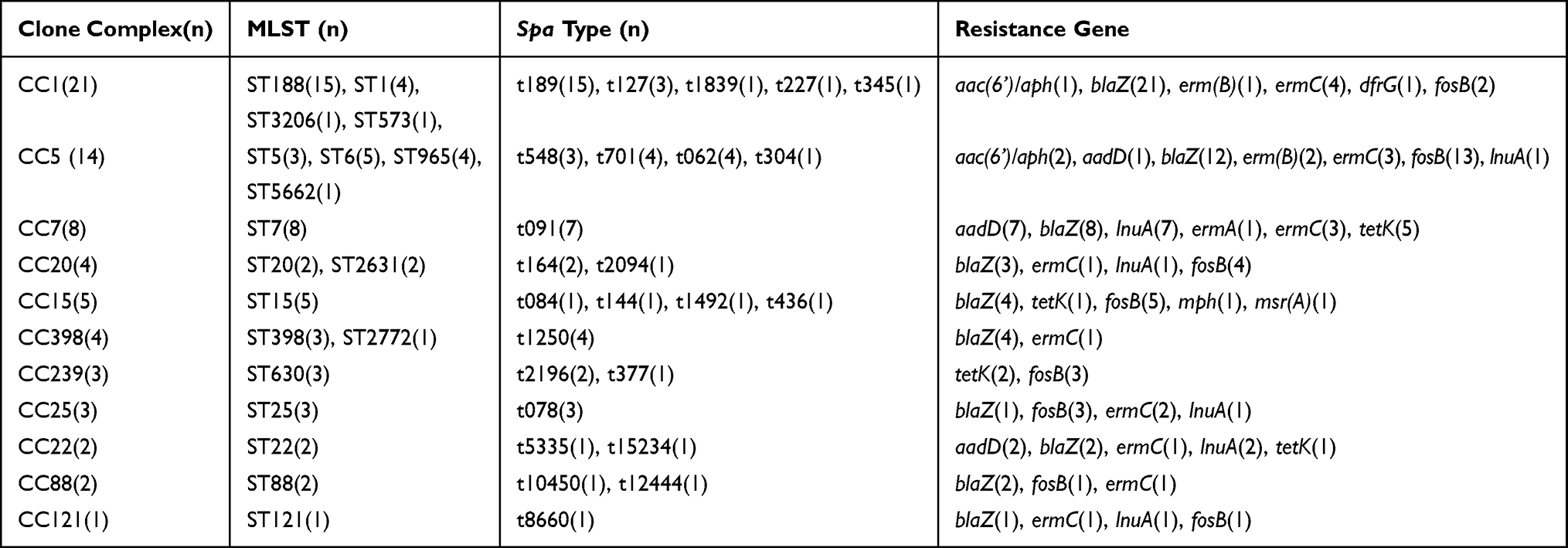 |
Table 4 Molecular Characteristics and Antibiotic Resistance Genes Profiles of 74 MSSA Isolates |
Using spa typing, 57 distinct types were found. The most prevalent spa types were t0437 (19.3%, 34/176), t0189 (9.1%, 16/176), t4549 (5.1%, 9/176), and t1250 (4.0%, 7/176). The MRSA isolates yielded 33 spa types, of which t437 was the most prevalent (Table 3). The MSSA isolates were assigned to 31 spa types, among which t189 was dominant (Table 4). Seven spa types—t002, t062, t10450, t1250, t127, t148, and t78—were identified in both MRSA and MSSA isolates.
When combined with MLST and spa typing, the most prevalent type was ST59-t437 (13.6%, 24/176), followed by ST188-t189 (8.5%, 15/176), ST630-t4539 (5.1%, 9/176), and ST7-t091 (4.0%, 7/176). It was interesting to note a significant association between certain STs and spa types: ST59 was associated primarily with t437 (57.1%, 24/42), ST188 with t189 (100.0%, 15/15), ST338 with t437 (83.3%, 5/6), and ST630 with t4549 (69.2%, 9/13). Remarkably, all ST59-t437 belonged to MRSA isolates, whereas all ST188-t189 belonged to MSSA isolates, indicated that some of the certain molecular types were closely correlated to methicillin resistance.
Sixteen clonal complexes (CCs) were obtained in five years (Supplementary Figure). The most abundant CCs were CC59, CC1, and CC5, representing 55.7% of the isolates. Specifically, the top three CCs in the MSSA group were CC1 (28.4%), CC5 (18.9%), and CC7 (10.8%), while the most dominant CCs in the MRSA group were CC59 (52.0%), CC239 (14.7%), and CC5 (5.9%).
Through SCCmec typing, five SCCmec types were identified in all MRSA strains, and two additional isolates were non-typeable. SCCmec IV, the most epidemic cassette type identified in this study (n = 62, 60. 8%) followed SCCmec V (n = 29, 28.4%). Traditional hospital-associated SCCmec types I–III were found in 7.8% of isolates, SCCmec III (n = 5, 4.9%), and SCCmec II (n = 3, 2.9%), but SCCmec type I was not observed. Almost all SCCmec V were associated with CC59, C239, CC398, and CC72. In contrast, all SCCmec III isolates were associated with CC239. Notably, one isolate carried the SCCmec type IX and belonged to ST398.
Association of Virulence Genes Among the Clonal Complexes
Toxin factors varied among the major 13 CCs (Table 5). The pvl gene was only found in CC22 (100%), CC88 (25%), CC398 (25%), CC59 (24.5%). The tst was considerably more common in CC72 and CC1 isolates. The prevalence of sep gene in CC7 and CC88 was significantly higher compared with other CCs . seb was mainly present in CC59 strains (96.2%), followed by CC25 (75.0%) and CC45 (60.0%). seb, sek, and seq were significantly higher in CC59 than in other CCs, clfA was considerably higher than in CC15. The lukE/D gene was more frequently possessed by isolates from the CC239 strains than by those from the CC59 (44.4% vs 0%), mainly existed in CC7, CC15, CC72, and CC88 isolates. Neither of the CC239 and CC7 isolates carried chp, and no CC1 found pvl-positive isolate. What’s more, we found an MSSA isolate that carried sek and seq gene.
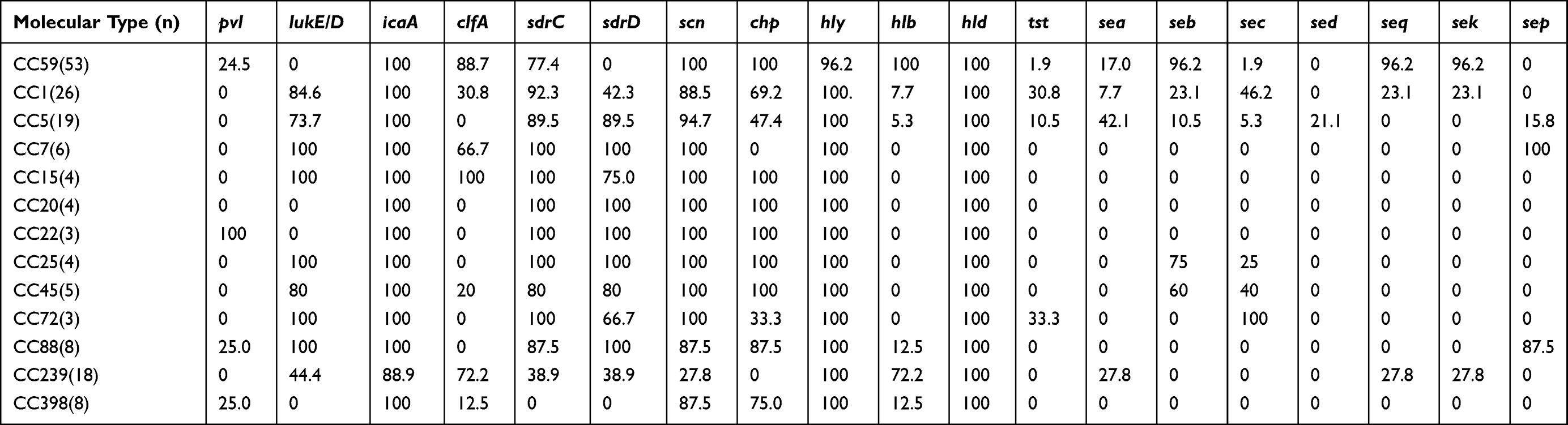 |
Table 5 Frequencies of Virulence Genes Among Different CC Clones (%) |
Discussion
S. aureus is considered one of the most representative pathogen that causes endemic and epidemic infections, and the wide distribution of S. aureus has been a severe problem worldwide. These epidemic staphylococcal strains have evolved, developing new genetic elements via horizontal gene transfer, and accumulated mutations that have altered gene expression and function.
The majority of the clinical MRSA cases were divided to the CC59 clone (ST59-IV/V-t439), a clone contributing to the most outbreaks of community-associated MRSA in China even in Asian.21,22 However, while previous studies found ST239 to be a major clone,23,24 only five of the MRSA isolates in this study were ST239. A recent study indicated that the ongoing replacement of ST239 by ST59 in China, is probably due to the carriage of the chp gene that enhances ST59 virulence activity.25 Up now, the transmission success of ST59 clones in hospital settings remains unclear. Jin et al suggested that clonal transmission was putatively associated with the intrinsic physiological properties of ST59, rather than with its resistance to antibiotics.26 ST630, the second most prevalent MRSA clone strain in this report, was also an emerging lineage in Nordic countries,27 and has spread to China28 and Japan.29 In addition, six types of SCCmec were identified, of which type IV accounted for 60.8% of the MRSA isolates, suggesting that cassette is frequently transferred. This is probably because the smaller size and simpler genetic makeup of SCCmec type IV are advantageous.30
The present study showed that the most predominant STs in MSSA were ST188 (20.3%), ST7 (10.8%), and ST6 (6.8%). ST188, with its significantly higher bacterial adhesion and biofilm formation ability than other STs, remained the most frequent MSSA clone in S. aureus infections among children and adults in China.31,32 Although most documented infections caused by ST188 have been linked to MSSA, ST188-MRSA has been reported.33–35 It has been reported that simultaneous acquisition of SCCmec may contribute to the emergence of ST188-MRSA.36 The diverse epidemiology between MRSA and MSSA might be associated with evolutionary processes that affect staphylococcal genetic.37 Furthermore, we found that up to seven STs included both MSSA and MRSA. A study performed in a Chinese hospital observed that roughly 14.2% of the MSSA isolates shared a common genetic background with MRSA lineages.38 These data support the notion that some MSSA lineages may provide a stable genomic environment for integrating the SCCmec element. Probably, compared to other lineages, these MSSA lineages possess fitness characteristics that promote their persistence in the host and additional potential transfer between hosts. Hence, this indicated that these MSSA isolates probably integrated the SCCmec element to promote MRSA infection and dissemination in communities and healthcare settings.
In vitro antimicrobial susceptibility testing of 176 isolates showed all strains were susceptible to linezolid and vancomycin, while the resistance rates observed for penicillin, erythromycin were over 56%. In the current study, the susceptibility rate of rifampicin in S. aureus was low (4.0%, 7/176). However, the resistance rate of S. aureus to rifampicin has been reported with increasing frequency in many regions.39,40 96.0% isolates of resistant to erythromycin carried one or more genes, which called ermA, ermB, ermC, and mph. According to previous studies, erm are major erythromycin resistance genes.41 In our study, we found that the ermB gene was detected in 54.5% of erythromycin-resistant strains, while the ermA gene was present in 7.1% of resistant strains. The efflux proteins, encoded by tetK gene, was detected from 84.1% of tetracycline-resistant strains.42 In addition, tetL and tetM genes are also resulted to tetracycline resistance, but their frequency is lower compared to tetK. Numerous studies have proved that WGS enables the rapid identification of infecting isolates and investigation of antimicrobial resistance determinants. Concordance between predicted antimicrobial resistance genotypes from WGS data and phenotypic susceptibility in S. aureus has been reported. In current study, there was a high correlation between phenotypic resistance and the presence of predicted genotype. A correlation above 95% was seen for all antimicrobials except penicillin and trimethoprim-sulfamethoxazole with a correlation of 92.9%, 93.6%.
In our study, differences in the contribution of virulence genes were found between the MSSA and MRSA strains. For the MRSA isolates, 64.7% carried the sek gene, while only 25.5% were splA/B-positive. However, only one MSSA isolate carried the sek gene, whereas 79.7% harbored the splA/B genes. sek is frequently reported to be present in MRSA strains and has been postulated to play a key role in the evolution of community lifestyle.43 In the present study, serine proteases A and B (splA and splB) were significantly more prevalent in MSSA than MRSA isolates. This observation similar to previous reports, as these genes are often isolated from countries with a low prevalence of MRSA.44
lukE/D has been reported as one of the most important pore-forming toxin genes in S. aureus isolates, which lyses host cells and promotes virulence of the bacteria.45,46 However, the characteristics of lukE/D-positive strains have not yet been well investigated in our region. According to our study, the lukE/D gene was detected in 78 isolates (44.3%; 51 MSSA and 27 MRSA), substantially lower than reported in previous reports.47,48 It is of interest to note that the distribution of lukE/D in CC clones was different from that in the previous reports. According to previous reports, the toxin locus of lukE/D was present in CC1, CC59, CC5, and CC7 but completely absent from CC22, CC30, and CC398.49,50 In this study, we did not find any CC59 (ST59) isolates that carried lukE/D, while the detection rate of CC59 strain lukE/D isolated by Liu et al was 100%.34 In a study by He et al,47 the detection rate of lukE/D in ST59 isolates was 66.7%. In addition, the prevalence of lukE/D was not present in CC20 and CC22 isolates, but was found in CC1, CC5, CC7, CC15, and CC88 isolates, which is vastly different from previous studies. One possible explanation for this difference is that lukE/D is the main virulence factor for S. aureus causing infection in the blood, however, the total number of blood samples in present MRSA isolates is relatively small, or it may be due to potential regional differences, and the relationship between lukE/D gene and MSSA infection requires further investigation. The study also found the discrepancies between the tst gene distribution and epidemic CCs in S. aureus. A multicenter study showed that roughly 31% of S. aureus isolates were test-positive in China, and the most predominant CCs were CC398, CC15 and CC188.51 However, the tst gene was present in 8.0% of isolates and was mostly identified in CC1 in our collection, significantly higher than the CC1-tst carrier rate in other areas. This is probably due to geographic heterogeneity or the dissemination of CC1-tst strains in our hospital even in our region.
In summary, our results showed that, although MRSA was still the major problem of S. aureus infections, efforts to fight infections caused by MSSA need to be intensified due to MSSA isolates carrying wide range of virulence factors. Comparative epidemiological studies of large populations of MSSA and MRSA will be necessary in the future to understand how MSSA and MRSA populations may co-evolve and interact in the future.
Abbreviations
MRSA, Methicillin-resistant S. aureus; SCCmec, Staphylococcal cassette chromosome mec; MSSA, methicillin-susceptible S. aureus; WGS, whole-genome sequencing; MLST, multilocus sequence typing; STs, sequence types; CCs, clonal complexes.
Ethics Statement
As the Staphylococcus aureus clinical isolation in this study was part of the routine hospital laboratory procedure, we have confirmed that the isolate has no identifiable patient data, the Second Affiliated Hospital of Nanchang University Medical Research Ethics Committee exempted this research for review.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Jiangxi Natural Science Foundation (No. 20181BAB205066), the Jiangxi Natural Science Foundation (No. 20202BABL216039), the Jiangxi Natural Science Foundation (20202BAB216021).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Murray CJ, Ikuta KS, Sharara F. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 2022;399(10325):629–655. doi:10.1016/S0140-6736(21)02724-0
2. Hiramatsu K, Ito T, Tsubakishita S, et al. Genomic basis for methicillin resistance in Staphylococcus aureus. Infect Chemother. 2013;45(2):117–136. doi:10.3947/ic.2013.45.2.117
3. Kaya H, Hasman H, Larsen J, et al. SCC mec finder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. MSphere. 2018;3(1). doi:10.1128/mSphere.00612-17
4. Wang X, Liu Q, Zhang H, et al. Molecular characteristics of community-associated Staphylococcus aureus isolates from pediatric patients with bloodstream infections between 2012 and 2017 in Shanghai, China. Front Microbiol. 2018;9:1211. doi:10.3389/fmicb.2018.01211
5. Rodriguez-Noriega E, Seas C, Guzman-Blanco M, et al. Evolution of methicillin-resistant Staphylococcus aureus clones in Latin America. Int J Infect Dis. 2010;14(7):e560–e566. doi:10.1016/j.ijid.2009.08.018
6. Oliveira DC, Tomasz A, de Lencastre H. The evolution of pandemic clones of methicillin-resistant Staphylococcus aureus: identification of two ancestral genetic backgrounds and the associated mec elements. Microb Drug Resist. 2001;7(4):349–361. doi:10.1089/10766290152773365
7. Zhang X, Marichannegowda MH, Rakesh KP, Qin H. Master mechanisms of Staphylococcus aureus: consider its excellent protective mechanisms hindering vaccine development! Microbiol Res. 2018;212–213:59–66. doi:10.1016/j.micres.2018.05.002
8. Zha G, Preetham HD, Rangappa S, et al. Benzimidazole analogues as efficient arsenals in war against methicillin-resistance Staphylococcus aureus (MRSA) and its SAR studies. Bioorg Chem. 2021;115:105175. doi:10.1016/j.bioorg.2021.105175
9. Verma R, Verma SK, Rakesh KP, et al. Pyrazole-based analogs as potential antibacterial agents against methicillin-resistance Staphylococcus aureus (MRSA) and its SAR elucidation. Eur J Med Chem. 2021;212:113134. doi:10.1016/j.ejmech.2020.113134
10. Kumar Verma S, Verma R, Xue F, Kumar Thakur P, Girish YR, Rakesh KP. Antibacterial activities of sulfonyl or sulfonamide containing heterocyclic derivatives and its structure-activity relationships (SAR) studies: a critical review. Bioorg Chem. 2020;105:104400. doi:10.1016/j.bioorg.2020.104400
11. Verma SK, Verma R, Kumar KS, et al. A key review on oxadiazole analogs as potential methicillin-resistant Staphylococcus aureus (MRSA) activity: structure-activity relationship studies. Eur J Med Chem. 2021;219:113442. doi:10.1016/j.ejmech.2021.113442
12. Rakesh KP, Ramesh S, Gowda DC. Effect of low charge and high hydrophobicity on antimicrobial activity of the quinazolinone-peptide conjugates. Russ J Bioorganic Chem. 2018;44(2):158–164. doi:10.1134/S1068162018020036
13. Li C, Sridhara MB, Rakesh KP, et al. Multi-targeted dihydrazones as potent biotherapeutics. Bioorg Chem. 2018;81:389–395. doi:10.1016/j.bioorg.2018.08.024
14. Jackson KA, Gokhale RH, Nadle J, et al. Public health importance of invasive methicillin-sensitive Staphylococcus aureus Infections: surveillance in 8 US counties, 2016. Clin Infect Dis. 2020;70(6):1021–1028. doi:10.1093/cid/ciz323
15. Karauzum H, Ferry T, de Bentzmann S, et al. Comparison of adhesion and virulence of two predominant hospital-acquired methicillin-resistant Staphylococcus aureus clones and clonal methicillin-susceptible S. aureus isolates. Infect Immun. 2008;76(11):5133–5138. doi:10.1128/IAI.01697-07
16. Tong SY, Holden MT, Nickerson EK, et al. Genome sequencing defines phylogeny and spread of methicillin-resistant Staphylococcus aureus in a high transmission setting. Genome Res. 2015;25(1):111–118. doi:10.1101/gr.174730.114
17. Highlander SK, Hulten KG, Qin X, et al. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 2007;7(1):99. doi:10.1186/1471-2180-7-99
18. Queck SY, Khan BA, Wang R, et al. Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 2009;5(7):e1000533. doi:10.1371/journal.ppat.1000533
19. Koser CU, Holden MT, Ellington MJ, et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med. 2012;366(24):2267–2275. doi:10.1056/NEJMoa1109910
20. Holden MT, Feil EJ, Lindsay JA, et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci USA. 2004;101(26):9786–9791. doi:10.1073/pnas.0402521101
21. Chen Y, Sun L, Ba X, et al. Epidemiology, evolution and cryptic susceptibility of methicillin-resistant Staphylococcus aureus in China: a whole-genome-based survey. Clin Microbiol Infect. 2022;28(1):85–92. doi:10.1016/j.cmi.2021.05.024
22. Song JH, Hsueh PR, Chung DR, et al. Spread of methicillin-resistant Staphylococcus aureus between the community and the hospitals in Asian countries: an ANSORP study. J Antimicrob Chemother. 2011;66(5):1061–1069. doi:10.1093/jac/dkr024
23. Liu Y, Wang H, Du N, et al. Molecular evidence for spread of two major methicillin-resistant Staphylococcus aureus clones with a unique geographic distribution in Chinese hospitals. Antimicrob Agents Chemother. 2009;53(2):512–518. Doi:10.1128/AAC.00804-08
24. Xiao M, Wang H, Zhao Y, et al. National surveillance of methicillin-resistant Staphylococcus aureus in China highlights a still-evolving epidemiology with 15 novel emerging multilocus sequence types. J Clin Microbiol. 2013;51(11):3638–3644. doi:10.1128/JCM.01375-13
25. Chen H, Yin Y, van Dorp L, et al. Drivers of methicillin-resistant Staphylococcus aureus (MRSA) lineage replacement in China. Genome Med. 2021;13(1):171. doi:10.1186/s13073-021-00992-x
26. Jin Y, Zhou W, Zhan Q, et al. Genomic epidemiology and characterization of methicillin-resistant Staphylococcus aureus from bloodstream infections in China. MSystems. 2021;6(6):e83721. doi:10.1128/mSystems.00837-21
27. Sieber RN, Overballe-Petersen S, Kaya H, Larsen AR, Petersen A. Complete genome sequences of methicillin-resistant Staphylococcus aureus strains 110900 and 128254, two representatives of the CRISPR-cas-carrying sequence type 630/ spa type t4549 lineage. Microbiol Resour Announc. 2020;9(41). doi:10.1128/MRA.00891-20
28. Chen W, He C, Yang H, et al. Prevalence and molecular characterization of methicllin-resistant Staphylococcus aureus with mupirocin, fusidic acid and/or retapamulin resistance. BMC Microbiol. 2020;20:183. doi:10.1186/s12866-020-01862-z
29. Kawamura K, Kitaoka K, Kimura K, et al. Spread of seb -positive methicillin-resistant Staphylococcus aureus SCC mec type II-ST764 among elderly Japanese in nonacute care settings. Microb Drug Resist. 2019;25(6):915–924. doi:10.1089/mdr.2018.0337
30. Lakhundi S, Zhang K. Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin Microbiol Rev. 2018;31(4). doi:10.1128/CMR.00020-18
31. Liang B, Mai J, Liu Y, et al. Prevalence and characterization of Staphylococcus aureus isolated from women and children in Guangzhou, China. Front Microbiol. 2018;9:2790. doi:10.3389/fmicb.2018.02790
32. Yu F, Li T, Huang X, et al. Virulence gene profiling and molecular characterization of hospital-acquired Staphylococcus aureus isolates associated with bloodstream infection. Diagn Microbiol Infect Dis. 2012;74(4):363–368. doi:10.1016/j.diagmicrobio.2012.08.015
33. Ho PL, Lai EL, Chow KH, Chow LS, Yuen KY, Yung RW. Molecular epidemiology of methicillin-resistant Staphylococcus aureus in residential care homes for the elderly in Hong Kong. Diagn Microbiol Infect Dis. 2008;61(2):135–142. doi:10.1016/j.diagmicrobio.2007.12.017
34. Liu C, Chen ZJ, Sun Z, et al. Molecular characteristics and virulence factors in methicillin-susceptible, resistant, and heterogeneous vancomycin-intermediate Staphylococcus aureus from central-southern China. J Microbiol Immunol Infect. 2015;48(5):490–496. doi:10.1016/j.jmii.2014.03.003
35. Ko KS, Lee JY, Baek JY, et al. Characterization of Staphylococcus aureus nasal carriage from children attending an outpatient clinic in Seoul, Korea. Microb Drug Resist. 2008;14(1):37–44. doi:10.1089/mdr.2008.0776
36. Wan TW, Liu YJ, Wang YT, et al. Potentially conjugative plasmids harboring Tn6636, a multidrug-resistant and composite mobile element, in Staphylococcus aureus. J Microbiol Immunol Infect. 2021;55(2):225–233.
37. Miko BA, Hafer CA, Lee CJ, et al. Molecular characterization of methicillin-susceptible Staphylococcus aureus clinical isolates in the United States, 2004 to 2010. J Clin Microbiol. 2013;51(3):874–879. doi:10.1128/JCM.00923-12
38. Gu F, He W, Xiao S, et al. Antimicrobial resistance and molecular epidemiology of Staphylococcus aureus causing bloodstream infections at Ruijin hospital in Shanghai from 2013 to 2018. Sci Rep. 2020;10(1):6019. doi:10.1038/s41598-020-63248-5
39. Bongiorno D, Mongelli G, Stefani S, Campanile F. Burden of rifampicin- and methicillin-resistant Staphylococcus aureus in Italy. Microb Drug Resist. 2018;24(6):732–738. doi:10.1089/mdr.2017.0299
40. Udo EE, Boswihi SS. Antibiotic resistance trends in methicillin-resistant Staphylococcus aureus Isolated in Kuwait hospitals: 2011–2015. Med Princ Pract. 2017;26(5):485–490. doi:10.1159/000481944
41. Kaur DC, Chate SS. Study of antibiotic resistance pattern in methicillin resistant Staphylococcus aureus with special reference to newer antibiotic. J Glob Infect Dis. 2015;7(2):78–84. doi:10.4103/0974-777X.157245
42. Guay GG, Rothstein DM. Expression of the tetK gene from Staphylococcus aureus in Escherichia coli: comparison of substrate specificities of TetA(B), TetA(C), and TetK efflux proteins. Antimicrob Agents Chemother. 1993;37(2):191–198. doi:10.1128/AAC.37.2.191
43. Thurlow LR, Joshi GS, Richardson AR. Virulence strategies of the dominant USA300 lineage of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). FEMS Immunol Med Microbiol. 2012;65(1):5–22. doi:10.1111/j.1574-695X.2012.00937.x
44. Rasmussen G, Monecke S, Brus O, Ehricht R, Soderquist B. Long term molecular epidemiology of methicillin-susceptible Staphylococcus aureus bacteremia isolates in Sweden. PLoS One. 2014;9(12):e114276. doi:10.1371/journal.pone.0114276
45. Poddighe D, Vangelista L. Staphylococcus aureus infection and persistence in chronic rhinosinusitis: focus on leukocidin ED. Toxins. 2020;12(11):678. doi:10.3390/toxins12110678
46. Vasquez MT, Lubkin A, Reyes-Robles T, et al. Identification of a domain critical for Staphylococcus aureus LukED receptor targeting and lysis of erythrocytes. J Biol Chem. 2020;295(50):17241–17250. doi:10.1074/jbc.RA120.015757
47. He C, Xu S, Zhao H, et al. Leukotoxin and pyrogenic toxin superantigen gene backgrounds in bloodstream and wound Staphylococcus aureus isolates from eastern region of China. BMC Infect Dis. 2018;18(1):395. doi:10.1186/s12879-018-3297-0
48. Morinaga N, Kaihou Y, Noda M. Purification, cloning and characterization of variant LukE-LukD with strong leukocidal activity of staphylococcal bi-component leukotoxin family. Microbiol Immunol. 2003;47(1):81–90. doi:10.1111/j.1348-0421.2003.tb02789.x
49. Chao G, Bao G, Cao Y, et al. Prevalence and diversity of enterotoxin genes with genetic background of Staphylococcus aureus isolates from different origins in China. Int J Food Microbiol. 2015;211:142–147. doi:10.1016/j.ijfoodmicro.2015.07.018
50. McCarthy AJ, Lindsay JA. Staphylococcus aureus innate immune evasion is lineage-specific: a bioinfomatics study. Infect Genet Evol. 2013;19:7–14. doi:10.1016/j.meegid.2013.06.012
51. He W, Chen H, Zhao C, et al. Population structure and characterisation of Staphylococcus aureus from bacteraemia at multiple hospitals in China: association between antimicrobial resistance, toxin genes and genotypes. Int J Antimicrob Agents. 2013;42(3):211–219. doi:10.1016/j.ijantimicag.2013.04.031
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.