")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 15
Comparative Structural Analysis of Human ACE2 Receptor with Spike Protein of SARS-CoV-2 Variants: Implications to Understand Infectivity of the Virus
Authors Koley T, Goswami A, Kumar M, Upadhyay N , Hariprasad G
Received 2 February 2022
Accepted for publication 1 June 2022
Published 16 June 2022 Volume 2022:15 Pages 21—27
DOI https://doi.org/10.2147/AABC.S360787
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr James Briggs
Tirthankar Koley, Arunima Goswami, Manoj Kumar, Neelam Upadhyay, Gururao Hariprasad
Department of Biophysics, All India Institute of Medical Sciences, New Delhi, 110029, India
Correspondence: Gururao Hariprasad, Tel +91-11-26594240, Fax +91-11-26588663, Email [email protected]
Purpose: Spike protein on SARS-CoV-2 virus plays an integral part during infection as cell entry depends on binding of this protein to human ACE2 receptor. Understanding of infectivity by these variants necessitates a comparative structural analysis of complexes of spike protein-receptor binding domain (RBD) of these variants to receptor.
Methodology: Wild type SARS-CoV-2 spike protein sequence was retrieved from the UniProt database, and mutations of five variants at receptor binding domain were manually incorporated and aligned using Clustal Omega. Crystal structure complexes of human ACE2 receptor with spike protein RBD domain of SARS-CoV-2 variants of wild type, α, β, and δ were extracted from the RCSB database. Wild type SARS-CoV-2 complex with receptor was used as template to generate model complexes of receptor with spike protein RBD of γ and omicron variants through WinCoot program. These were energy minimized and validated and molecular dynamic simulation was performed using Desmond simulation program.
Results: Mutations are distributed across the entire length of RBD, but the maximum number of mutations are seen at 11 positions within binding interface motifs of six variant sequences. Interface of spike protein RBDs with human ACE2-receptor shows different mix of hydrogen bonded and ionic interactions. Alpha and β variants have few interactions, while γ and δ variants have higher number of interactions compared to wild type variant. Omicron variant, with 10 polar interactions including two ionic bonds, has the highest binding energy.
Conclusion: Different mutations on RBD of spike protein results in varying quantity and quality of interactions, thereby affecting potency of each variant. Variations in binding are due to interactions of mutant residues and induced conformational changes on loops of RBDs. Variants α and β have a low potency, while, γ, δ, and omicron have a higher potency. These results correlate with viral infectivity and place clinical observations in the right perspective.
Keywords: SARS-CoV-2, variants, omicron, mutations, spike protein, human ACE2 receptor, interactions, binding, infectivity
Introduction
COVID-19 is a global pandemic that started in the early 2020, is caused by the SARS-CoV-2 virus that has so far affected more than 500 million people and caused more than six million deaths.1 Mutations of wild type of SARS-CoV-2 genome has led to emergence of new variants over the last two years, and based upon the risk to public health, it has prompted the characterization of variants such as α (B.1.1.7), β (B.1.351) δ (P.1), as “variants being monitored” (VBM), and, variants such as γ (B.1.617.2) and omicron (B.1.1.529) as “variants of concern” (VOC).2
COVID-19 was first reported in Wuhan in China, before being declared a global pandemic by WHO in March, 2020. This infection by the wild type variant had infected close to 44.8 million people and resulted in 1.1 million deaths worldwide.3 Alpha-variant, with seven spike protein mutations was first reported in September 2020 in the UK, and was two times more transmissible than the wild type.4–6 Beta-variant, with nine spike protein mutations, was first reported in May 2020 in Republic of South Africa, and is 1.7 times more transmissible than the wild type.4–6 Delta-variant with 17 spike protein mutations was first reported in November 2020 in Brazil, and is 2.4 times more transmissible than the wild type.4,5,7 Gamma-variant that has 12 spike protein mutations was first reported in October 2020 in India, and is four times more transmissible than the wild type.4,5,8 The latest variant of SARS-CoV-2, omicron, with 30 spike protein mutations, was first reported in November 2021 in multiple countries and is 16 times more transmissible than the wild type.9,10
Spike protein plays an integral part during the pathogenesis of SARS-CoV-2 as the cell entry of the virus depends on the binding of this protein to human ACE2 receptor.11 Structural stability of spike protein and its affinity to human ACE2 receptor determines the transmissibility of the SARS-CoV-2 variant.12–14 In this study, comparative structural analysis of the binding of spike protein RBD of all these six variants to human ACE2 receptor has been carried out.
Methodology
Sequence Alignment
Amino acid sequence of the wild type SARS-CoV-2 spike protein (P0DTC2) was retrieved from the UniProt database. Mutations of spike protein RBD of five SARS-CoV-2 variants of α (B.1.1.7), β (B.1.351), γ (P.1), δ (B.1.617.2), and omicron (B.1.1.529) were manually incorporated in the retrieved wild type spike protein RBD sequence. Multiple sequence alignment of these six sequences was performed using the online sequence alignment tool Clustal Omega.15 Based on the interaction analysis of wild type RBD with human ACE2 receptor (PDB Id 6M0J), binding sequence motifs on the spike protein RBD sequences of these six variants were identified.
Molecular Modeling of Spike Protein and its Complex with Human ACE2 Receptor
Crystal structure complexes of human ACE2 receptor with spike protein RBD domain of SARS-CoV-2 variants of wild type (PDB Id 6M0J), α (PDB Id: 7FEM), β (PDB Id: 7VXD), and δ (PDB Id: 7V8A), were extracted from the Research Collaboratory Structural Bioinformatics database. Crystal structure complex of human ACE2 receptor with spike protein RBD of wild type SARS-CoV-2 was used as a template to generate model complexes of human ACE2 receptor with spike protein RBD of γ and omicron variants. Mutations K417T, E484K, N501Y, and mutations G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H were introduced manually on the template through WinCoot visualization program.16 This generated complexes of human ACE2 receptor with spike protein RBD of γ and omicron variants, respectively. Rotamer corrections of the introduced amino acids were performed, energy minimized and validated. These six complexes were taken for molecular dynamic studies and analysis.
Molecular Dynamic Simulation
Complexes were solvated using TIP3P water model in an orthorhombic box extending to 10 Å from protein atoms in each direction. They were then neutralized by 12 Na+ ions and energy minimized with convergence threshold of 1 kcal/mol/Å. MD simulation was performed using Desmond simulation program with the help of force field parameters of OPLS_2005.17–19 They were then equilibrated at 300 K and 1 atmospheric pressure for 200 ps under NTP condition with the help of Nosé–Hoover thermostat and Martyna–Tobias–Klein barostat under periodic boundary condition. It was followed by recording the production trajectory for 100 ns under similar NTP condition and other run parameters.20 Simulation was performed with step size of 2 fs in the presence of LINCS harmonic constraints and motion was integrated by RESPA dynamics integrator.17 Long range electrostatic interactions were calculated by particle mesh Ewald (PME) algorithm. This protocol was used for all the complexes for the sake of comparison.
Results and Discussion
Sequence Analysis
Sequence alignment of spike protein-receptor binding domain (RBD) of SARS-CoV-2 variants show α variant to be having maximum homology with wild type sequence, while omicron variant which has 15 mutations has least homology (Figure 1). Though mutations are distributed across entire length of RBD, maximum number of mutations are seen at 11 positions within receptor binding interface motifs of six sequences. Some of the notable mutations include: (1) Tyr501 in α, β, γ, and omicron; (2) Asn417 in β and omicron; (3) Thr417 in γ; (4) Lys484 in γ variant; (5) Arg452 in δ variant, and (6) Ser446, Asn477, Arg493, Ser496, Arg498, and His505 in omicron. It is therefore clear that with respect to interface binding sequence motifs, α is same as wild type variant. Beta and δ have two variations; γ has three variations, and omicron has 10 variations compared to wild type binding sequence motifs. These primary structure variations could have bearing on the fold, binding, and potency of RBD to human ACE2 receptor.
 |
Figure 1 Sequence homology studies of SARS-CoV-2 variants. The sequences in order are wild type (green), alpha (blue), beta (pink), gamma (yellow), delta (brown), and omicron (cyan). Human ACE2 receptor binding motifs are shown underlined and in boxes. Mutations are shown in bold. The residue numbers and percentage homology to wild type are given at the end of the sequences. The symbol “*” indicates identical residues, “:” indicates conserved substitution and “.” indicates semi-conserved substitution. |
Overall Structural Analysis of Spike Protein RBD Complex with Human ACE2 Receptor
Structure of spike protein RBD comprises of five β-sheets at the core surrounded by five short helices, and loops connecting these secondary structural elements along with a β-wing at the center forms bulk of the binding interface in all six complexes. The human ACE2 receptor comprises 19 helices and single β-wing, of which two long helices and β-wing, forms interacting interface. Illustration of six superimposed complexes is provided in Figure 2. For purpose of description, interacting helices will henceforth be designated as IH1 and IH2, and β-strands will be designated as Iβ1 and Iβ2. IH1 and IH2 that lie adjacent to one another form a concave surface along with β-wing at one of its ends. Interacting surface of ACE2 receptor has aspartate, glutamate, histidine, lysine, tyrosine, and glutamine residues that provide ample opportunities for interactions with spike protein RBD of variants.21
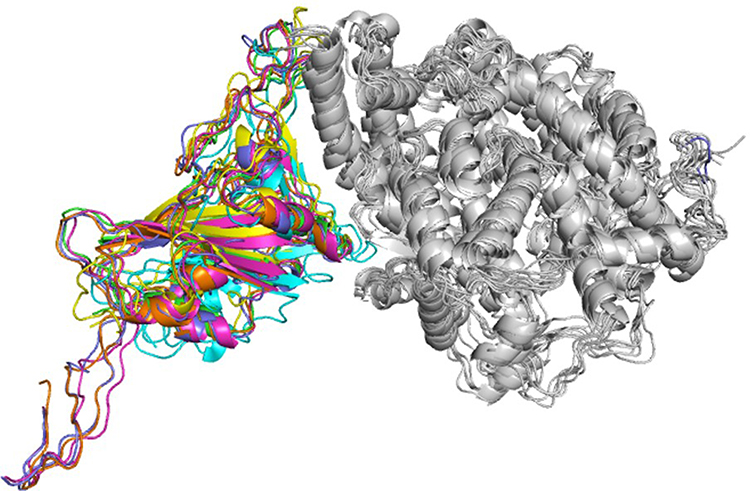 |
Figure 2 Ribbon diagram showing superimposed structure complexes of spike protein RBDs with human ACE2 receptor. Spike protein RBDs of variants wild type (green), alpha (blue), beta (pink), gamma (yellow), delta (brown), and omicron (cyan) are shown with human ACE2 receptor (white). |
Effect of Mutations on Binding of Spike Protein of SARS-CoV-2 Variants on Human ACE2 Receptor
Interface of spike protein RBDs of the six variants with human ACE2 receptor shows different mix of interactions primarily owing to the mutations and secondly, due to induced conformational changes. Details of participating residues and their array of interactions in the six complexes of human ACE2 receptor with spike protein RBD of wild type, α, β, γ, δ, and omicron is presented in Figure 3. Salient features of these interactions and effects of the mutations is described. (A) RBD of wild type with human ACE2 receptor: all the interacting residues from RBD interact with residues on IH1, except for Asn487 that makes a polar interaction with Tyr83, the first residue on IH2. The middle of interface is stabilized by three interactions of which one is an ionic interaction Nζ Lys417 … Oδ2 Asp30=2.9Å. There are an array of hydrogen bonded interactions from the loops and short helix of RBD with residues from lower part of IH1. In addition, glycine on a loop interacts with lysine on β-turn of receptor (O Gly496 … Nζ Lys353=3.0Å) (Figure 3A); (B) RBD of α with human ACE2 receptor: There are five hydrogen bonded interactions involving four non-mutated residues of the RBD (Figure 3B). The mutation Asn501Tyr does not result in any interaction, but a change in the loop conformation results in fewer interactions compared to the wild type; (C) RBD of β with human ACE2 receptor: There are five hydrogen bonds, two at either end of the interface and one in the middle (Figure 3C). Mutation Lys417Asn makes no interactions with the receptor. (D) RBD of γ with human ACE2 receptor: There are nine polar interactions across the length of interacting surfaces (Figure 3D). Of particular interest are ionic interactions Nζ Lys484 … Oε1 Glu75=2.8Å and Nζ Lys484 … Oε2 Glu75=3.3Å made by the mutation Glu484Lys at the upper end of the interface that exaggerates the proximity of the loop towards the interacting helices. As a reason, RMSD of main chain Cα of this complex is 2.1Å as compared with wild type; (E) RBD of δ with human ACE2 receptor: There are 10 hydrogen bonded interactions at the interface (Figure 3E). Mutations Leu452Arg on Iβ1 of the β-wing, and Thr478Lys on the loop have their respective side chains oriented away from the interface, and thereby are not a part of interactions with receptor; (F) RBD of omicron with human ACE2 receptor: There are 10 polar interactions between RBD and receptor of which two are ionic interactions from the mutation Gln493Arg (NH2 Arg493 … OE1 Glu35 and NH2 Arg493 … Oδ1 Asp38) (Figure 3F). Two other mutations Gly496Ser and Asn501Tyr are involved in hydrogen bonded interactions (Oγ 496Ser … Nζ Lys353; OH 501Tyr … Nζ Lys353). Gln498 that makes a hydrogen bond with Gln42 in the wild type, is mutated to Arg498 that influences a conformational change in loop, wherein Thr500 makes polar interactions with Arg357 and Asp355. This results in an exaggerated proximity of the loop to the receptor as compared to the other five variants. As a reason, RMSD of main chain Cα of this complex is 3Å as compared with wild type. In addition, mutation Tyr505His makes hydrophobic interactions with Lys353 (Cβ Hi505 … C Lys353=4Å; Cε Hi505 … Cα Lys353=4Å, Cε Hi505 … Cγ Lys353=3.7Å, and Cδ Hi505 … Cα Lys353=4Å). These interactions account for high potency of omicron RBD to the receptor, which is validated by the high binding energy, and increased RMSD in comparison with wild type counterpart.12
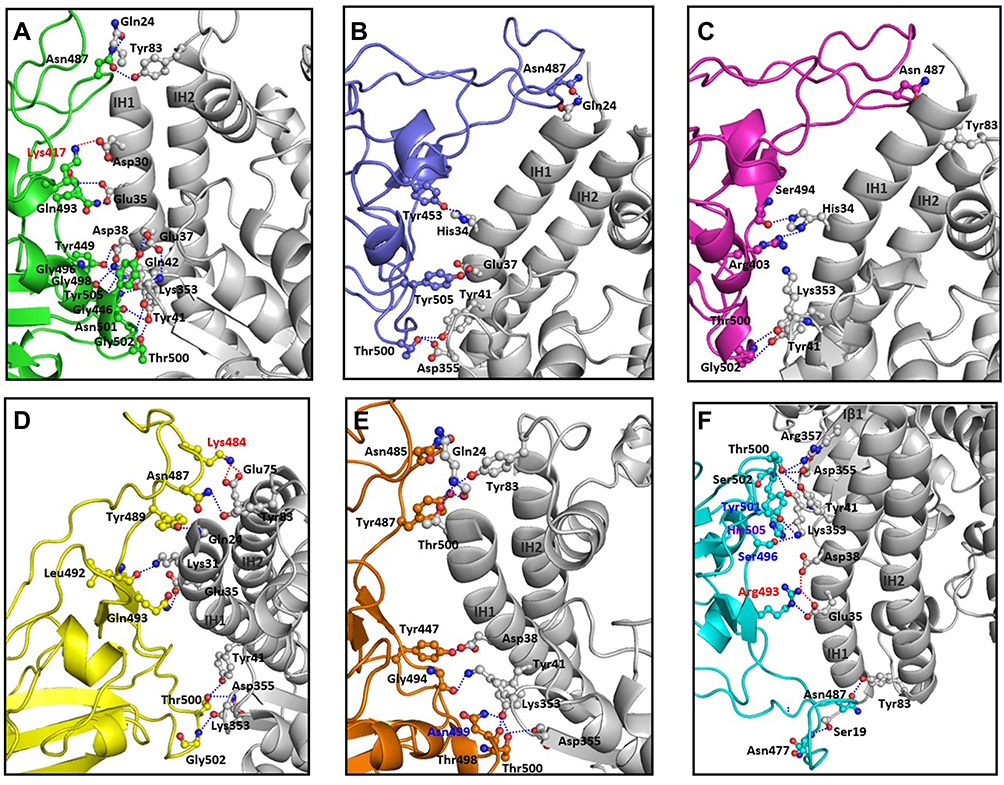 |
Figure 3 Interacting interface between RBD domains of spike protein from SARS-CoV-2 variants with human ACE2 receptor. Details of the interactions between residues of spike protein RBDs from (A) wild type (green), (B) alpha (blue), (C) beta (pink), (D) gamma (yellow), (E) delta (brown), and (F) omicron (cyan) with human ACE2 receptor (white) are shown. Red dotted lines indicate ionic interactions, blue dotted lines indicate hydrogen bonded interactions and purple dotted lines indicate hydrophobic interactions. Mutated residues on the spike protein participating in these interactions are labeled in red, blue, and purple, respectively. |
Potency of Spike Protein RBD Binding with Human ACE2 Receptor and Virus Infectivity
Interaction of RBD with the human ACE2 receptor is very integral step in infection and transmissibility of COVID-19 in the community.12 Variations in quantity and quality of mutations, and nature of their interactions as detailed in the above discussion therefore dictates the potencies that are reflected by binding energies (Table 1). In accordance, α and β variants with few interactions have a low infectivity, while γ and δ variants, with higher number of interactions, have infectivity more than twice the infectivity of wild type variant. The current variant omicron, with 10 polar interactions including two ionic bonds has highest potency, which is validated by high binding energy, and increased RMSD in comparison wild type counterpart. This explains the extremely high rate of infectivity of the omicron variant.
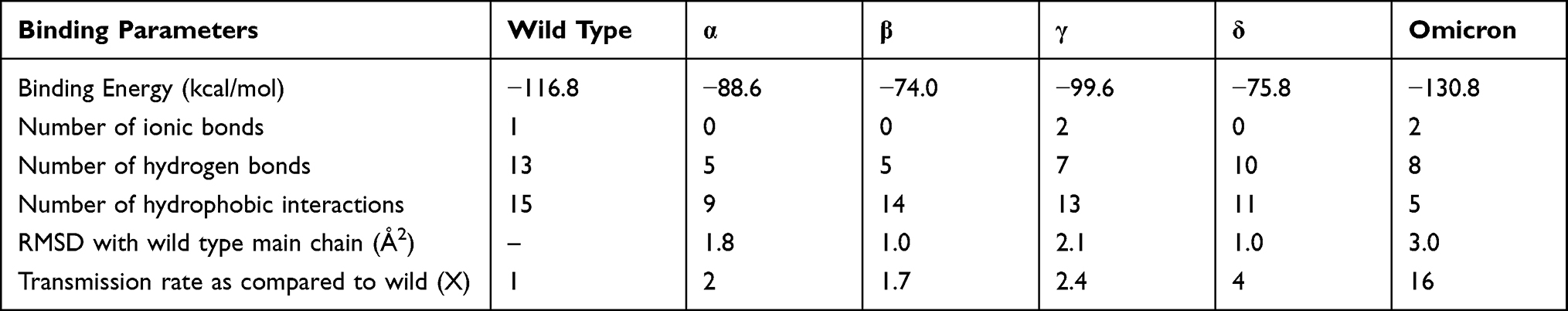 |
Table 1 Comparative Analysis of Biophysical Parameters Governing the Binding of Spike Protein RBD to Human ACE2 Receptor and Transmission Rates |
Interestingly, neutralizing antibodies have been known to target specific sites SARS-CoV-2, especially the residues on interfacial surface of spike protein RBD.22–24 Structural modeling done in this study is an ideal platform to understand antibody binding and vaccine efficacy. There have been few other recent studies such as ours that have explained the binding interactions of RBD of spike protein to ACE2 receptor.25,26 In comparison to these, our study has the following advantages: (1) binding studies of RBD from α, β, γ, δ and the most recent omicron variants to human ACE2 receptor have been delineated; (2) binding parameters have been correlated to the infectivity of each of the five strains; (3) intermolecular interactions between RBD and ACE2 receptor have been highlighted in detail, and (4) holistic approach of understanding the effect of mutations at level of sequence, secondary structural motifs and three-dimensional fold has been adopted.
Conclusion
Comparative structural analysis of the interactions between the wild type and mutated proteins from five variants with the human ACE2 receptor reveals the following features: (1) Different mutations on RBD of spike protein results in varying quantity and quality of interactions, thereby affecting binding potency for each of the variants. (2) variations in binding are due to interactions of mutant residues and induced conformational changes on loops of the RBDs. (3) Ionic interactions play a major role in determining potency. (4) While the alpha and beta have few interactions, and low potency. (5) gamma, delta, and omicron have a higher number of interactions, and thereby a higher potency. (6) These results validate the clinical parameter of viral infectivity and places the observations in the right perspective.
Abbreviations
RBD, receptor binding domain; ACE2, angiotensin converting enzyme-2; RMSD, root mean square deviation.
Acknowledgments
The work was funded by All India Institute of Medical Sciences, New Delhi to Gururao Hariprasad (A-COVID-2).
Disclosure
The authors report no conflicts of interest in this work.
References
1. World Health Organisation. Available from: https://covid19.who.int/.
2. Parums DV. Revised World Health Organization (WHO) terminology for variants of concern and variants of interest of SARS-CoV-2. Int Med J Exp Clin Res. 2021;27:e933622–1.
3. World Health Organisation. Coronavirus disease (COVID-19) situation reports (who.int).
4. Mohammadi M, Shayestehpour M, Mirzaei H. The impact of spike mutated variants of SARS-CoV2 [Alpha, Beta, Gamma, Delta, and Lambda] on the efficacy of subunit recombinant vaccines. Braz J Infect Dis. 2021;25(4):101606. doi:10.1016/j.bjid.2021.101606
5. Lou F, Li M, Pang Z, et al. Understanding the secret of SARS-CoV-2 variants of concern/interest and immune escape. Front Immunol. 2021:4326. doi:10.3389/fimmu.2021.744242
6. Duong D. Alpha, Beta, Delta, Gamma: what’s important to know about SARS-CoV-2 variants of concern? Can Med Assoc J. 2021;193(27):E1059–E1060. doi:10.1503/cmaj.1095949
7. Faria NR, Mellan TA, Whittaker C, et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science. 2021;372(6544):815–821. doi:10.1126/science.abh2644
8. Katella K. Things to know about the delta variant. Yale Med. 2021;72(2):727. doi:10.51253/pafmj.v72i2.7088.
9. Callaway E Heavily mutated Omicron variant puts scientists on alert. Nature; 2021. Available from: https://www.nature.com/articles/d41586-021-03552-w.
10. Kollmeyer B. Japan study confirms omicron variant’s high transmissibility: report. International news. Japan study confirms omicron variant’s high transmissibility: report – MarketWatch.
11. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Nat Acad Sci. 2020;117(21):11727–11734. doi:10.1073/pnas.2003138117
12. Koley T, Kumar M, Goswami A, et al. Structural modeling of Omicron spike protein and its complex with human ACE-2 receptor: molecular basis for high transmissibility of the virus. Biochem Biophys Res Commun. 2022;592:51–53. doi:10.1016/j.bbrc.2021.12.082
13. Kumar V, Singh J, Hasnain SE, et al. Possible link between higher transmissibility of alpha, Kappa and delta variants of SARS-CoV-2 and increased structural stability of its spike protein and hACE2 affinity. Int J Mol Sci. 2021;22(17):9131. doi:10.3390/ijms22179131
14. Koley T, Madaan S, Chowdhury SR, et al. Structural analysis of COVID-19 spike protein in recognizing the ACE2 receptor of different mammalian species and its susceptibility to viral infection. 3 Biotech. 2021;11(2):1–6. doi:10.1007/s13205-020-02599-2
15. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7(1):539. doi:10.1038/msb.2011.75
16. Emsley P, Lohkamp B, Scott WG, et al. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66(4):486–501. doi:10.1107/S0907444910007493
17. Bowers KJ, Chow DE, Xu H, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters.
18. Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–11236. doi:10.1021/ja9621760
19. Banks JL, Beard HS, Cao Y, et al. Integrated modeling program, applied chemical theory (IMPACT). J Comput Chem. 2005;26(16):1752–1780. doi:10.1002/jcc.20292
20. Kumar M, Dahiya S, Sharma P, et al. Structure based in silico analysis of quinolone resistance in clinical isolates of Salmonella Typhi from India. PLoS One. 2015;10(5):e0126560. doi:10.1371/journal.pone.0126560
21. Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi:10.1126/science.abb2762
22. Starr TN, Czudnochowski N, Liu Z, et al. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature. 2021;597(7874):97–102. doi:10.1038/s41586-021-03807-6
23. Bertoglio F, Fühner V, Ruschig M, et al. A SARS-CoV-2 neutralizing antibody selected from COVID-19 patients binds to the ACE2-RBD interface and is tolerant to most known RBD mutations. Cell Rep. 2021;36(4):109433. doi:10.1016/j.celrep.2021.109433
24. Min L, Sun Q. Antibodies and vaccines target RBD of SARS-CoV-2. Front Mol Biosci. 2021;8:247. doi:10.3389/fmolb.2021.671633
25. Mandal N, Padhi AK, Rath SL. Molecular insights into the differential dynamics of SARS-CoV-2 variants of concern (VOCs). bioRxiv. 2021. doi:10.1101/2021.10.22.465272
26. Bhadane RN, Salo-Ahen OM. High-throughput molecular dynamics-based alchemical free energy calculations for predicting the binding free energy change associated with the common mutations in the spike receptor-binding domain of SARS-CoV-2. bioRxiv. 2022. doi:10.1101/2022.03.07.483402
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.