")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Comparison of beta-endorphin and CGRP levels before and after treatment for severe schizophrenia
Authors Urban-Kowalczyk M , Smigielski J, Strzelecki D
Received 1 December 2015
Accepted for publication 22 January 2016
Published 15 April 2016 Volume 2016:12 Pages 863—868
DOI https://doi.org/10.2147/NDT.S101647
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Małgorzata Urban-Kowalczyk,1 Janusz Śmigielski,2 Dominik Strzelecki1
1Department of Affective and Psychotic Disorders, 2Department of Geriatrics, Healthy Aging Research Centre (HARC), Medical University of Lodz, Lodz, Poland
Objectives: Links between endorphins and dopaminergic transmission have not been fully explored in schizophrenia. Both endorphins excess and deficiency were postulated. CGRP is probably involved in dopaminergic transmission. The aim of this study was the evaluation of beta-endorphin (BE) and CGRP blood concentrations before and after treatment of severe schizophrenia.
Methods: Seventy patients treated with various antipsychotics, with severe symptoms of schizophrenia (51 with positive symptoms, 19 with negative symptoms), 15 first-degree relatives, and 44 healthy controls were included in the study. BE and CGRP blood concentrations were measured during patients severe schizophrenia and in their stable mental state after treatment. The results were compared with relatives and controls.
Results: BE and CGRP concentrations in patients with negative symptoms were higher than in relatives and in controls. BE levels in patients with positive symptoms were lower than in patients with negative symptoms (P<0.0000) and controls (P<0.0006). No significant changes in CGRP concentration were found in patient samples. CGRP levels in these samples were independent of treatment, but they were significantly higher than in relatives and controls. After the treatment, BE level decreased in patients with negative symptoms (P<0.0001) and increased in patients with positive symptoms (P<0.0000). No differences in BE concentration between patients in stable mental state, their relatives, and controls were found.
Conclusion: Effective antipsychotic treatment results in “normalization” of BE level. Specific changes in BE concentration could be involved in dopaminergic transmission and related to some symptoms of schizophrenia.
Keywords: schizophrenia, negative symptoms, neuroleptics, β-endorphin, CGRP
Introduction
Since the discovery of neuropeptides, several attempts have been made to identify the significance of these compounds for the etiology and symptomatology of schizophrenia. Nevertheless, the understanding of their roles remains incomplete, and much remains to be confirmed.1,2 Samples of peripheral blood, cerebrospinal fluid (CSF), and peripheral blood mononuclear cells, as well as postmortem brain samples, from schizophrenic patients have been exhaustively examined, the majority of tests being focused on opioids such as beta-endorphin (BE). BE is the best-known endogenous opioid peptide with strong analgesic properties, but it also has numerous biological functions.3 Neurons containing endorphins modulate the regulation of dopaminergic neuronal activity, suggesting that pathogenesis of schizophrenia may be associated with an imbalance in the modulatory effect of opioids on the dopaminergic system. Most of the scientific enthusiasm for studying the potential importance of opioid and opiate signaling in schizophrenia was evident in the late 1970s and 1980s. Opioid agonists effectively alter dopamine (DA) release, reuptake, and metabolism in the striatum and substantia nigra. Conversely, chronic neuroleptic treatment enhances the synthesis and release of pituitary BE and prolactin through DA receptor blockade. The mesolimbic–mesocortical DA reward–motivation circuitries in the major psychiatric disorders involve the endogenous opioids and opiates active at local μ-receptors that modulate the activity of the mesolimbic–mesocortical DA.4 As both the mesolimbic–mesocortical DA systems and the mesostriatal DA system play important roles in schizophrenia, μ-receptor functioning may also exert an impact. Morphine immunoreactivity has been found in the hippocampus, which is known to be neuronally disarrayed in schizophrenia cases.5 Endogenous opioids may be downstream products of the dopaminergic system and neuromodulators of reward and motivation circuitries. They may be involved in the pathophysiology of schizophrenia. In this case, the regulation of opioid synthesis by DA, which can be measured, is highly significant.6,7
The hypotheses concern three main mechanisms: endorphin deficiency, the presence of abnormal endorphins, and the excess of endorphins in schizophrenia.8 Some authors postulate that BE may have therapeutic effects in patients with schizophrenia,9 but their findings are contradictory. Gerner et al10 observed no significant improvement after BE administration. Moreover, six of eight patients worsened after treatment. One uncontrolled trial suggested that hemodialysis might decrease symptoms in patients with schizophrenia.11 It was found that the leucine form of BE decreased in dialysates over time as patients improved, indicating that leu-endorphin caused psychotic symptoms.12 However, Emrich et al failed to confirm the effectiveness of dialysis in schizophrenia.13
Nevertheless, most studies suggest the presence of a possible excess of opioid in schizophrenia. Domschke et al14 observed increased CSF levels of BE in patients with acute psychosis and decreased levels in those with chronic symptoms compared to controls. A series of reports suggest that the opioid antagonist naloxone can induce improvement in patients with schizophrenia. While some studies have found that low doses of naloxone reduced auditory hallucinations in some individuals,15,16 others have failed to replicate those findings.17 Moreover, intracerebral administration of BE was seen to produce rigid immobility in rats,18 which is regarded as an analog of human catatonia. These previous studies had some limitations, such as small sample sizes and heterogenous groups of patients, and they used different types of biological materials, including CSF, plasma, cells, and tissue, to identify endogenous opioids. In addition, some studies failed to perform sufficient evaluations of mental state.
CGRP is a 37-amino-acid peptide widely distributed throughout the brain, which has been associated with dopaminergic neurotransmission. It is present in the frontal cortex, occipital cortex, striatum, and hippocampus where it plays a role in several central activities, including olfaction, audition, learning, and feeding.19 CGRP acts on neurons in the amygdala, central tegmental area, hippocampus, and hypothalamus.20 Given intracerebroventricularly, CGRP affects behaviors related to DA: it induces catalepsy and decreases apomorphine-induced hypermotility.21 The involvement of CGRP in psychiatric disorders is also suggested by altered CGRP levels in the CSF of patients with dementia and depression.22,23 We hypothesize that BE and CGRP are etiologically related to symptomatology of schizophrenia and their concentrations can change during effective treatment.
Methods
Participants
Seventy inpatients who met the International Classification of Diseases, Revision 10 (ICD-10) criteria for schizophrenia with exacerbated symptoms were included in the study. They were distinguished into two subgroups: 51 individuals with predominant positive symptoms (aged 38.78±11.83 years, median =38.00 years; 31 females and 20 males) and 19 with predominant negative symptoms (aged 39.83±10.88 years, median =36.00 years; 13 females and six males). Patients were classified into negative and positive psychopathological subtypes using the Positive and Negative Syndrome Scale (PANSS) composite index.24 The composite index is obtained by subtracting the PANSS negative symptoms subscale score from the positive symptoms subscale score. The classification into the psychopathological subtypes was done using the original PANSS validation data. According to these, the 25th and 75th percentile limits (score of −8 and +3) were used as cutoff levels for defining, respectively, the positive and negative subgroups (Table 1).
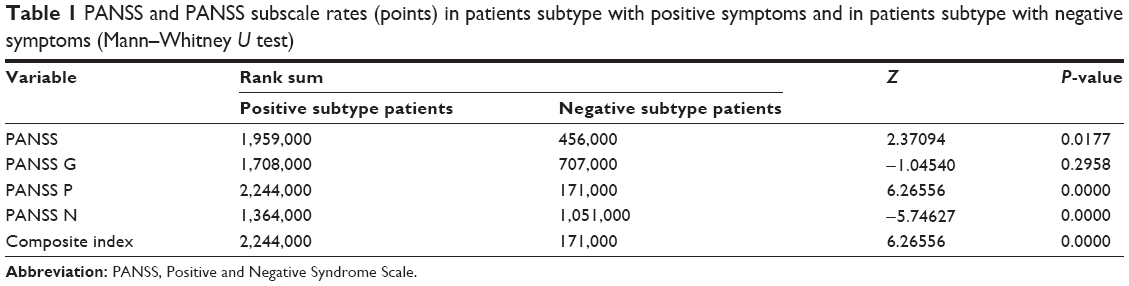 | Table 1 PANSS and PANSS subscale rates (points) in patients subtype with positive symptoms and in patients subtype with negative symptoms (Mann–Whitney U test) |
All patients were taking first-generation antipsychotic (haloperidol, perazine) or second-generation antipsychotic (SGA) (risperidone, olanzapine, quetiapine, amisulpride, aripiprazole), or were undergoing combined treatment, in therapeutic doses. The exclusion criteria for all groups were: 1) use of a psychoactive substance currently or in the past and 2) mental disorders (other than schizophrenia for patients sample). Additionally, a sample from first-degree relatives of 15 patients was evaluated (aged 36.00±13.99 years, median =31.00 years; eleven females and four males). The patients were clinically evaluated by a senior psychiatrist, and the PANSS was administered. Controls were 44 volunteers (aged 38.59±7.37 years, median =36.00 years; 26 females and 18 males) free of any drug treatment, with no record of mental disorder. The Kruskal–Wallis test showed no statistically significant age difference between study groups (H=1.27, P=0.7362). There were also no sex differences among study groups (χ2 =2.42, P=0.4894). Mean hospitalization period was 11.94 weeks for patients with positive symptoms subtype and 12.72 weeks for patients with negative subtype. All subjects were in good physical health. Two assessments were performed of BE and CGRP concentration in peripheral blood samples of patients and one in the samples of controls. Individuals with severe schizophrenia were assessed during the first 5 days of hospitalization and during a stable mental state after treatment on the day of discharge. All participants gave their written informed consent prior to their inclusion in the study. This research was approved by the Ethics Committee of the Medical University of Lodz (Nr RNN/91/12/KE).
Biochemical assay
Plasma CGRP concentration was measured by radioimmunoassay. Two 4 mL peripheral blood samples were collected from all participants and placed in two clot tubes. The samples were then centrifuged. The plasma for the CGRP assay was stored at −70°C until needed. The CGRP assay itself was performed using Human CGRP Kit (EK-015-02; Bachem, Bubendorf, Switzerland). To measure BE plasma concentration, 3 mL peripheral blood samples were collected from all participants. All blood samples were drawn directly into ethylenediaminetetraacetic acid. After centrifugation, the plasma samples were stored at −20°C until assay. The BE assay was performed using a DRG Diagnostics β-Endorphin Kit (RIA 3023). All blood samples from patients and controls were withdrawn between 7 and 8 am.
Statistical analysis
In order to describe the structure of the study group, indexes were calculated for qualitative characteristics, whereas arithmetic mean ( ) and median were calculated for quantitative traits. Standard deviation was used to measure dispersion. Moreover, the range of the tested variables was specified, that is, minimum and maximum values and asymmetry coefficient.
) and median were calculated for quantitative traits. Standard deviation was used to measure dispersion. Moreover, the range of the tested variables was specified, that is, minimum and maximum values and asymmetry coefficient.
Statistical analysis was based on nonparametric tests. The Shapiro–Wilk test was used to analyze the empirical distributions of the studied parameters. The Mann–Whitney U test was applied for measurable characteristics (for unpaired samples) as their distribution was non-normal. The Wilcoxon test was performed for paired samples.
The differences between more than two groups were tested by the Kruskal–Wallis test. P-values <0.05 were regarded as statistically significant. Statistical and graphical analysis was performed using Statistica (StatSoft, Tulsa, OK, USA) and MS Office 2007 (Microsoft Corporation, Redmond, WA, USA).
Results
BE assay
The concentration of BE at the beginning of hospitalization was significantly higher in patients with negative symptoms than in those with positive symptoms (mean 30.71 vs 15.34 pmol/L, P=0.0000). It was also higher than in the sample of relatives and controls (mean 19.59 and 18.82 pmol/L, P<0.001), but the concentration among patients with positive symptoms was lower than in controls (P=0.0006). After effective treatment, BE levels significantly decreased in the group with negative symptoms (mean 19.78 pmol/L, P=0.0001) and increased in patients with predominantly positive symptoms (mean 19.73 pmol/L, P=0.0000). The final BE concentrations in both patient groups did not differ from those in first-degree relatives and controls. No difference in BE concentrations was observed between first-degree relatives and controls (Figure 1).
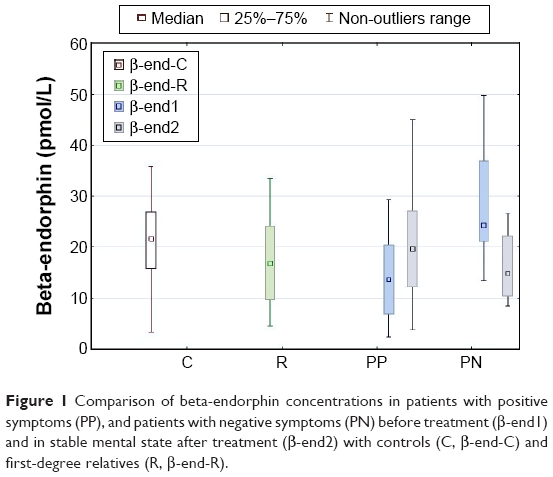 | Figure 1 Comparison of beta-endorphin concentrations in patients with positive symptoms (PP), and patients with negative symptoms (PN) before treatment (β-end1) and in stable mental state after treatment (β-end2) with controls (C, β-end-C) and first-degree relatives (R, β-end-R). |
CGRP assay
No difference was observed in CGRP concentrations between patient groups at the beginning of hospitalization (5.85 ng/mL in patients with negative symptoms vs 6.46 ng/mL in those with positive symptoms, P=0.24). Both patient samples had higher levels of CGRP than relatives (mean 3.64 ng/mL) and controls (mean 4.19 ng/mL) in the first assessment. CGRP concentration in patients with positive symptoms slightly increased after effective treatment (mean 6.72 ng/mL, P=0.043), but it did not change in patients with negative symptoms (P=0.54). The concentrations of CGRP remained higher in patient samples after treatment than in relatives and controls. No difference in CGRP concentrations was observed between first-degree relatives (mean 3.64 ng/mL) and controls (mean 4.19 ng/mL) (Figure 2).
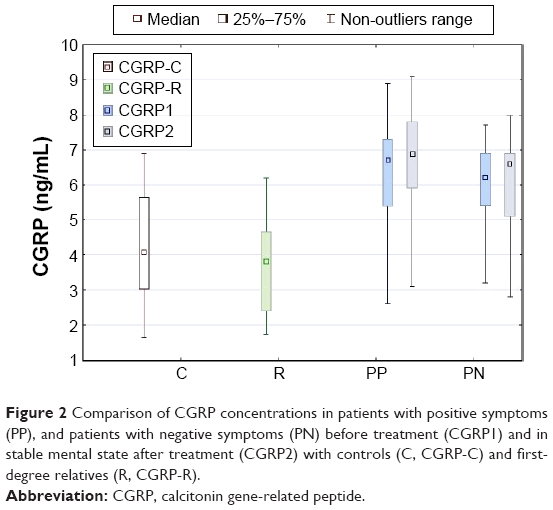 | Figure 2 Comparison of CGRP concentrations in patients with positive symptoms (PP), and patients with negative symptoms (PN) before treatment (CGRP1) and in stable mental state after treatment (CGRP2) with controls (C, CGRP-C) and first-degree relatives (R, CGRP-R). |
Discussion
DA agonists such as amphetamine and phencyclidine have been suggested to increase CGRP levels in rat limbic forebrain,25 and this effect could be prevented by pretreatment with D1 and D2 receptor antagonists.26,27 In the present study, CGRP concentrations were found to be significantly higher in all patients with schizophrenia than in a sample of relatives and a group of controls, and remained so even after effective treatment. Angelucci et al28 explored the possible effects of haloperidol and risperidone on CGRP in rats. They found that both antipsychotic agents increased CGRP-like immunoreactivity in the hippocampus. This effect is most likely secondary to changes in the DA system following D2 receptor blockade. A later study29 assessed the brain levels of CGRP-like immunoreactivity in rats treated with first-generation antipsychotic (haloperidol) and SGAs (risperidone and olanzapine). While haloperidol and higher risperidone doses decreased CGRP-like immunoreactivity in the striatum, higher olanzapine doses significantly increased the concentration of CGRP in the striatum and hypothalamus. Both lower and higher doses of olanzapine were found to increase the level of CGRP in the frontal cortex, which could be due to the different receptor profiles of olanzapine and the way it acts on multiple DA and serotonin receptors. As the patients in the present study were treated with various individual and combined antipsychotics, it was impossible to analyze the effect of a particular agent on changes in CGRP concentrations. Nevertheless, our findings confirm previous reports which suggest that one of the effects of antipsychotic drugs on DA could be mediated in part by reduced synthesis of CGRP, and that CGRP level is altered in schizophrenia. Further studies with specific antipsychotics, especially SGAs, might help to clarify this phenomenon.
The prevailing view in the literature is that an excess of endogenous opioids is present in schizophrenia. Although the nature of the “hypermorphinergic” pathology is unclear, it might be associated with the presence of some psychopathological symptoms.30 In older research, study groups tended to be small and nonhomogenous, and lacked any accurate assessment of psychopathology. In later reports, BE concentration was also investigated in drug-naïve patients and in those after pharmacological treatment. Mauri et al31 failed to find significant differences in the BE content of peripheral blood mononuclear cells between 30 drug-naïve schizophrenics and controls, although there was a trend toward higher values in the patients. Among a group of nine patients treated with haloperidol, BE tended to be lower in patients with percent amelioration >40% as measured by the Brief Psychiatric Rating Scale and Scale for the Assessment of Positive Symptoms, and in patients with percent amelioration >40% on the Scale for the Assessment of Negative Symptoms. Panza et al32 found no difference in BE concentration between 15 drug-naïve schizophrenics and controls. The level of BE increased after 2 weeks of haloperidol treatment.
Some reports suggest that opioid antagonists may bestow an antipsychotic effect on the subject. The majority of these studies used naloxone, a short-acting opioid antagonist which can reduce psychotic symptoms by a short-lasting blockade of the opioid receptors. Repeated naloxone infusions may not decrease positive psychotic symptoms to the same degree as a single dose.33 Rapaport et al34 describe decreases in Bunney–Hamburg psychosis ratings and the Brief Psychiatric Rating Scale thinking disturbance score after neuroleptic treatment augmentation using nalmefene. Eleven patients were treated with nalmefene for an average of ~37 days. This long-acting opioid antagonist has a half-life of ~12 hours and is 50 times more potent than naloxone which can lead to clinical improvement. Our findings indicate increased levels of BE in exacerbated patients with predominantly negative symptoms. In contrast, BE concentrations in patients with positive symptoms were lower than in controls. Finally, after drug treatment, BE levels decreased in those with negative symptoms and increased in those with positive symptoms. These changes occurred as a result of effective neuroleptic treatment despite the use of various individually tailored drugs. BE concentrations in patients at the end point of the study (clinical improvement, discharge from the hospital) did not differ from those observed in first-degree relatives and in healthy controls. These findings suggest that patients with schizophrenia display a different pattern of BE concentration which is dependent on the predominant psychopathology.
Effective antipsychotic treatment results in the “normalization” of BE level. Nevertheless, in our study, plasma BE was measured by a commercial enzyme-linked immunosorbent assay (ELISA) kit without pretreatment of the plasma samples as solid phase extraction which usually removes unspecific factors present in plasma. Moreover, peripheral tissues, for example, gastrointestinal tissues, contain BE that may be released to circulation, so the assessment result probably does not present direct BE levels.
Limitations
One of the limitations of this study is the non-homogeneity of the treatments in the patient samples. Therefore, we cannot exclude that the altered levels of BE and CGRP might have resulted from the specific psychopathology and applied pharmacotherapy. In addition, the patients achieved clinical improvement in different periods of time, and it is not known whether the neuropeptide concentrations remained stable after improvements in mental state. Furthermore, the group of patients with predominant negative symptoms was significantly fewer than those with positive symptoms. Finally, the study does not address patients with first-episode psychosis which might have been particularly interesting in the context of the increased and almost stable CGRP level. Regarding BE, the increased level seems to be a state rather than a trait marker.
Conclusion
In general, we did not observe increased BE in the first-degree relatives of the patients. Healthy subjects reveal diurnal rhythm of BE release with the highest plasma concentration early in the morning.35 Our study was based only on single daily blood samples withdrawn between 7 and 8 am, but diurnal BE secretion pattern in subjects with schizophrenia may differ from controls.
Further studies should directly investigate the impact of the levels of particular antipsychotics and take into account the clinical symptomatology, severity of symptoms, and duration of illness.
Acknowledgment
This study was supported by grant from Medical University of Lodz (No 502-03/1-155-02/502-14-107).
Disclosure
The authors report no conflicts of interest in this work.
References
De Wied D, Sigling HO. Neuropeptides involved in the pathophysiology of schizophrenia and major depression. Neurotox Res. 2002;4:453–468. | ||
Kream RM, Stefano GB, Ptáček R. Psychiatric implications of endogenous morphine: up-to-date review. Folia Biol. 2010;56(6):231–241. | ||
Stefano GB, Goumon Y, Casares F, et al. Endogenous morphine. Trends Neurosci. 2000;23:436–442. | ||
Fricchione GL, Mendoza A, Stefano GB. Morphine and its psychiatric implications. Adv Neuroimmunol. 1994;4(2):117–131. | ||
Lipska BK, Weinberger DR. Delayed effects of neonatal hippocampal damage on haloperidol-induced catalepsy and apomorphine-induced stereotypic behaviors in the rat. Brain Res Dev Brain Res. 1993;15(75):213–222. | ||
Neri C, Ghelardini C, Sotak B, et al. Dopamine is necessary to endogenous morphine formation in mammalian brain in vivo. J Neurochem. 2008;106:2337–2344. | ||
Stefano GB, Kream R. Endogenous opiates, opioids, and immune function: evolutionary brokerage of defensive behaviors. Semin Cancer Biol. 2008;18:190–198. | ||
Volavka J, Davis LG, Ehrlich YH. Endorphins, dopamine, and schizophrenia. Schizophr Bull. 1979;5(2):227–239. | ||
Kline NS, Li CH, Lehmann HE, Lajtha A, Laski E, Cooper T. Beta-endorphin – induced changes in schizophrenic and depressed patients. Arch Gen Psychiatry. 1977;34:1111–1113. | ||
Gerner RH, Catlin DH, Gorelick DA, Hui KK, Li CH. beta-Endorphin. Intravenous infusion causes behavioral change in psychiatric inpatients. Arch Gen Psychiatry. 1980;37(6):642–647. | ||
Wagemaker H, Cade R. The use of hemodialysis in chronic schizophrenia. Am J Psychiatry. 1977;134:684–685. | ||
Palmour R, Ervin F, Wagemaker H, Cade R. Characterization of a peptide derived from the serum of psychiatric patients. In: Usdin E, Bunney WE Jr, Kline NS, editors. Endorphins in Mental Health Research. New York: Macmillan; 1979:581–593. | ||
Emrich HM, Kissling W, Fischler M, Zerssen DF, Riedhammer H, Edel HH. Hemodialysis in schizophrenia: three failures with chronic patients. Am J Psychiatry. 1979;136:1095. | ||
Domschke W, Dickschas A, Mitznegg P. C.S.F. beta-endorphin in schizophrenia. Lancet. 1979;12:1024. | ||
Gunne LM, Lindstrom L, Terenius L. Naloxone-induced reversal of schizophrenic hallucinations. J Neural Transm. 1977;40:13–19. | ||
Pickar D, Vartanian F, Bunney WE, et al. Short-term naloxone administration in schizophrenic and manic patients: a World Health Organization Collaborative Study. Arch Gen Psychiatry. 1982;39(3):313. | ||
Janowsky DS, Segal DS, Bloom F, Abrams A, Guillemin R. Lack of effect of naloxone on schizophrenic symptoms. Am J Psychiatry. 1977;134:926–927. | ||
Bloom F, Segal D, Ling N, Guillemin R. Profound behavioral effects in rats suggest new etiological factors in mental illness. Science. 1976;194:630–632. | ||
Van Rossum D, Hanisch UK, Quirion R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci Biobehav Rev. 1997;21:649–678. | ||
Drumheller A, Menard D, Fournier A, Jolicoeur FB. Neurochemical effects of CGRP. Ann NY Acad Sci. 1992;30(657):546–548. | ||
Clementi G, Grassi M, Valerio C, Prato A, Fiore CE, Drago F. Effects of calcitonin gene-related peptide on extrapyramidal motor system. Pharmacol Biochem Behav. 1992;42:545–548. | ||
Mathé AA, Agren H, Lindström L, Theodorsson E. Increased concentration of calcitonin gene-related peptide in cerebrospinal fluid of depressed patients. A possible trait marker of major depressive disorder. Neurosci Lett. 1994;5(182):138–142. | ||
Mathé AA, Agren H, Wallin A, Blennow K. Calcitonin gene-related peptide and calcitonin in the CSF of patients with dementia and depression: possible disease markers. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:41–48. | ||
Kay SR. Positive and negative syndromes in schizophrenia. In: Van Praag H, editor. Clinical and Experimental Psychiatric. Monograph No 5. New York: Brunnel/Mazer; 1991. | ||
Mathé AA, Hertel SP, Nomikos GG, Gruber PAS, Mathé EJM. The psychotomimetic drugs D-amphetamine and phencyclidine release calcitonin gene related peptide in the limbic forebrain of the rat. J Neurosci Res. 1996;46:316–323. | ||
Gruber SHM, Mathé AA. Effects of amphetamine and dopamine receptor antagonists on calcitonin gene-related peptide, neuropeptide Y and neurotensin in rat brain. Neuropsychopharmacology. 2000;23:2–118. | ||
Gruber SH, Nomikos GG, Mathé AA. Dopamine receptor antagonists prevent the d-amphetamine-induced increase in calcitonin gene-related peptide levels in ventral striatum. J Neurosci Res. 2001;15(64):606–611. | ||
Angelucci F, Gruber SH, Mathé AA. A pilot study of rat brain regional distribution of calcitonin, katacalcin and calcitonin gene-related peptide before and after antipsychotic treatment. Neuropeptides. 2001;35:285–291. | ||
Angelucci F, Gruber SH, Caltagirone C, Mathé AA. Differential effects of olanzapine, haloperidol and risperidone on calcitonin gene-related peptide in the rat brain. Neuropeptides. 2008;42:535–541. | ||
Laux-Biehlmann A, Mouheiche J, Vérièpe J, Goumon Y. Endogenous morphine and its metabolites in mammals: history, synthesis, localization and perspectives. Neuroscience. 2013;13(233):95–117. | ||
Mauri MC, Rudelli R, Vanni S, et al. Cholecystokinin, beta-endorphin and vasoactive intestinal peptide in peripheral blood mononuclear cells of drug-naive schizophrenic patients treated with haloperidol compared to healthy controls. Psychiatry Res. 1998;20(1–2):45–50. | ||
Panza G, Monzani E, Sacerdote P, Penati G, Panerai AE. Beta-endorphin, vasoactive intestinal peptide and cholecystokinin in peripheral blood mononuclear cells from healthy subjects and from drug-free and haloperidol treated schizophrenic patients. Acta Psychiatr Scand. 1992;85:207–210. | ||
Pickar D, Bunney WE Jr, Douillet P, et al. Repeated naloxone administration in schizophrenia: a phase II World Health Organization Study. Biol Psychiatry. 1989;25:440–448. | ||
Rapaport MH, Wolkowitz O, Kelsoe JR, Pato C, Konicki PE, Pickar D. Beneficial effects of nalmefene augmentation in neuroleptic-stabilized schizophrenic patients. Neuropsychopharmacology. 1993;9(2):111–115. | ||
Gil-Ad I, Dickerman Z, Amdursky S, Laron Z. Diurnal rhythm of plasma beta endorphin, cortisol and growth hormone in schizophrenics as compared to control subjects. Psychopharmacology. 1986;88:496–499. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.