")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Comparison of Bioavailability and Bioequivalence of Generic and Brand Name Formulations of Escitalopram Oxalate Tablets in Healthy Chinese Population Under Fasting and Fed Conditions
Authors Li Q, Huo H, Hu W, Sui Y, Tang Y
Received 22 July 2020
Accepted for publication 28 October 2020
Published 24 November 2020 Volume 2020:14 Pages 5167—5177
DOI https://doi.org/10.2147/DDDT.S271970
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Qiuying Li, Hua Huo, Wenli Hu, Yin Sui, Yunbiao Tang
Department of Drug Clinical Trial Management Agency, General Hospital of Northern Theater Command, Shenyang 110840, People’s Republic of China
Correspondence: Yunbiao Tang; Hua Huo
Department of Drug Clinical Trial Management Agency, General Hospital of Northern Theater Command, Shenyang, People’s Republic of China
Tel/Fax +86-24-28897206
Email [email protected]; [email protected]
Purpose: This study compared the bioequivalence of two formulations of escitalopram oxalate 20 mg tablets in terms of bioavailability and tolerability in healthy Chinese male and female subjects.
Patients and Methods: A randomized, single-blind, two-period, two-sequence crossover study was performed under fasting and fed conditions, with a 21-day washout period. In total, 24 healthy subjects (18 males and 6 females) were enrolled in the fasting test and the fed test, respectively. Blood samples were collected over 168 h post-dose in each period. The concentrations of escitalopram in plasma were determined by high-performance liquid chromatography coupled with a tandem mass spectrometry. Pharmacokinetic parameters used for bioequivalence assessment were determined from the drug concentration data using noncompartmental analysis.
Results: All subjects showed good medication compliance. The 90% confidence intervals (CIs) for the geometric mean ratios of AUC0-t, AUC0-∞, and Cmax were within the bioequivalence acceptance criteria (80.00% to 125.00%). Adverse events were recorded and no deaths or serious adverse events (SAEs) occurred.
Conclusion: Escitalopram oxalate 20 mg tablets produced in China were bioequivalent to the reference formulation (Lexapro®) in healthy Chinese male and female subjects under fasting and fed conditions.
Keywords: escitalopram, escitalopram oxalate tablets, clinical trials, LC-MS/MS, pharmacokinetics, bioequivalence
Introduction
Depressive disorder is a common mental health condition that seriously affects the physical health and quality of life of patients. It has been reported that over 264 million people of all ages worldwide experience depressive disorders, in which females are more commonly affected than males.1 The prevalence of depressive disorder has increased by 14.3% from 2007 to 2017.1 In particular, chronic moderate or major depressive disorder can become a severe mental illness and people with depressive disorder have emerged as a high-risk group for suicide. The World Health Organization (WHO) has warned that depressive disorder has become one of the leading causes of disability.1 Rapid control of suicidal symptoms has become an essential part of the treatment strategy for this condition.
Clinical first-line antidepressants mainly include selective serotonin reuptake inhibitors (SSRIs) and serotonin and norepinephrine reuptake inhibitors (SNRIs). At present, SSRIs have become the most widely prescribed antidepressants in many countries since the first SSRI was introduced in 1987.2 Escitalopram oxalate (CAS 219,861–08-2) is a new type of SSRI and has a rapid-onset antidepressant activity.3 Escitalopram is a single S-enantiomer of citalopram, a dicyclo hydride derivative. The chemical structure of the escitalopram oxalate tablet is shown in Figure 1. The mechanism of the antidepressant effect of escitalopram may be associated with the inhibition of 5-hydroxytryptamine (5-HT) reuptake by neurons in the central nervous system and by enhancing the function of central serotoninergic nerves. In vitro and animal studies have shown that escitalopram is a highly selective SSRI, and effects on norepinephrine and dopamine reuptake are limited. It is well tolerated, with almost no anticholinergic effects, and exerts no effects on the cardiovascular system.4 Escitalopram oxalate was developed by Lundbeck, Switzerland, under the trade name Lexapro®. It was launched in the United States in September 2002. In July 2006, Lexapro® was approved for use in China.
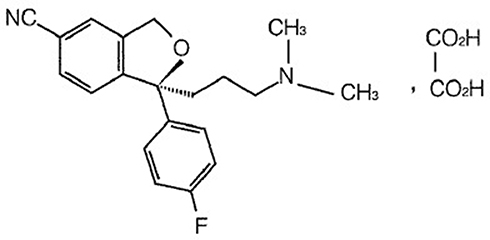 |
Figure 1 Chemical structure of escitalopram oxalate. |
Escitalopram’s oral absorption is complete and is unaffected by food. Absolute bioavailability is about 80%.5 The plasma protein binding rate is about 56%.6 Escitalopram is mainly metabolized by demethylation in the liver. N-demethylation of escitalopram is metabolized by CYP3A4, CYP2C19, and, to a lesser extent, CYP2D6 enzymes.6 The polymorphic CYP2C19 enzyme has been shown to affect the metabolism of escitalopram.6 Elimination half-life is approximately 30 hours after multiple dosing and metabolites may have a longer half-life.5 Escitalopram and its metabolites are mainly eliminated by the liver and kidney and are excreted from urine primarily in the form of metabolites.7,8
A randomized, double-blind, controlled clinical study conducted by Lepola et al of over 8 weeks duration demonstrated the safety and efficacy of escitalopram in treating major depressive disorder.9 The long-term clinical tolerability study and treatment response are based on a 12-month open-label study of 486 patients treated with escitalopram. After more than 12 months of treatment, escitalopram (10 to 20 mg) showed good safety and tolerability.10 The treatment of panic disorder with escitalopram is safe and well tolerated, and the incidence of common adverse events (AE) was similar to that of citalopram and placebo.11 The incidence of adverse reactions was lower, and the degree was mild, and mainly manifested as drowsiness, dizziness, and weight gain; in addition, compliance was also better.12 Although thought to be generally safe, it has been reported that escitalopram is linked to drug-induced liver injury and the measurement of liver function tests should be considered after initiation of antidepressant treatment.13
As a non-fatal disease, depressive disorder occurs in 80% of low- and middle-income countries.14 It carries a heavy burden, both psychologically and economically, which the families of patients have to bear. Therefore, an indigenous generic formulation with the same quality and efficacy is urgently needed to use interchangeably with the original formulation, which would be more beneficial for patients, especially for the Chinese population, which is also affordable in terms of cost and medication compliance.
As previously reported, the CYP2C19 genotype has a substantial impact on exposure and therapeutic endogenous levels of escitalopram.15 There is a high incidence of CYP2C19 poor metabolizers (PMs) within the Chinese population, and exposure to escitalopram in the Chinese is higher than in other populations.16 To date, the evidence relative to the exposure for escitalopram in Chinese male and female populations under fed conditions is very sparse. Thus, we investigated the pharmacokinetic profiles of escitalopram oxalate 20 mg tablets, both under fasting and fed conditions and the pharmacokinetic parameters were compared to demonstrate that the test formulation is bioequivalent to the reference formulation in terms of rate and extent of absorption.
Patients and Methods
Subjects and Trial Design
The study was designed as a randomized, single-blind, two-period, two-sequence crossover study, performed under fasting and fed conditions, with a 21-day washout period. In total, 24 healthy adult subjects (18 males and 6 females) were enrolled in the fasting test and the fed test, respectively. The same person was allowed to participate in only one test.
Before the study was carried out, the independent institutional ethics committee of the General Hospital of Northern Theater Command reviewed and approved the clinical protocol, which was performed in compliance with the Declaration of Helsinki guidelines and Good Clinical Practice. A letter of non-objection was obtained with the number (2016) 85. Since December 1, 2015, National Medical Products Administration (NMPA) adopted archival filing management of bioequivalence study for chemical drugs. As per the NMPA requirements, the content of this study was recorded in the online filing system and obtained registration number CTR20170237.
The aim, content, process, and possible adverse reactions of the study were fully introduced to the subjects, and written informed consent was obtained prior to study participation. Subject screening was performed to assess eligibility within 2 weeks before the day of enrollment. It included demographic data, physical examination, diseases history, vital signs (blood pressure, pulse, and body temperature), electrocardiography (ECG), screening for addictive drugs, and laboratory tests for liver function, renal function, coagulation function, hematology, blood glucose, routine urinalysis, and seroimmunology (HBsAg, anti-HCV, anti-TP, and anti-HIV), and a breath alcohol test. Pregnancy tests (for women) were performed just before dosing in each period. The inclusion criteria were healthy male or female subjects aged between 18 and 65 years at the screening who had signed the informed consent. The weight of males was not less than 50 kg and weight was not less than 45 kg for females, with the body mass index (BMI) between 19 and 26 kg/m2. Women had had no pregnancy plan or availability of adequate contraception for 6 months; women could not be enrolled in the study if they were pregnant or lactating. Further individuals with known contraindication or hypersensitivity to escitalopram oxalate, liver dysfunction, renal insufficiency, abnormal hematology with clinical significance, positive test results for HBsAg, anti-HCV, anti-TP and/or anti-HIV; any living condition or special diet that might significantly alter the absorption, distribution, metabolism or excretion of the study preparations; any disease that increases the risk of bleeding such as hemorrhoids, acute gastritis or gastric and duodenal ulcer; history of epilepsy; history of prolonged QTc interval; drug or alcohol abuse; smoking more than 5 cigarettes per day; or intake of any chocolate, food-containing caffeine and/or xanthine within 48 hours of the first dosing day were excluded from the study.
Trial Conduct
Eligible subjects arrived at the Phase I clinical trial ward in the afternoon the day before dosing. All subjects were required to fast overnight for at least 10 hours except for mineral water after dinner. In the dosing day’s morning, an intravenous in-dwelling needle was placed and a venous blood sample was collected before dosing. The blood samples of fed test were collected after having ingested a high-fat, high-calorie diet and before dosing. Subjects of fasting arm were given 240 mL water, and fed test subjects were given with 240 mL water 30 minutes after consuming a high-fat, high-calorie diet. Subjects were not allowed to drink 1 hour prior to and after dosing and had lunch and dinner 4 hours and 10 hours after dosing. Each subject was required to remain seated and to restrict physical activity for 30 minutes prior to and 10 hours after dosing. Vital signs were performed before dosing and at 2, 4, 6, 12, 24, 48, 72, 120, and 168 hours after dosing and AEs were assessed before and during the entire study.
Venous blood samples (4 mL) were collected in tubes containing K2EDTA anticoagulant at 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 6.0, 8.0, 12, 24, 36, 48, 72, 120, and 168 hours after dosing. The blood samples were centrifuged at 2500×g for 10 minutes at 4°C to separate the plasma, and then the plasma was transferred to two clean tubes (analytical sample and backup). All plasma samples were stored at −60°C prior to analysis. The date and time of collecting each sample were correctly recorded. After the washout period, the same operation was repeated with the alternate study preparation. Safety examinations were performed under fasting conditions 168 hours after dosing in the second period, and included physical examination, vital signs, ECG and laboratory tests for liver function, renal function, coagulation function, hematology, blood glucose, routine urinalysis, and pregnancy tests for women.
Study Preparations
Test formulation: Esitalopram oxalate 20 mg tablet (batch number: 1,606,916, content: 98.6%), valid until May 2019, produced by Shandong Jingwei Pharmaceutical co., LTD (China).
Reference formulation: Lexapro® containing escitalopram oxalate 20 mg tablets (Batch No.: A474374, content: 98.0%), valid until October 2020, FOREST PHARMACEUTICALS. INC.
Safety Evaluation
Safety evaluation parameters included AEs, severe adverse events (SAE), concomitant medication, changes in clinical laboratory results (hematology, blood chemistry and urinalysis), clinical symptoms, vital signs, ECGs, and physical examination. The severity of AEs was determined according to the CTCAE4.03 standard. The safety evaluation parameters were listed and summarized with descriptive statistics. Baseline data, post-administration data and change from baseline were summarized on each follow-up and every treatment; the shift table was used to describe the change from baseline to each follow-up after administration for the clinical significance of the examination results as the classification data. All statistical evaluations were conducted by SAS® (version 9.3; SAS Institute, Cary, NC, USA).
Method of Analysis
The method determining the concentrations of escitalopram in plasma was validated in accordance with the United States Food and Drug Administration (FDA) bioanalytical method validation guidance and the National Medical Products Administration (NMPA) guidelines. Quantitation was achieved with high-performance liquid chromatography using a Venusil XBP C18 column (3 µm, 2.1×50 mm; Agela Corporation, China) coupled to an API 4000 tandem mass spectrometry detector (AB Sciex, USA) with an electrospray ionization interface operated in positive ionization mode method. The column temperature was set at 40°C. The mobile phase was 0.1% formic acid in acetonitrile. The total run time of each sample was 5.0 min with gradient elution. The mass spectrometry detector was used with the mass transition ion pair value of 325.2/262.2 for escitalopram and 331.2/262.2 for R-enantiomer of citalopram-d6 (as the internal standard). The method was validated for selectivity, sensitivity, linearity, accuracy and precision, recovery, matrix effect, dilution integrity, and stability.
Pharmacokinetic Concentration Analysis
A 100 µL volume of plasma sample was placed in plastic tubes and was vortex-mixed with 50 µL of 50 ng/mL of IS for 1 minute. The content of the tube with 450 µL of acetonitrile solvent was shaken for 10 minutes at room temperature and centrifuged at 2880×g for 10 minutes at 4°C. A 300 µL volume of supernatant was transferred to another tube and evaporated under nitrogen gas until dryness. The residue was reconstituted with 200 µL 50% methanol, shaken for 10 minutes, centrifuged and 10 µL was injected into the LC-MS/MS system. Calibration standards, controls, and samples were processed in batches. Data processing was performed using the Analyst 1.6.2 software package (AB Sciex, USA).
Sample Size Estimation
The intra-individual variation of pharmacokinetic parameters (ie, AUC, Cmax) was the main analysis index for sample size estimation. According to the Biopharmaceutics Classification System (BCS) escitalopram oxalate was classified as a Class 1 agent. According to reported literature, the maximum intra-individual variation of escitalopram pharmacokinetic parameters (AUC, Cmax) was 17.91%. Using the geometric mean ratio between 0.95 and 1.05 and an alpha=0.05 (0.10 for both sides) of the one-sided test for calculation, the sample size of this two-period cross study was determined to be 20 subjects to meet the standard requirements of bioequivalence with PASS (Version 11) software. With consideration of the consent withdrawal or dropouts, this study included a total of 24 subjects for the fasting and fed tests, respectively.
Pharmacokinetic Analysis
Based on the plasma concentration data, the pharmacokinetic parameters of escitalopram were calculated using noncompartmental methods by WinNonlin® 6.4 software (Pharsight Corporation, Mountain View, CA, USA) and comprehensively reflected the characteristics of escitalopram absorption, distribution, metabolism, and excretion in the human body. The highest plasma concentration of the drug (Cmax, ng/mL) and the time to arrive to the Cmax (Tmax, hours) were directly obtained from the plasma concentration–time profiles. The elimination rate constant (λz) was obtained by taking the log-linear regression of the elimination phase concentration point and the elimination half-life (t1/2) was calculated as 0.693/λz. The area under the plasma concentration–time curve (AUC0–t) from time zero to the last quantifiable concentration time was calculated using the trapezoidal method. AUC0-∞ was the AUC0–t extrapolated to infinite time, and was calculated as follows: AUC0-∞=AUC0-t+Ct/λz, where Ct is the last quantifiable concentration. The relative bioavailability (F) was calculated based on the total absorption of the test preparation (T) and the reference preparation (R). F=AUC0-t(T)/AUC0-t (R) ×100%.
Statistical Analysis
Descriptive statistical analysis was performed for pharmacokinetic parameters. Quantitative indicators were summarized with descriptive statistics by arithmetic mean, standard deviation, median, minimum, maximum and geomean. Qualitative indicators were used for descriptive statistics using frequency or number and percentage of subjects. After transformation of Cmax, AUC0-t and AUC0-∞ to their logarithmic (Ln) values, analysis of variance (ANOVA) was conducted using a linear-mixed model to evaluate the effects of period, treatment, sequence, and subjects with WinNonlin® 6.4 (Pharsight Corporation, Mountain View, CA, USA). Additionally, the bioequivalence between the two formulations was assessed by two one-sided t-tests and the 90% confidence intervals (CIs) analysis. The Wilcoxon signed-rank test was performed on the nonparametric nature of Tmax. The acceptance criteria for bioequivalence were that the 90% CIs of the geometric mean ratios of the test formulation to the reference formulation were completely within 80.0–125.0% for the AUC and Cmax.
Results
Method Validation
The clinical dosage of escitalopram oxalate was small and the plasma concentrations were very low. The lower limit of quantification (LLOQ) was improved to 0.20 ng/mL in human plasma. The standard calibration curves showed good linearity within the range of 0.20 to 50 ng/mL using least-squares regression analysis for escitalopram (r >0.99). The five concentration levels of the quality control samples were 0.20 ng/mL (LLOQ), 0.60 ng/mL (LOQ), 3.0 ng/mL (GMOQ), 25 ng/mL (MOQ), and 45 ng/mL (HOQ). The intra-batch and inter-batch precision assayed from QC samples ranged from 0.40% to 3.6% and 1.1% to 2.6%, expressed as the coefficient of variation (CV), respectively. The intra-batch and inter-batch accuracy ranged from 94.0% to 99.7% and 95.0% to 98.7% of the actual value with the same QC samples, respectively. The stability studies indicated that escitalopram oxalate was stable under collection to post-treatment conditions. The results indicated that this validated method was accurate and sensitive for the determination of escitalopram in human plasma.
Demography and Compliance
We enrolled 24 healthy subjects for the fasting and fed tests, respectively. All subjects met the eligibility criteria for the protocol. All showed good medication compliance. The investigator examined their mouth, water bottle, and hands to ensure that the subjects were taking the drugs. In the fasting test, 22 subjects completed all treatments according to the protocol. One subject withdrew consent on the day before the first dosing due to personal reasons and one dropped out due to vomiting during the first test period. In the fed test, 22 subjects completed the test protocol, two subjects dropped out due to vomiting during the first period and were not treated in the second period. The demographic characteristic and other baseline values under fasting and fed conditions are shown in Table 1.
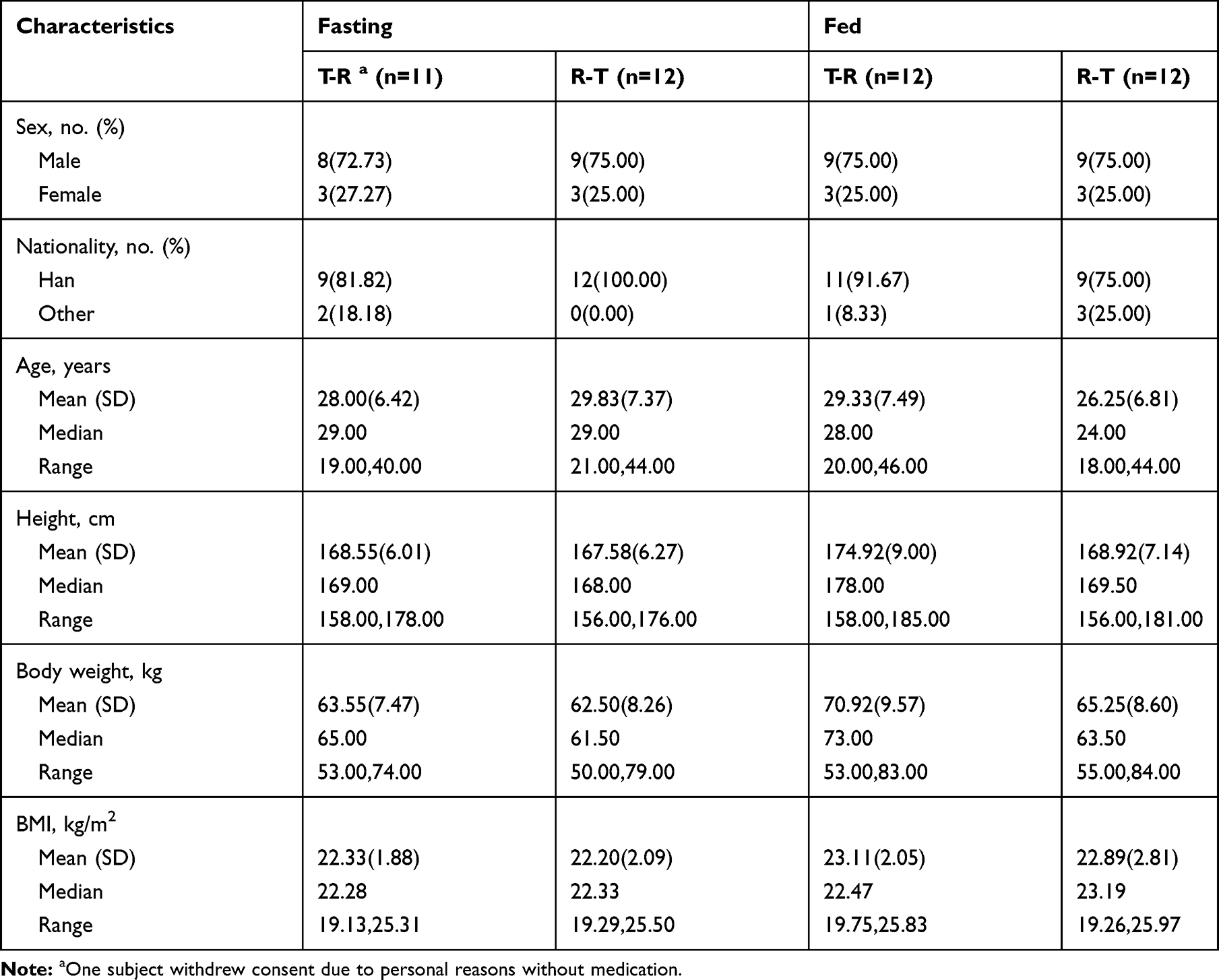 |
Table 1 Demographic Characteristics and Other Baseline Values of Test-Reference Treatment (T-R) and Reference-Test Treatment (R-T) Groups Under Fasting and Fed Conditions |
Pharmacokinetics and Bioequivalence Analysis
The mean plasma concentration-time profiles after oral administration of escitalopram oxalate 20 mg tablets of the test and reference formulations under fasting conditions are presented in Figure 2. Likewise, the profiles of the fed test are presented in Figure 3. The values of the pharmacokinetic parameters for the test and reference formulations under fasting and fed conditions are summarized in Table 2. Statistical comparison after Ln-transformed Cmax, AUC0-t, and AUC0-∞ of the test and reference formulations under fasting and fed conditions are reported in Tables 3 and 4, respectively. The results of ANOVA test under fasting and fed conditions are shown in Table 5. The results show that period and sequence did not have a significant influence on the results. But the p-values of the effect of subjects were less than 0.05, which did not affect the bioequivalence decision.
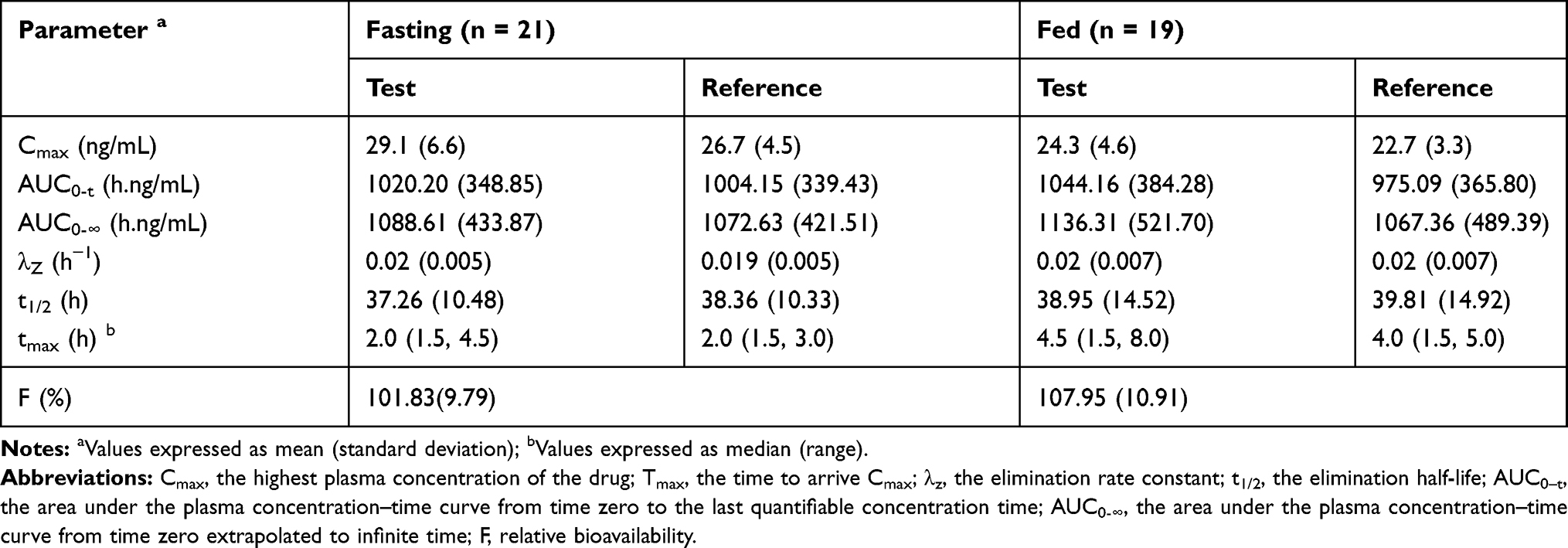 |
Table 2 The Main Pharmacokinetic Parameters After Oral Administration of 20 Mg Escitalopram Oxalate Tablets Under Fasting and Fed Conditions |
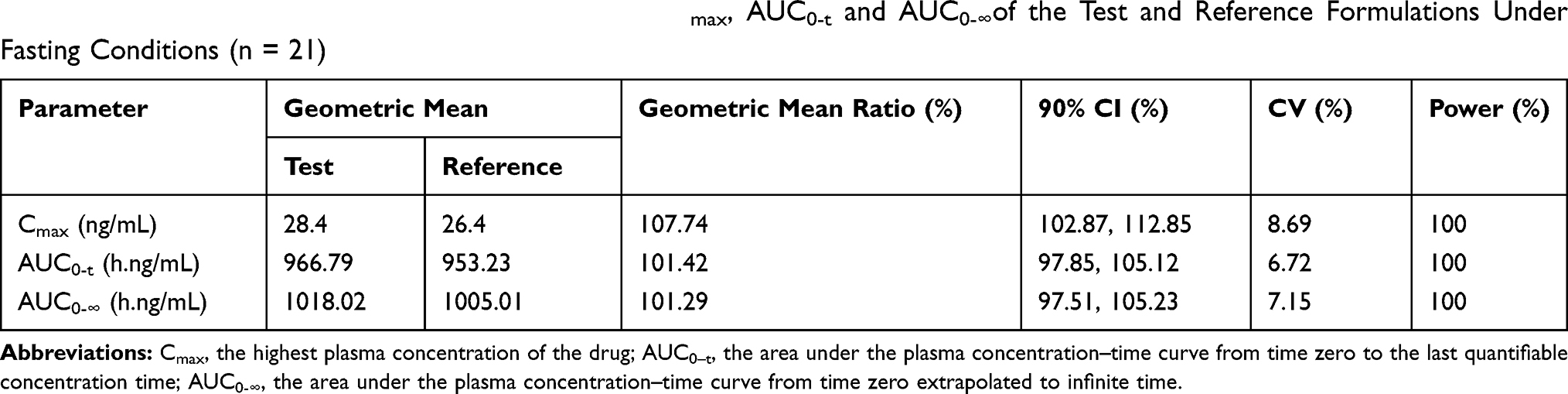 |
Table 3 Statistical Comparison After Ln-Transformed Cmax, AUC0-t and AUC0-∞of the Test and Reference Formulations Under Fasting Conditions (n = 21) |
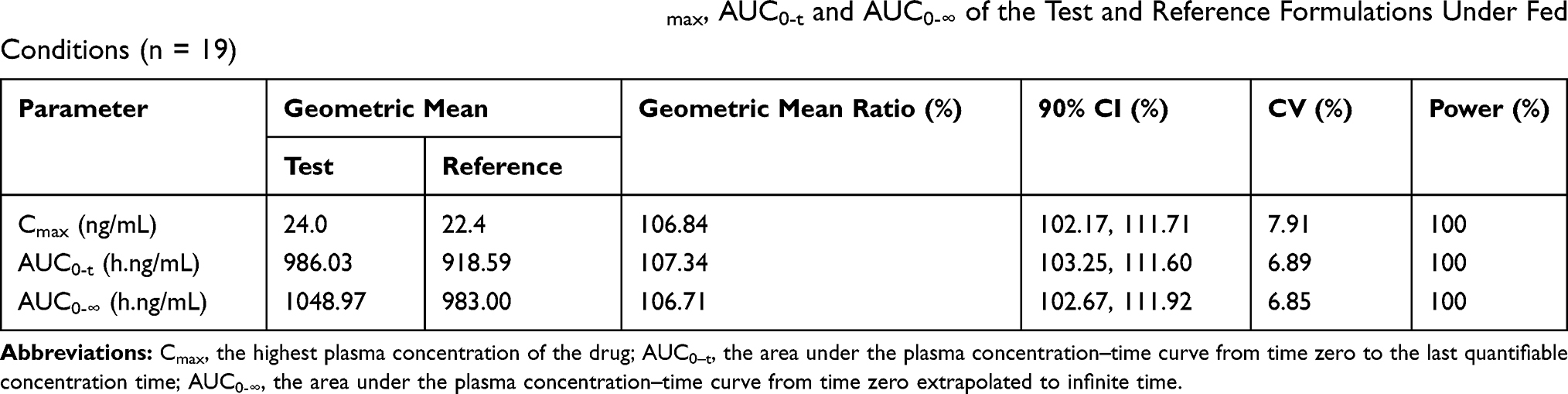 |
Table 4 Statistical Comparison After Ln-Transformed Cmax, AUC0-t and AUC0-∞ of the Test and Reference Formulations Under Fed Conditions (n = 19) |
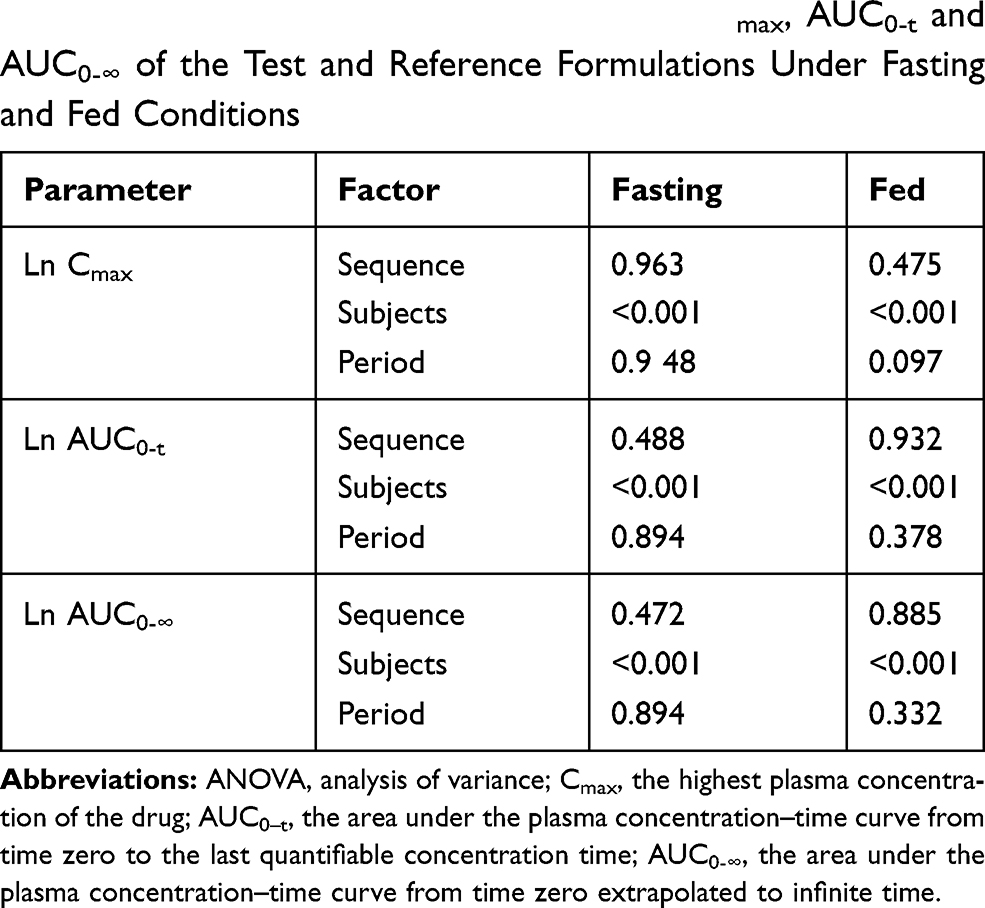 |
Table 5 ANOVA Test After Ln-Transformed Cmax, AUC0-t and AUC0-∞ of the Test and Reference Formulations Under Fasting and Fed Conditions |
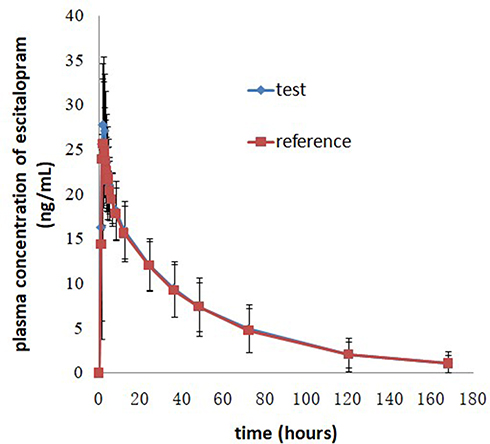 |
Figure 2 Mean plasma concentration-time profiles after oral administration of 20 mg escitalopram oxalate tablets of the test and reference formulations under fasting conditions. |
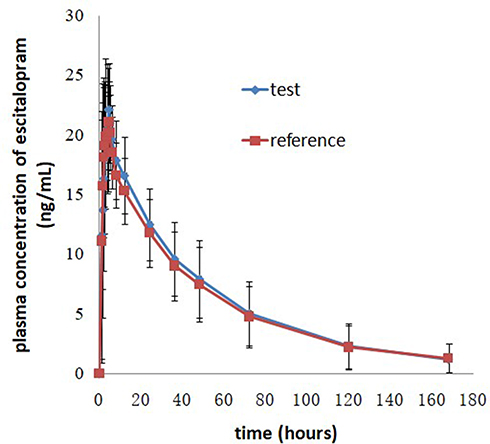 |
Figure 3 Mean plasma concentration-time profiles after oral administration of 20 mg escitalopram oxalate tablets of the test and reference formulations under fed conditions. |
Safety
There were no significant changes in laboratory tests, results of physical examinations, or vital signs of enrolled subjects before and after oral administration of escitalopram oxalate 20 mg tablets of the test or reference formulation under fasting and fed conditions. No deaths or serious SAEs occurred. During the fasting test, 9 subjects experienced 26 AEs, of which 15 AEs were judged as “possibly” related to the drugs. During the fed test, 10 subjects experienced 12 AEs, of which 11 AEs were judged as “possibly” related to the drugs. One subject of the fasting test group and two subjects of the fed test group dropped out due to vomiting during the first period. The outcomes of all AEs improved were cured or disappeared. The incidence rate and comparison of AEs for the two formulations under fasting and fed conditions are shown in Table 6. The summary of AEs for the test and reference formulations is presented in Table 7.
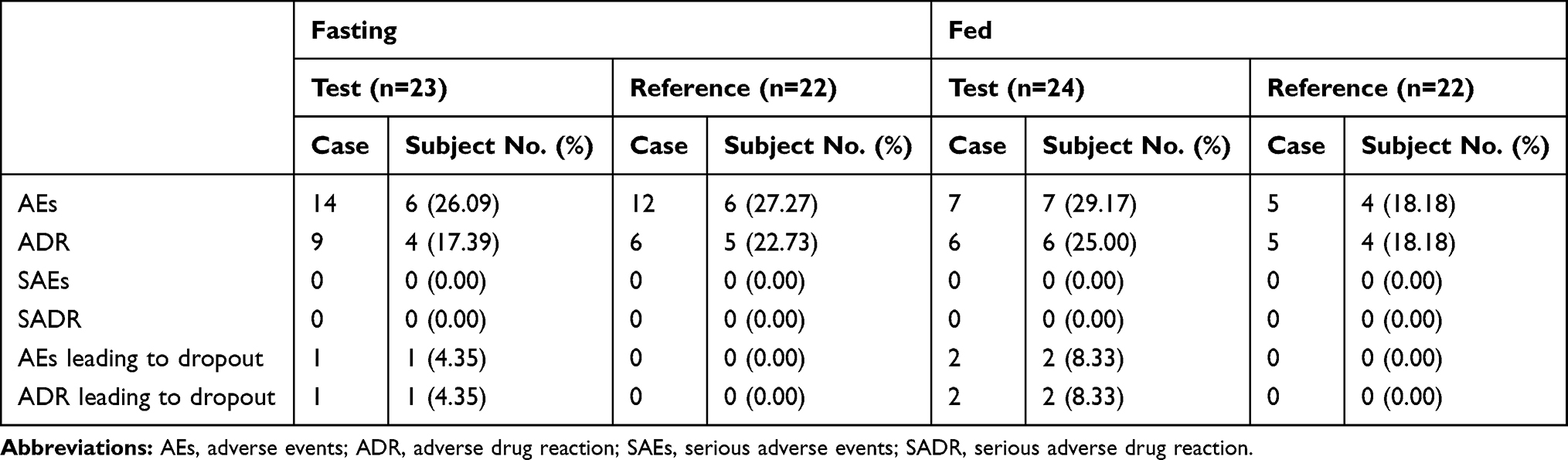 |
Table 6 The Incidence Rate and Comparison of AEs for the Two Formulations Under Fasting and Fed Conditions |
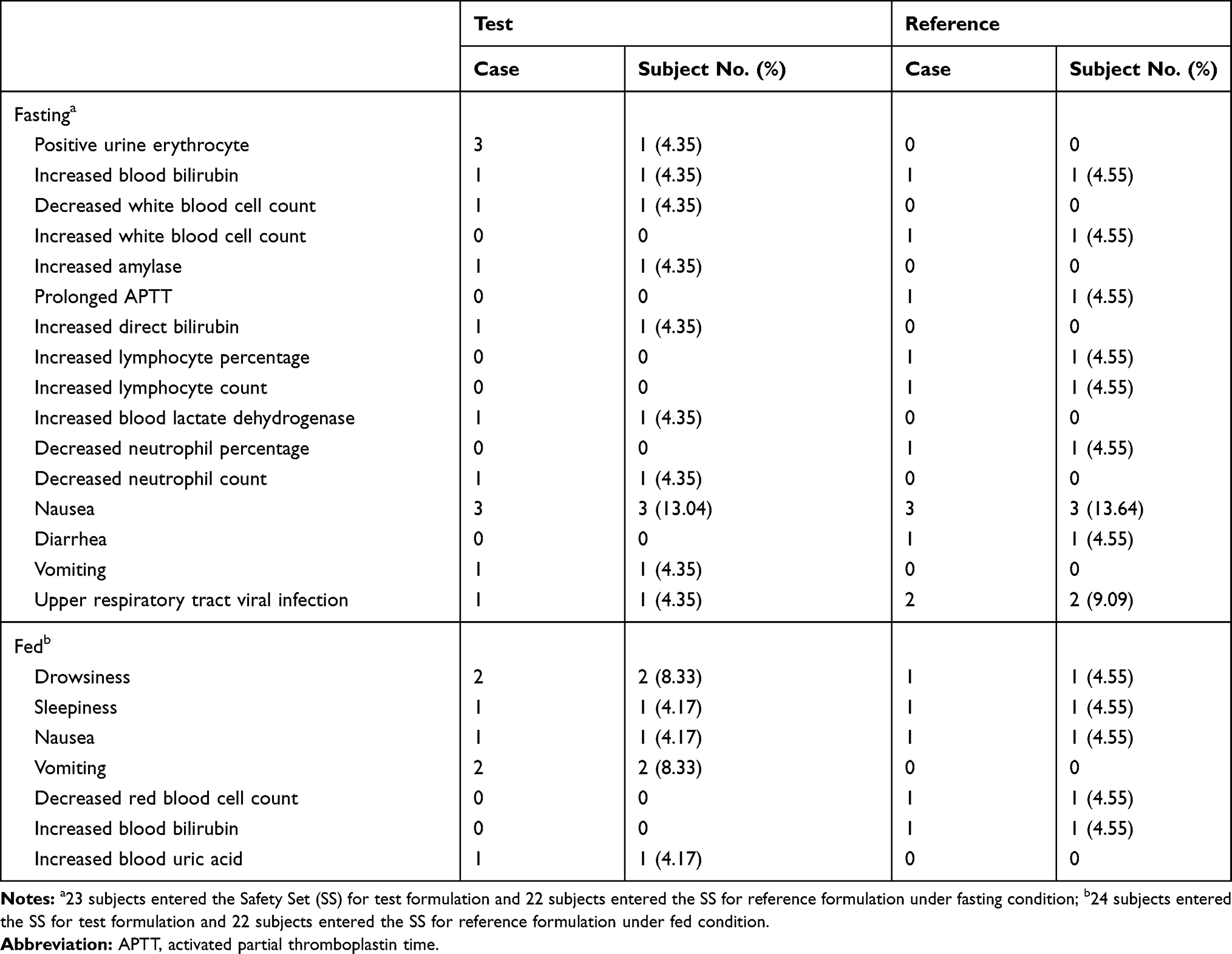 |
Table 7 Summary of AEs for the Test and Reference Formulations Under Fasting and Fed Conditions |
Discussion
Compliance and Tolerability
During the study, the subjects were all in good health with no concomitant disease before and after the study, no drug combination occurred in the subjects, so there was no potential influence on pharmacokinetics parameters or bioequivalence test results. After received the drugs, the side effects observed, including vomiting, nausea, diarrhea, and drowsiness, were all in compliance with the package insert for escitalopram, which have also been frequently reported in the literature.4,17,18 During the short duration of the study, both the test and reference preparations were well tolerated after twice dosing in the healthy subjects. Further study is needed order to determine whether the test formulation has good safety and tolerability in healthy Chinese subjects and patients.
Rate of Absorption
The pharmacokinetic parameters Cmax and tmax were used to estimate the rate of absorption after oral administration of escitalopram oxalate 20 mg tablets of the test and reference formulations. The plasma concentrations of escitalopram increased gradually, and then declined up to 168 hours. In the present study, the two one-sided 90% CIs for the ratios of the Ln-transformed means of Cmax and AUC were within the range of 80.0–125.0% under the fasting and fed conditions, which satisfied the acceptance criteria of bioequivalence. In one case in the fasting test group, the first sample concentration achieved a Cmax in the early stage (5–15 minutes after administration), while the same occurred in three cases of the fed test. In considering whether to adding these subjects’ data would influence the pharmacokinetic parameters and bioequivalence conclusion, we carried out sensitivity analysis and the results showed that there was no effect on the bioequivalence outcomes. Few studies have reported the pharmacokinetic behavior of escitalopram in healthy Chinese male and female subjects under fed conditions. Compared with the median of tmax of the fasting test, the tmax under the fed condition was slightly delayed, it indicated that food could delay the time to reach the maximum concentration but did not affect the maximum concentration in vivo.
Extent of Absorption
AUC0-t and AUC0-∞ were used to reflect the extent of absorption in the circulatory system after oral administration of escitalopram oxalate 20 mg tablets. In the fasting and fed test, the pharmacokinetics parameters (AUC0-t and AUC0-∞) of 19 subjects were included in the bioequivalence analysis. The results obtained from the present study indicated that the sample size met the statistical requirement. To date, there are limited data relative to the exposure of escitalopram in Chinese populations under the fed condition. The mean Cmax and AUC of escitalopram oxalate 20 mg tablets achieved in the fed test conditions was similar to that achieved in the fasting test, which indicated that food did not affect the rate and extent of absorption of escitalopram in vivo. The pharmacokinetic characteristics of escitalopram were fully comprised in the study design, given the sampling time of 168 h and a washout period of 21 days.
Pharmacokinetic Parameter Comparison
In the fasting and fed tests, the intra-individual variation of escitalopram oxalate was less than 8.69% and 7.91%, respectively. This provides information to be used in sample size estimations of Chinese population in future clinical trials.
Pharmacokinetic endpoints (Cmax and AUC) are usually used to evaluate the exposure of the formulation in the body. The demethylation of escitalopram is mainly metabolized by cytochrome CYP2C19, which is known to have genetic polymorphisms. It has been reported that the proportion of CTP2C19 PMs in the Chinese population is higher than the Caucasian population.19,20 The phenotypic frequency of PMs showed a significant ethnic difference in about 25% in Chinese,21,22 which was significantly higher than that in Caucasians (2–5%).23,24 In the present study, the mean Cmax and AUC0-∞ of escitalopram were higher than those reported in previous studies conducted in American, European and other Caucasian populations7,25 after oral administration of escitalopram oxalate 20 mg tablets for reference and test formulations under fasting conditions. As previously reported, CYP2C19 activity is a better predictor of escitalopram clearance,26 in our study, the mean t1/2 for the reference and test formulations was longer than those obtained in previous studies conducted in White populations and were similar to studies conducted in Chinese and Indian populations.27–29
Compared with previous reports under the same fasting conditions,7,25 the median of time required to reach peak plasma concentrations occurred at 2 hours with both formulations and was generally short in the present study. But under the fed condition, the median of tmax was similar to other previous reports conducted under the fasting conditions. By comparing the pharmacokinetic parameters, the exposure of escitalopram in the Chinese population was higher than that of Caucasian populations and the drug required a longer time to be metabolized in the Chinese population. Therefore, consideration should be made as to whether a dosage reduction is required in clinical practice. In addition, therapeutic drug monitoring should be conducted to minimize the incidence of adverse reactions and toxicity in the Chinese population.
Conclusion
Based on the results of this study, it may be concluded that the escitalopram oxalate 20 mg tablets produced in China are bioequivalent to the reference formulation (Lexapro®) in terms of the rate and extent of absorption under fasting and fed conditions. Both formulations were well tolerated in these small samples of Chinese male and female healthy subjects.
Acknowledgment
The authors thank the subjects who participated in this trial and the members of the Phase I Clinical Trial Center team of General Hospital of Northern Theater Command for their hard work. This study was financially sponsored by Jewim Pharmaceutical Co., Ltd.
Disclosure
The authors have no conflict of interests in this work.
References
1. James SL, Abate D, Abate KH, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. 2018;392(10159):1789–1858.
2. Lochmann D, Richardson T. Selective serotonin reuptake inhibitors. Handb Exp Pharmacol. 2019;250:135–144.
3. Murdoch D, Keam SJ. Spotlight on escitalopram in the management of major depressive disorder. CNS Drugs. 2006;20(2):167–170. doi:10.2165/00023210-200620020-00007
4. Burke WJ. Escitalopram. Expert Opin Investig Drugs. 2002;11(10):1477–1486. doi:10.1517/13543784.11.10.1477
5. Søgaard B, Mengel H, Rao N, Larsen F. The pharmacokinetics of escitalopram after oral and intravenous administration of single and multiple doses to healthy subjects. J Clin Pharmacol. 2005;45(12):1400–1406. doi:10.1177/0091270005280860
6. Rao N. The clinical pharmacokinetics of escitalopram. Clin Pharmacokinet. 2007;46(4):281–290. doi:10.2165/00003088-200746040-00002
7. Almeida S, Pedroso P, Filipe A, Neves RI, Tanguay M, Torns A. Bioequivalence of two formulations of escitalopram. Arzneimittelforschung. 2012;62(7):307–312.
8. Mendes GD, Babadopulos T, Bau FR, Chen LS, De Nucci G. Comparative bioavailability of two escitalopram formulations in healthy human volunteers. Int J Clin Pharmacol Ther. 2010;48(8):554–562. doi:10.5414/CPP48554
9. Lepola UM, Loft H, Reines EH. Escitalopram (10–20 mg/day) is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol. 2003;18(4):211–217. doi:10.1097/01.yic.0000074988.54339.7c
10. Wade A, Despiegel N, HeldboReines E. Escitalopram in the long-term treatment of major depressive disorder. Ann Clin Psychiatry. 2006;18(2):83–89. doi:10.1080/10401230600614447
11. Stahl SM, Gergel I, Li D. Escitalopram in the treatment of panic disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2003;64(11):1322–1327. doi:10.4088/JCP.v64n1107
12. Boulenger JP, Huusom AK, Florea I, Baekdal T, Sarchiapone M. A comparative study of the efficacy of long-term treatment with escitalopram and paroxetine in severely depressed patients. Curr Med Res Opin. 2006;22(7):1331–1341. doi:10.1185/030079906X115513
13. Ng QX, Yong CSK, Loke W, Yeo WS, Soh AYS. Escitalopram-induced liver injury: a case report and review of literature. World J Hepatol. 2019;11(10):719–724. doi:10.4254/wjh.v11.i10.719
14. Stickley A, Leinsalu M, DeVylder JE, Inoue Y, Koyanagi A. Sleep problems and depression among 237 023 community-dwelling adults in 46 low- and middle-income countries. Sci Rep. 2019;9(1):12011. doi:10.1038/s41598-019-48334-7
15. Jukić MM, Haslemo T, Molden E, Ingelman-Sundberg M. Impact of CYP2C19 genotype on escitalopram exposure and therapeutic failure: a retrospective study based on 2087 patients. Am J Psychiatry. 2018;175(5):463–470. doi:10.1176/appi.ajp.2017.17050550
16. Zhou L, Sharma P, Yeo KR, et al. Assessing pharmacokinetic differences in Caucasian and East Asian (Japanese, Chinese and Korean) populations driven by CYP2C19 polymorphism using physiologically-based pharmacokinetic modelling. Eur J Pharm Sci. 2019;139:105061. doi:10.1016/j.ejps.2019.105061
17. Chenu F, Batten LA, Zernig G, Ladstaetter E, Hébert C, Blier P. Comparison of pharmacokinetic profiles of brand-name and generic formulations of citalopram and venlafaxine: a crossover study. J Clin Psychiatry. 2009;70(7):958–966. doi:10.4088/JCP.09m05315
18. Bareggi SR, Mundo E, Dell’Osso B, Altamura AC. The use of escitalopram beyond major depression: pharmacological aspects, efficacy and tolerability in anxiety disorders. Expert Opin Drug Metab Toxicol. 2007;3(5):741–753. doi:10.1517/17425255.3.5.741
19. Bishop JR, Najjar F, Rubin LH, et al. Escitalopram pharmacogenetics: CYP2C19 relationships with dosing and clinical outcomes in autism spectrum disorder. Pharmacogenet Genomics. 2015;25(11):548–554.
20. Jiang T, Rong Z, Xu Y, et al. Pharmacokinetics and bioavailability comparison of generic and branded citalopram 20 mg tablets: an open-label, randomized-sequence, two-period crossover study in healthy Chinese CYP2C19 extensive metabolizers. Clin Drug Investig. 2013;33(1):1–9. doi:10.1007/s40261-012-0010-8
21. de Leon J, Armstrong SC, Cozza KL. Clinical guidelines for psychiatrists for the use of pharmacogenetic testing for CYP450 2D6 and CYP450 2C19. Psychosomatics. 2006;47(1):75–85. doi:10.1176/appi.psy.47.1.75
22. Chen L, Qin S, Xie J, et al. Genetic polymorphism analysis of CYP2C19 in Chinese Han populations from different geographic areas of mainland China. Pharmacogenomics. 2008;9(6):691–702. doi:10.2217/14622416.9.6.691
23. World Medical Association General Assembly. World medical association declaration of Helsinki: ethical principles for medical research involving human subjects. J Int Bioethique. 2004;15(1):124–129.
24. Horai Y, Nakano M, Ishizaki T, et al. Metoprolol and mephenytoin oxidation polymorphisms in far eastern oriental subjects: Japanese versus mainland Chinese. Clin Pharmacol Ther. 1989;46(2):198–207. doi:10.1038/clpt.1989.126
25. Nilausen DØ, Zuiker RG, van Gerven J. The perception and pharmacokinetics of a 20-mg dose of escitalopram orodispersible tablets in a relative bioavailability study in healthy men. Clin Ther. 2011;33(10):1492–1502.
26. Areberg J, Christophersen JS, Poulsen MN, Larsen F, Molz KH. The pharmacokinetics of escitalopram in patients with hepatic impairment. AAPS J. 2006;8(1):E14–E19. doi:10.1208/aapsj080102
27. Agarwal S, Gowda KV, Selvan PS, Chattaraj TK, Pal TK. Bioequivalence of two commercial preparations of escitalopram oxalate/clonazepam using a liquid chromatography-electrospray mass spectrometry method. Arzneimittelforschung. 2008;58(11):551–556.
28. Li J, Tian Y, Zhang ZJ, Wang N, Ren X, Chen Y. Pharmacokinetics and bioequivalence study of escitalopram oxalate formulations after single-dose administration in healthy Chinese male volunteers. Arzneimittelforschung. 2009;59(5):228–232.
29. Singh SS, Shah H, Gupta S, et al. Liquid chromatography–electrospray ionisation mass spectrometry method for the determination of escitalopram in human plasma and its application in bioequivalence study. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;811(2):209–215. doi:10.1016/S1570-0232(04)00703-2
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.