")
Back to Journals » Journal of Blood Medicine » Volume 8
Comparative safety of intravenous Ferumoxytol versus Ferric Carboxymaltose for the Treatment of Iron Deficiency Anemia: rationale and study design of a randomized double-blind study with a focus on acute hypersensitivity reactions
Authors Adkinson NF, Strauss WE, Bernard K, Kaper RF, Macdougall IC, Krop JS
Received 20 May 2017
Accepted for publication 9 August 2017
Published 26 September 2017 Volume 2017:8 Pages 155—163
DOI https://doi.org/10.2147/JBM.S142236
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
N Franklin Adkinson,1 William E Strauss,2 Kristine Bernard,2 Robert F Kaper,2 Iain C Macdougall,3 Julie S Krop2
1Department of Medicine, Division of Allergy and Clinical Immunology, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 2AMAG Pharmaceuticals, Inc., Waltham, MA, USA; 3Department of Renal Medicine, King’s College Hospital, London, UK
Background: Intravenous (IV) iron is often used to treat iron deficiency anemia in patients who are unable to tolerate or are inadequately managed with oral iron. However, IV iron treatment has been associated with acute hypersensitivity reactions. The comparative risk of adverse events (AEs) with IV iron preparations has been assessed by a few randomized controlled trials, which are most often limited by small patient numbers, which lack statistical power to identify differences in low-frequency AE such as hypersensitivity reactions.
Materials and methods: Ferumoxytol versus Ferric Carboxymaltose for the Treatment of Iron Deficiency Anemia (FIRM) is a randomized, double-blind, international, multicenter, Phase III study designed to compare the safety of ferumoxytol and ferric carboxymaltose (FCM). The study includes adults with hemoglobin <12.0 g/dL (females) or <14.0 g/dL (males), transferrin saturation ≤20% or ferritin ≤100 ng/mL within 60 days of dosing, and a history of unsatisfactory or nontolerated oral iron therapy or in whom oral iron therapy is inappropriate. Patients are randomized (1:1) to ferumoxytol 510 mg or FCM 750 mg, each given intravenously on days 1 and 8. Primary end points are the incidence of moderate-to-severe hypersensitivity reactions, including anaphylaxis, and moderate-to-severe hypotension. All potential hypersensitivity and hypotensive reactions will be adjudicated by a blinded, independent Clinical Events Committee. A secondary safety end point is the composite frequency of moderate-to-severe hypersensitivity reactions, including anaphylaxis, serious cardiovascular events, and death. Secondary efficacy end points include mean change in hemoglobin and mean change in hemoglobin per milligram of iron administered from baseline to week 5. Urinary excretion of phosphorus and the occurrence of hypophosphatemia after IV iron administration will be examined as well as the mechanisms of such hypophosphatemia in a substudy.
Conclusion: FIRM will provide data on the comparative safety of ferumoxytol and FCM, two IV iron preparations with similar dosing schedules, focusing on moderate-to-severe hypersensitivity reactions, including anaphylaxis, and moderate-to-severe hypotension. The study plans to enroll 2000 patients and is expected to complete in 2017.
Keywords: anaphylaxis, ferumoxytol, ferric carboxymaltose, hypersensitivity, hypotension, iron deficiency anemia
Introduction
Anemia is a common condition, with an estimated 1.6 billion individuals affected worldwide,1 of whom 500–800 million have the most common form, iron deficiency anemia (IDA).2 The burden of disease is higher in underdeveloped countries, but there are approximately 5 million people in the USA with IDA.2 IDA can result from insufficient iron absorption, inadequate dietary iron, blood loss, or an increased physiological requirement for iron (eg, during pregnancy).1 Although IDA frequently coincides with chronic kidney disease (CKD), it is associated with a number of other conditions, such as heavy menstruation, certain gastrointestinal diseases affecting iron absorption, and cancer.1 Therapy with oral iron is not always suitable for managing IDA because of gastrointestinal side effects, poor absorption, and often poor adherence to treatment.3
Intravenous (IV) iron therapy is commonly used for the treatment of IDA as an alternative to oral iron to restore iron stores, raise hemoglobin levels, and reduce erythropoiesis-stimulating agent doses in those receiving treatment.4 However, IV iron administration is associated with a number of safety concerns, including the potential for inducing iron overload, oxidative stress, an increased risk of infection, and hypersensitivity reactions.5 Of these safety issues associated with IV iron treatment, severe acute hypersensitivity reaction – or reactions resembling hypersensitivity reactions – is one that concerns clinicians the most.4 Symptoms such as dyspnea or respiratory compromise and hypotension can also raise suspicion for hypersensitivity reactions, but true anaphylaxis may not be present. For example, while hypotension can be commonly encountered with IV iron administration, profound hypotension as a manifestation of hypersensitivity reactions is uncommon and rarely the only sign of this severe type of reaction.7 Of recent attempts to define anaphylaxis, the consensus model published by Sampson et al7 has gained acceptance because of its specific criteria for a range of clinical settings.8,9 In addition, ancillary measurements of serum tryptase can be helpful in the diagnosis of severe hypersensitivity reactions, particularly if measured 1–2 hours after symptom onset and compared with an appropriate baseline.9,10
The risk of hypersensitivity reactions appears to be highest with iron dextrans (0.6%–0.7%), particularly high molecular weight iron dextran,4 which has been withdrawn from the market due to safety concerns. Hypersensitivity reactions to high molecular weight iron dextran are generally believed to be mediated by immunoglobulin E;6 however, nonimmune mechanisms may be involved in acute reactions with more recently developed IV iron preparations in which immunoglobulin E antibodies have not been found. Alternative mechanisms include the induction of oxidative stress by release of free reactive iron and what has more recently been called complement activation-related pseudoallergy (CARPA),4,6,11,12 and differences in the ability of IV iron preparations to induce CARPA may exist.11
While hypersensitivity reactions, iron overload, oxidative stress, and infections have generally received the greatest attention,5,13 hypophosphatemia is a more recently recognized adverse event (AE) following treatment with a number of IV iron preparations.14–18 The incidence of hypophosphatemia, as reported from several clinical trials of patients with IDA treated with ferric carboxymaltose (FCM), ranged from 41% to 70%.19–21 While often transient and without apparent clinical sequelae,19 symptoms of hypophosphatemia can include bone pain, confusion, and muscle weakness.14,16,18,22 A recent case report described a young woman who developed severe symptoms of fatigue and diffuse muscle pain and weakness associated with profound hypophosphatemia after receiving FCM 1000 mg.23
The mechanism of hypophosphatemia following administration of IV iron is not fully understood. However, recent data have shown that hypophosphatemia after IV iron infusion can be mediated by an increase in intact fibroblast growth factor 23 (FGF-23), which in turn reduces tubular reabsorption and leads to renal phosphate wasting.21,24,25 While the data implicating FGF-23, vitamin D, and bone metabolism in the development of hypophosphatemia following IV iron administration are of interest, a knowledge gap remains when encountering this potential AE in clinical practice.
There are limited data comparing rates of acute adverse reactions associated with various IV iron preparations. Prior direct comparative studies of IV iron preparations were generally small, open-label, or not statistically powered to detect small but meaningful differences in rare AEs between agents (Table 1).26–31 Conversely, a number of epidemiologic studies that retrospectively analyzed large claims databases have examined the relative risk of AEs with ferumoxytol versus other IV iron products (Table 1).32–34 For example, a retrospective cohort analysis of Medicare Part A and Part B claims for 2009–2012 found no significant difference in hypersensitivity symptoms (hazard ratio [HR], 1.04; 95% confidence interval [CI], 0.94–1.16) or hypotension (HR, 0.83; 95% CI, 0.52–1.34) between non-CKD ferumoxytol users and matched users of other iron compounds; results were similar for patients with nondialysis-dependent CKD.34 Data from an evaluation of the US Renal Data System found that patients receiving dialysis in clinics that switched from iron sucrose or ferric gluconate to ferumoxytol due to formulary reasons had similar incidences of major AEs as those patients receiving dialysis in clinics that continued to provide iron sucrose or ferric gluconate. Specifically, the risks were similar in the ferumoxytol group compared with the iron sucrose/ferric gluconate group for all-cause (HR, 0.95; 95% CI, 0.85–1.07), cardiovascular (HR, 0.99; 95% CI, 0.83–1.19), and infectious mortality (HR, 0.88; 95% CI, 0.61–1.25).32 A third study assessed the risk of anaphylaxis at first administration among patients treated with various IV iron products from the Medicare claims database; iron dextran had a risk of 85 per 100,000 persons, while ferric gluconate, ferumoxytol, and iron sucrose had risks of 47, 34, and 16 per 100,000 persons, respectively.33 Although such retrospective epidemiologic studies may provide more rigorous information than spontaneous reports of postmarketing surveillance data, they are often affected by various sources of confounding and bias, especially involving patient selection. Prospective investigations with substantial sample sizes are needed to precisely compare the incidence of important AEs among available IV iron products.
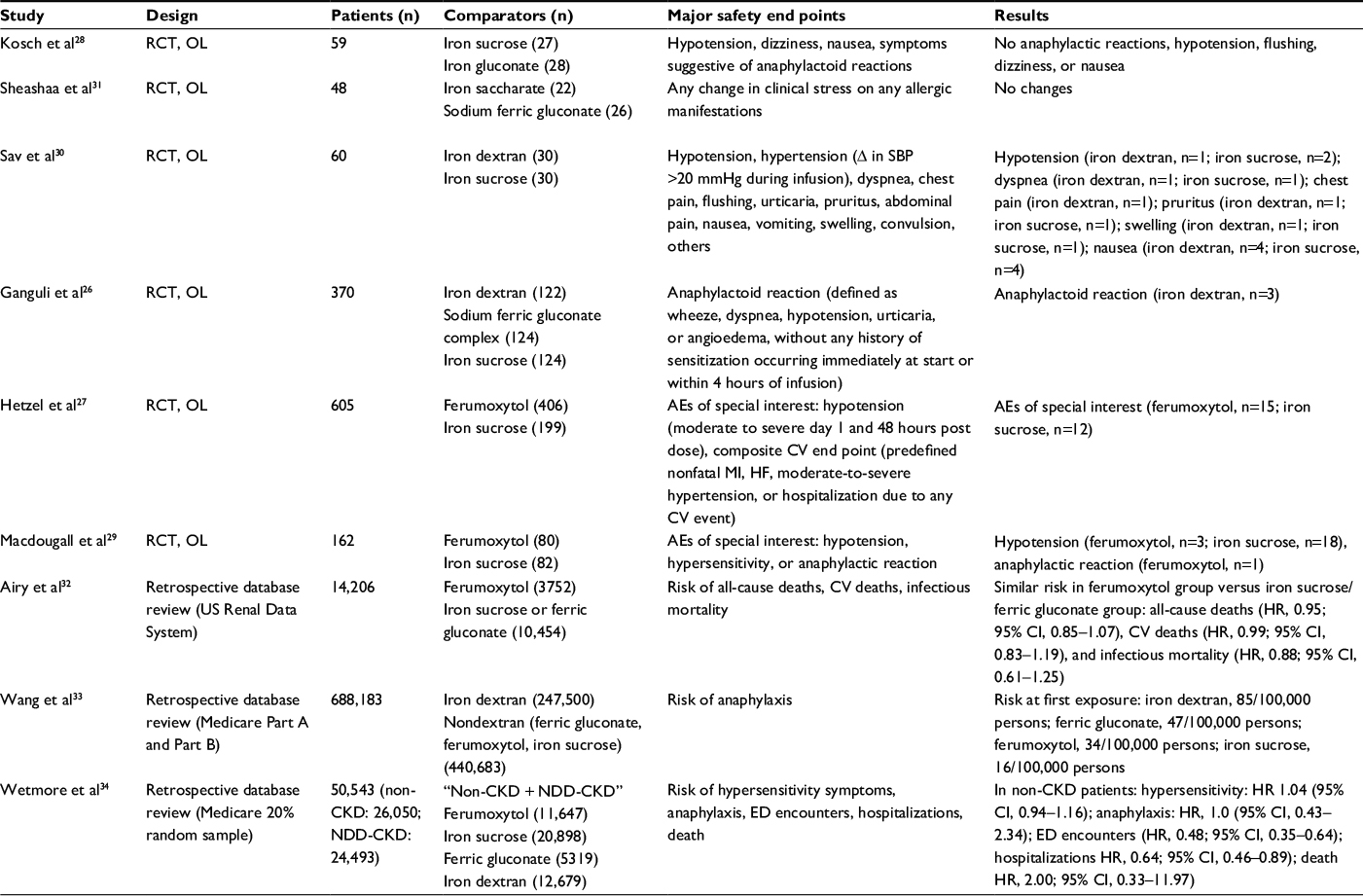 | Table 1 Comparative safety studies of IV iron Abbreviations: AEs, adverse events; CI, confidence interval; CKD, chronic kidney disease; CV, cardiovascular; ED, emergency department; HF, heart failure; HR, hazard ratio; IV, intravenous; MI, myocardial infarction; NDD, non-dialysis dependant; OL, open label; RCT, randomized controlled trial; SBP, systolic blood pressure. |
Ferumoxytol has been approved in the USA, Canada, and Europe for the treatment of IDA in adult subjects with CKD and is available under the trade name Feraheme® (ferumoxytol) injection or Rienso®. The marketing of Rienso® has been discontinued in Europe and Canada for business reasons. FCM is currently registered in 46 countries and marketed in 37 countries worldwide and is also the only other IV iron approved in the USA that may deliver a complete course of therapy in two doses. The dose of FCM to be administered in this trial is 1.500 g (delivered as 2 × 750 mg), which is consistent with the approved and marketed doses of FCM in the USA (Injectafer®).
Currently, no large prospective comparative trials evaluating the risk of hypersensitivity reactions and/or hypotensive events with ferumoxytol versus other iron formulations have been published. The Ferumoxytol Compared to Ferric Carboxymaltose for the Treatment of Iron Deficiency Anemia (FIRM) study addresses this need.
Materials and methods/design
Main study
FIRM is a randomized, double-blind, parallel-design, Phase III study (ClinicalTrials.gov identifier: NCT02694978) taking place at approximately 130 study sites in North America (USA and Canada) and Eastern Europe (Latvia, Lithuania, Poland, and Hungary). The study plans to enroll approximately 2000 patients. The study duration will be approximately 9 weeks, which includes a screening period of up to 30 days and a treatment period of approximately 5 weeks, with follow-up visits at weeks 2 and 5 (Figure 1). Male and female patients aged ≥18 years with IDA of any etiology, with the exception of dialysis-dependent CKD, for whom oral iron is unsatisfactory, not tolerated, or is medically inappropriate are eligible (Table 2). Exclusion criteria include known hypersensitivity to ferumoxytol or FCM, hemoglobin ≤7.0 g/dL, or low blood pressure (BP).
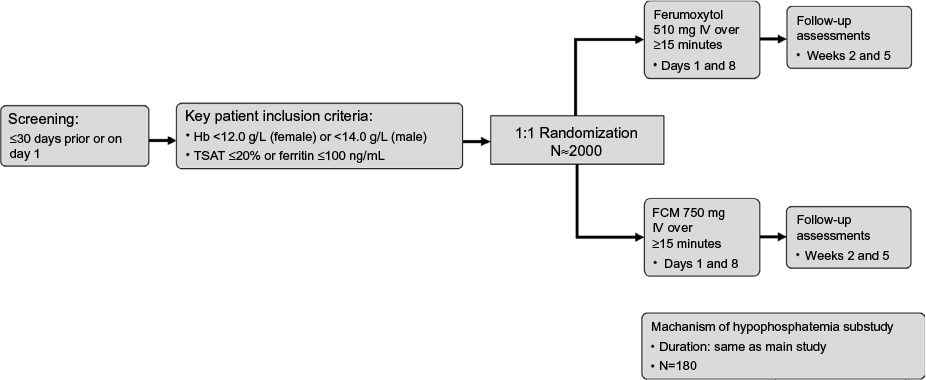 | Figure 1 Study design. Abbreviations: FCM, ferric carboxymaltose; Hb, hemoglobin; IV, intravenous; TSAT, transferrin saturation. |
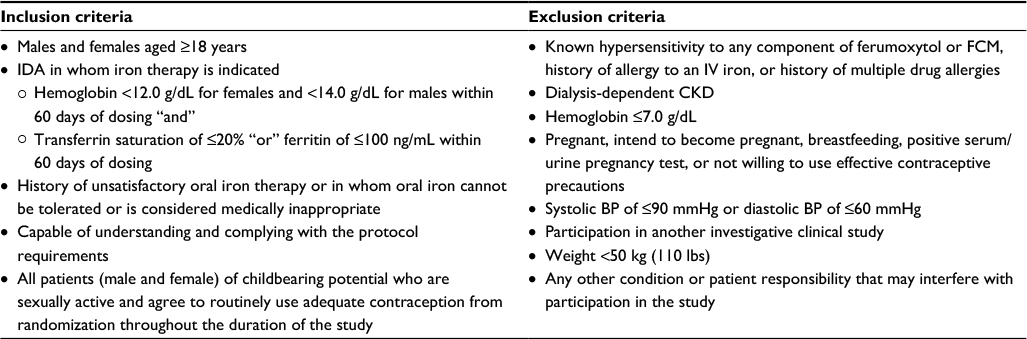 | Table 2 Key inclusion and exclusion criteria in the FIRM trial Abbreviations: BP, blood pressure; CKD, chronic kidney disease; FIRM, Ferumoxytol versus Ferric Carboxymaltose for the Treatment of Iron Deficiency Anemia; FCM, ferric carboxymaltose; IDA, iron deficiency anemia; IV, intravenous. |
This study is being planned in accordance with Good Clinical Practice guidelines, the Declaration of Helsinki, and any and all other applicable regulatory requirements. The majority of sites are utilizing a central Institutional Review Board (IRB), Quorum Review IRB, which reviewed and approved the protocol. A small number of sites (two in Canada, one in the USA) are using local IRBs (Joint Group Health Centre/Sault Area Hospital, Comité d’éthique de la recherche du CIUSSS de l’Est-de-l’Île-de-Montréal, and Brooke Army Medical Center, respectively) which also reviewed and approved. All patients will provide written informed consent before enrollment.
Hypophosphatemia substudy
A hypophosphatemia substudy that will enroll up to 180 patients and be conducted at selected sites in the USA will enroll concurrently with the main study.
Study objectives
This study aims to examine the safety of IV ferumoxytol compared with IV FCM for the treatment of IDA. The primary safety objective is to assess the incidence of moderate-to-severe hypersensitivity reactions, including anaphylaxis, and moderate-to-severe hypotension with ferumoxytol versus FCM. The secondary safety objectives include assessing the incidence of the composite safety end point of moderate-to-severe hypersensitivity reactions, serious cardiovascular events, and death with ferumoxytol versus FCM, as well as assessing the incidence of treatment-emergent serious AEs following administration of ferumoxytol or FCM. The primary efficacy objective is to measure the mean change in hemoglobin from baseline to week 5; an additional efficacy objective is to measure the mean change in hemoglobin per milligram of iron from baseline to week 5.
The objective of the substudy is to investigate the mechanism(s) associated with any increase in urinary excretion of phosphorus and resultant hypophosphatemia following administration of ferumoxytol or FCM.
Randomization and interventions
After a screening period of up to 30 days, patients are randomized (1:1) to receive either ferumoxytol 1.02 g, administered as two doses of 510 mg, or FCM 1.50 g, administered as two doses of 750 mg, each delivered as an IV infusion over at least 15 minutes (Figure 1). Randomization is carried out via a centralized interactive web response system. The study drug is admixed by an unblinded test article preparer into a final volume of 250 mL of normal saline. Patients and study staff at the clinical sites remain blinded to study drug allocation. Doses are administered by blinded staff, with the second dose administered 7 days after day 1. Up to 180 patients who participate in the main study will participate in the optional hypophosphatemia substudy. Patients may discontinue and withdraw from the study at any time.
Study end points
Table 3 provides a complete description of study end points. The primary end point is to characterize the incidence of moderate-to-severe hypersensitivity reactions, including anaphylaxis, and moderate-to-severe hypotension. A secondary safety end point is the composite of moderate-to-severe hypersensitivity reactions, including anaphylaxis, serious cardiovascular events, and death. Secondary efficacy end points will examine the mean change in hemoglobin and the mean change in hemoglobin per milligram of iron from baseline to week 5. The hypophosphatemia substudy will examine changes in blood and urine markers of phosphate and bone metabolism, including fractional excretion of phosphate, parathyroid hormone (PTH), FGF-23, and vitamin D.
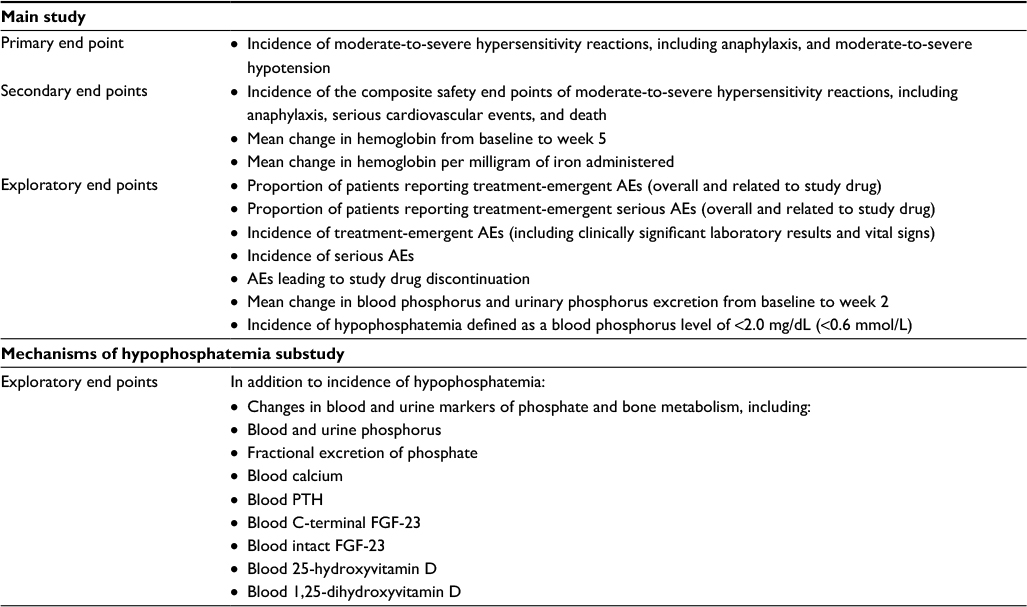 | Table 3 Study end pointsa Note: aIncidences of AEs and distributions of biochemical end points will be compared statistically to determine whether the data differ by treatment product. Abbreviations: AEs, adverse events; FGF-23, fibroblast growth factor 23; PTH, parathyroid hormone. |
Assessments
Safety assessments
All events considered representing possible hypersensitivity reactions and hypotension will be adjudicated by an independent, blinded Clinical Events Committee (CEC) using prespecified criteria described in the protocol. The CEC is composed of five clinicians familiar with IV iron therapy and its potential associated AEs, allergic/hypersensitivity reactions, and adjudication of clinical events in trials. Each event considered to represent a possible hypersensitivity reaction or hypotension will have an adjudication packet created, which includes all relevant clinical information available from the case report forms, source documents (including the Targeted Hypersensitivity Questionnaire), and requested information (eg, hospitalization discharge summaries). For all patients with a possible hypersensitivity event, blood samples for measurement of serum tryptase are collected 1–2 hours after the start of the event and collected again during the week 2 visit to obtain a baseline value, as tryptase levels are expected to normalize within 2 weeks following a hypersensitivity reaction. Each event is adjudicated by two members of the CEC (one immunologist and one internal medicine specialist) as well as by the CEC Chair who is an allergist/immunologist. In the event, the members are not in agreement as to whether or not the event represents a hypersensitivity reaction and/or hypotension, a teleconference will be held to review the case and achieve consensus of all available CEC members.
Hypersensitivity reactions will be recorded and adjudicated by the CEC. Hypersensitivity is defined as a local or generalized response following exposure to a putative allergen, which includes one or more objective signs and symptoms potentially suggesting hypersensitivity, including generalized erythema or pruritus, urticaria (hives, welts, or wheals), flushing, dyspnea, wheezing, bronchospasm, tachypnea, stridor, rapid-onset edema (facial, laryngeal, or pharyngeal) syncope, and dizziness. Anaphylaxis is defined as a severe, systemic, potentially fatal allergic reaction with a sudden onset and will be categorized based on the criteria of Sampson et al7 (Table 4). Hypotension is defined as a >30% decrease from baseline in systolic BP or a decrease of >20 mmHg in systolic BP along with symptoms (eg, dizziness/lightheadedness, fatigue, syncope, lack of consciousness, or blurred vision).
 | Table 4 Clinical criteria for anaphylaxis7 Note: aFor the FIRM study, only the first criteria were applicable because patients with a prior allergic reaction to either study medication were excluded. FIRM, Ferumoxytol versus Ferric Carboxymaltose for the Treatment of Iron Deficiency Anemia. Adapted from J Allergy Clin Immunol, 117(2), Sampson HA, Munoz-Furlong A, Campbell RL, et al, Second symposium on the definition and management of anaphylaxis: summary report – Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium, 391–397, Copyright (2006), with permission from Elsevier.7 Abbreviations: BP, blood pressure; PEF, peak expiratory flow. |
AEs are collected while the patient is present in the clinic during the 1-hour postdosing observation period and at follow-up visits at weeks 2 and 5. Postdose vital signs are obtained at approximately 5, 10, and 30 minutes following completion of the infusion. AEs are assessed by the investigator for severity and relationship to study medication; any signs or symptoms potentially representing hypersensitivity reactions, even if felt to be unlikely, are captured and adjudicated by the independent CEC.
Laboratory tests
Laboratory tests including chemistry, hematology, iron panel, and spot urine (for creatinine, phosphorus, albumin, and glucose) are obtained at baseline and at specific follow-up visits.
Assessments are completed on day 1 (dose 1), day 8 (dose 2), week 2, and week 5. The end points for the hypophosphatemia substudy are the temporal associations of C-terminal and intact FGF-23, 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and PTH, with changes in blood phosphorus and fractional excretion of phosphate. Patients participating in the substudy will have an additional sample of serum obtained at the time of the routine blood draws for the main study on day 1, day 8, week 2, and week 5.
Statistical analyses
The safety population will include any randomized patient who received any amount of study drug. All safety analyses will be performed on the safety population; the treatment group is based on actual treatment received. The intention-to-treat population will include all randomized patients who had any exposure to study drug and will serve as the primary efficacy analysis population. The evaluable population will include all randomized patients who met all inclusion criteria and did not violate any exclusion criteria or have any significant protocol violations/deviations considered to impact study integrity.
For the primary and secondary end point analyses, the proportion of patients who meet the criteria of treatment-emergent moderate-to-severe hypersensitivity reactions including anaphylaxis and/or hypotension for the primary end point, and the composite safety end point of moderate-to-severe hypersensitivity reactions including anaphylaxis, serious cardiovascular events, and deaths for the secondary end point, will be presented by treatment group. Based on an assumed AE rate of 3.3% for the primary safety end point in both treatment groups, a sample size of 2000 patients will provide approximately 90% power for the noninferiority test using a noninferiority margin of 2.64%. The assumed AE rate for the primary end point is based on the rate of these events that occurred in two earlier pivotal randomized controlled trials conducted in a similar population of patients who were unable to tolerate or had inadequate response to oral iron therapy. Noninferiority with respect to the primary end point will be concluded if the upper limit of the 95% CI for the difference in rates (ferumoxytol minus FCM) does not exceed the noninferiority margin of 2.64%.
Descriptive statistics, graphic approach, and statistical modeling methods will be used for the exploratory analysis of treatment-related hypophosphatemia and the temporal association of C-terminal and intact FGF-23, 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and PTH with changes in blood and urine phosphorus and fractional excretion of phosphate.
Discussion
The relative safety of IV iron preparations regarding acute infusion-related hypersensitivity reactions is not well characterized. As described previously, a few retrospective epidemiological studies have explored the relative risks of serious AEs associated with ferumoxytol and various IV iron products.32–34 However, while such analyses provide useful information, the data may be prone to known and unknown confounding and ultimately require confirmation in prospective randomized controlled studies.
The FIRM study will provide a direct comparative assessment of the relative risks of hypersensitivity reactions and hypotension associated with ferumoxytol and FCM, two commonly used IV iron products that have a similar dosing schedule. In addition, data will be generated on the relative efficacy of these agents for improving hemoglobin concentrations. The study differs from other previous comparative trials in that its main focus is a comparison of hypersensitivity reactions and hypotension with two IV iron agents in a large-scale randomized controlled trial. Another major strength of this study will be the use of an independent CEC that will review all reported hypersensitivity reactions and deaths in a blinded fashion using prespecified criteria. Finally, the hypophosphatemia substudy will provide data on the incidence of IV iron-related hypophosphatemia and contribute to understanding the mechanisms implicated in the development of this condition.
The FIRM study will provide valuable data regarding the association between IV irons and hypersensitivity reactions, hypotension, and hypophosphatemia in IDA of any etiology among patients with nondialysis-dependent CKD, as well as contribute to the comparative safety literature on IV iron products.
Acknowledgments
The FIRM study and development of this manuscript were supported by AMAG Pharmaceuticals, Inc. The authors thank Jennifer L Giel and Christine M O’Leary who provided medical writing and editorial support, respectively, on behalf of inScience Communications and Springer Healthcare (Philadelphia, PA, USA), funded by AMAG.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
NFA is a paid consultant for AMAG Pharmaceuticals, Inc. WES, KB, RK, and JK are employees of AMAG Pharmaceuticals, Inc. ICM has received speakers fees, honoraria, and consultancy fees from several IV iron manufacturers, including AMAG Pharmaceuticals, Inc., Pharmacosmos, Roche, Takeda, and Vifor Pharma. The authors report no other conflicts of interest in this work.
References
WHO/CDC. Worldwide Prevalence of Anemia 1993–2005: WHO Global Database on Anemia. Geneva, Switzerland: WHO Press; 2008. | ||
Miller JL. Iron deficiency anemia: a common and curable disease. Cold Spring Harb Perspect Med. 2013;3(7):a011866. | ||
Ford DC, Dahl NV, Strauss WE, et al. Ferumoxytol versus placebo in iron deficiency anemia: efficacy, safety, and quality of life in patients with gastrointestinal disorders. Clin Exp Gastroenterol. 2016;9:151–162. | ||
Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl. 2012;2(4):280–335. | ||
Macdougall IC, Bircher AJ, Eckardt KU, et al. Iron management in chronic kidney disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int. 2016;89(1):28–39. | ||
Macdougall IC, Vernon K. Complement activation-related pseudo-allergy: a fresh look at hypersensitivity reactions to intravenous iron. Am J Nephrol. 2017;45(1):60–62. | ||
Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report – Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network Symposium. J Allergy Clin Immunol. 2006;117(2):391–397. | ||
Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: Report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol. 2004;113(5):832–836. | ||
Lieberman P, Kemp SF, Oppenheimer J, Lang DM, Bernstein IL, Nicklas RA. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 suppl 2):S483–S523. | ||
Lieberman P, Nicklas RA, Randolph C, et al. Anaphylaxis – a practice parameter update 2015. Ann Allergy Asthma Immunol. 2015;115(5):341–384. | ||
Hempel JC, Poppelaars F, Gaya da Costa M, et al. Distinct in vitro complement activation by various intravenous iron preparations. Am J Nephrol. 2017;45(1):49–59. | ||
Szebeni J. Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology. 2005;216(2–3):106–121. | ||
Bircher AJ, Auerbach M. Hypersensitivity from intravenous iron products. Immunol Allergy Clin North Am. 2014;34(3):707–723. | ||
Blazevic A, Hunze J, Boots JM. Severe hypophosphataemia after intravenous iron administration. Neth J Med. 2014;72(1):49–53. | ||
Mani LY, Nseir G, Venetz JP, Pascual M. Severe hypophosphatemia after intravenous administration of iron carboxymaltose in a stable renal transplant recipient. Transplantation. 2010;90(7):804–805. | ||
Sato K, Nohtomi K, Demura H, et al. Saccharated ferric oxide (SFO)-induced osteomalacia: in vitro inhibition by SFO of bone formation and 1,25-dihydroxy-vitamin D production in renal tubules. Bone. 1997;21(1):57–64. | ||
Sato K, Shiraki M. Saccharated ferric oxide-induced osteomalacia in Japan: iron-induced osteopathy due to nephropathy. Endocr J. 1998;45(4):431–439. | ||
Schouten BJ, Doogue MP, Soule SG, Hunt PJ. Iron polymaltose-induced FGF23 elevation complicated by hypophosphataemic osteomalacia. Ann Clin Biochem. 2009;46(pt 2):167–169. | ||
Van Wyck DB, Mangione A, Morrison J, Hadley PE, Jehle JA, Goodnough LT. Large-dose intravenous ferric carboxymaltose injection for iron deficiency anemia in heavy uterine bleeding: a randomized, controlled trial. Transfusion. 2009;49(12):2719–2728. | ||
Onken JE, Bregman DB, Harrington RA, et al. A multicenter, randomized, active-controlled study to investigate the efficacy and safety of intravenous ferric carboxymaltose in patients with iron deficiency anemia. Transfusion. 2014;54(2):306–315. | ||
Schaefer B, Wurtinger P, Finkenstedt A, et al. Choice of high-dose intravenous iron preparation determines hypophosphatemia risk. PLoS One. 2016;11(12):e0167146. | ||
Mannheim J [webpage on the Internet]. Hypophosphatemia; 2014. Available from: https://medlineplus.gov/ency/article/000307.htm. Accessed March 11, 2017. | ||
Anand G, Schmid C. Severe hypophosphataemia after intravenous iron administration. BMJ Case Rep. 2017;2017. | ||
Schouten BJ, Hunt PJ, Livesey JH, Frampton CM, Soule SG. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study. J Clin Endocrinol Metab. 2009;94(7):2332–2337. | ||
Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res. 2013;28(8):1793–1803. | ||
Ganguli A, Kohli HS, Jha V, Gupta KL, Sakhuja V. The comparative safety of various intravenous iron preparations in chronic kidney disease patients. Ren Fail. 2008;30(6):629–638. | ||
Hetzel D, Strauss W, Bernard K, Li Z, Urboniene A, Allen LF. A phase III, randomized, open-label trial of ferumoxytol compared with iron sucrose for the treatment of iron deficiency anemia in patients with a history of unsatisfactory oral iron therapy. Am J Hematol. 2014;89(6):646–650. | ||
Kosch M, Bahner U, Bettger H, Matzkies F, Teschner M, Schaefer RM. A randomized, controlled parallel-group trial on efficacy and safety of iron sucrose (Venofer) vs iron gluconate (Ferrlecit) in haemodialysis patients treated with rHuEpo. Nephrol Dial Transplant. 2001;16(6):1239–1244. | ||
Macdougall IC, Strauss WE, McLaughlin J, Li Z, Dellanna F, Hertel J. A randomized comparison of ferumoxytol and iron sucrose for treating iron deficiency anemia in patients with CKD. Clin J Am Soc Nephrol. 2014;9(4):705–712. | ||
Sav T, Tokgoz B, Sipahioglu MH, et al. Is there a difference between the allergic potencies of the iron sucrose and low molecular weight iron dextran? Ren Fail. 2007;29(4):423–426. | ||
Sheashaa H, El-Husseini A, Sabry A, et al. Parenteral iron therapy in treatment of anemia in end-stage renal disease patients: a comparative study between iron saccharate and gluconate. Nephron Clin Pract. 2005;99(4):c97–c101. | ||
Airy M, Mandayam S, Mitani AA, et al. Comparative outcomes of predominant facility-level use of ferumoxytol versus other intravenous iron formulations in incident hemodialysis patients. Nephrol Dial Transplant. 2015;30(12):2068–2075. | ||
Wang C, Graham DJ, Kane RC, et al. Comparative risk of anaphylactic reactions associated with intravenous iron products. JAMA. 2015;314(19):2062–2068. | ||
Wetmore JB, Weinhandl ED, Zhou J, Gilbertson DT. Relative incidence of acute adverse events with ferumoxytol compared to other intravenous iron compounds: a matched cohort study. PLoS One. 2017;12(1):e0171098. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.