")
Back to Journals » OncoTargets and Therapy » Volume 12
Comparative review of drug–drug interactions with epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small-cell lung cancer
Received 16 November 2018
Accepted for publication 12 March 2019
Published 9 July 2019 Volume 2019:12 Pages 5467—5484
DOI https://doi.org/10.2147/OTT.S194870
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Zi-Yi Xu, Jun-Ling Li
Department of Medical Oncology, National Cancer Center/National Clinical Research Center/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China
Abstract: The development of small-molecule tyrosine kinase inhibitors (TKIs) that target the epidermal growth factor receptor (EGFR) has revolutionized the management of non-small-cell lung cancer (NSCLC). Because these drugs are commonly used in combination with other types of medication, the risk of clinically significant drug–drug interactions (DDIs) is an important consideration, especially for patients using multiple drugs for coexisting medical conditions. Clinicians need to be aware of the potential for clinically important DDIs when considering therapeutic options for individual patients. In this article, we describe the main mechanisms underlying DDIs with the EGFR-TKIs that are currently approved for the treatment of NSCLC, and, specifically, the potential for interactions mediated via effects on gastrointestinal pH, cytochrome P450-dependent metabolism, uridine diphosphate-glucuronosyltransferase, and transporter proteins. We review evidence of such DDIs with the currently approved EGFR-TKIs (gefitinib, erlotinib, afatinib, osimertinib, and icotinib) and discuss several information sources that are available online to aid clinical decision-making. We conclude by summarizing the most clinically relevant DDIs with these EFGR-TKIs and provide recommendations for managing, minimizing, or avoiding DDIs with the different agents.
Keywords: drug metabolism, pharmacokinetics, CYP450, UDP-glucuronosyltransferase, P-glycoprotein
Introduction
The occurrence of drug–drug interactions (DDIs) is a serious problem for the use of anticancer drugs. DDIs can exacerbate the risk of serious or fatal adverse events, and/or lead to reductions in therapeutic efficacy.1 In particular, inducers of drug-metabolizing enzymes are known to increase the systemic clearance of many anticancer agents,2 for example, long-term anticonvulsant therapy increases the systemic clearance of several antileukemic agents, thereby reducing their clinical efficacy.3
Tyrosine kinase inhibitors (TKIs) are effective for a wide variety of solid and hematologic malignancies, and are now established as standard therapeutic options;1 more than 20 different TKIs are currently in use.4 In particular, the development of small-molecule TKIs that target the epidermal growth factor receptor (EGFR) – EGFR-TKIs – has revolutionized the management of locally advanced/metastatic non-small-cell lung cancer (NSCLC).5 This article reviews clinically relevant DDIs with EGFR-TKIs approved for the treatment of NSCLC.
The EGFR (HER1; ErbB1) is a member of the ErbB receptor family, which also includes HER2 (Neu, ErbB2), HER3 (ErbB3), and HER4 (ErbB4).6,7 The main physiological role of ErbB-linked tyrosine kinases (TKs) is regulation of cellular proliferation.6 Somatic EGFR mutations are important oncogenic drivers of NSCLC,8–10 and occur in 10–15% of the Caucasian patients and around 50% of the Asian patients with metastatic NSCLC and adenocarcinoma histology.11 The most common EGFR mutations are deletions in exon 19 (del19) and L858R point mutations in exon 21.12 These “activating” mutations lead to activation of intracellular signaling by EGFR in a ligand-independent manner.13,14 NSCLC patients with activating EGFR mutations become dependent on EGFR activity to stimulate downstream signaling pathways and maintain the malignant phenotype (“oncogene addiction”).15,16 Consequently, blocking ErbB family pathways with EGFR TKIs can inhibit tumor cell proliferation and initiate apoptosis.
EGFR-TKIs widely available for the treatment of advanced NSCLC include the first-generation reversible TKIs, erlotinib and gefitinib; the second-generation irreversible ErbB family blocker, afatinib; and the third-generation EGFR-wild-type sparing, irreversible EGFR/T790M inhibitor, osimertinib.17 In China, another first-generation EGFR-TKI, icotinib, is available.18 EGFR-TKIs are recommended as first-line treatment options for advanced EGFR mutation-positive (EGFRm+) NSCLC,19,20 having demonstrated robust benefit in terms of progression-free survival (PFS, erlotinib: median 9.7–13.1 months;21–23 gefitinib: 8.4–10.9 months;24–28 icotinib 4.0–12.4 months;29 afatinib: 11.0–13.6 months;25,30,31 osimertinib: 17.7 months).32
The first-generation EGFR-TKIs, gefitinib and erlotinib, bind reversibly to EGFR TK and thereby inhibit both mutant and (to a lesser extent) wild-type EGFR.33 In randomized phase III trials, both agents demonstrated improved PFS and response rates, but not overall survival (OS) compared with platinum-doublet chemotherapy.21–24,27,28,34 However, patients with EGFRm+ NSCLC inevitably develop resistance to first-generation EGFR-TKIs.35 In the majority (50–60%), resistance develops due to development of a novel T790M missense mutation in exon 20 of the EGFR,36–38 which impairs binding of the EGFR-TKIs to the kinase domain of the receptor.39
Another first-generation EGFR-TKI, icotinib has a similar chemical structure and physicochemical properties to erlotinib and displays similar clinical efficacy.29 A randomized, double-blind trial in Chinese patients with advanced NSCLC who had failed to respond to chemotherapy concluded that icotinib was non-inferior to gefitinib with regard to PFS; median PFS was 4.6 months for icotinib and 3.4 months for gefitinib.40 In a meta-analysis of pooled data for icotinib in patients with NSCLC (11 studies), the pooled mean PFS was 7.34 months (95% confidence interval: 5.60–9.07). EGFRm+ patients had longer PFS (median 11.0 months) than EGFRm- patients (1.97 months).29
Further “generations” of EGFR-TKIs have also been developed, the aim being to improve efficacy and tolerability, and to overcome treatment resistance; data from head-to-head trials suggest improved outcomes with second-/third-generation versus first-generation TKIs.
In contrast to gefitinib and erlotinib, afatinib is an ErbB receptor family inhibitor. Afatinib binds covalently and irreversibly blocks signaling via all hetero- and homodimers formed by ErbB1, but also by HER2, HER3, and HER4.41,42 It was postulated that this broad irreversible inhibition might delay or avoid the development of resistance.43 In two phase III trials conducted in treatment-naïve NSCLC patients, afatinib demonstrated significant improvements in PFS versus platinum-doublets,30,31 while pre-specified analyses of both trials identified significant improvements in OS with afatinib versus chemotherapy in patients with del19 mutations.44 In a phase IIb study of treatment-naïve patients with advanced EGFRm+ NSCLC, afatinib significantly improved PFS, time to treatment failure (TTF) and objective response rate (ORR) versus gefitinib, with a trend towards improvement in overall survival (OS).25,45
Osimertinib is an oral, third-generation, irreversible EGFR-TKI inhibitor46 that selectively inhibits EGFR harboring both activating mutations and EGFR T790M resistance mutations, with less activity versus wild-type EGFR.47 In a double-blind phase III trial in patients with previously untreated, EGFRm+ (del19 or L858R) advanced NSCLC, osimertinib demonstrated efficacy superior to that of standard EGFR-TKIs (gefitinib or erlotinib). Median PFS for osimertinib was significantly longer than for standard EGFR-TKIs (18.9 months versus 10.2 months).32 In a phase I study in patients harboring the EGFR T790M mutation having progressed during therapy with EGFR-TKIs, second-line (or later) osimertinib was highly active, with median PFS in T790M-positive patients (9.6 months) being substantially longer than in T790M-negative patients (2.8 months).48 A single-arm study in patients harboring both activating EGFR mutations and the T790M mutation and with progression after prior EGFR-TKI therapy also showed antitumor efficacy of osimertinib (objective response rate 64%; disease control rate 90%) suggesting that osimertinib can overcome T790M-mediated acquired resistance.49 Another second-generation EGFR-TKI, dacomitinib, recently became available in the US and EU, but to date there is little published evidence on potentially clinically relevant DDIs with dacomitinib.
In the treatment of NSCLC, EGFR-TKIs are commonly used together with other types of medication. Consequently, the risk of serious DDIs should be taken into consideration when selecting appropriate treatment.5 As the increasing molecular stratification of lung cancer has provided more options for targeted intervention and rational combination therapy, a clear understanding of the DDI profiles of different TKIs has become essential. Clinicians need to consider the potential implications of clinically important DDIs when formulating individualized therapeutic strategies for their patients. In this article, we review the key pharmacologic properties of the EGFR-TKIs currently approved for the treatment of NSCLC and the clinically relevant DDIs associated with each agent.
Literature search strategy
We searched the published literature (English language only) relating to established and potential DDIs between the EGFR-TKIs of interest (ie, those currently approved for the treatment of NSCLC) and other prescription drugs, over-the-counter drugs, and herbal medicines. Relevant publications were identified by means of searches of US National Library of Medicine (NLM) PubMed, using the search terms [interaction] OR [drug-drug] AND [Drug name (for each EGFR-TKI)]. Other relevant publications were identified from citations in the key publications identified via NLM PubMed. Further information was obtained from the US and EU prescribing information for each agent (not available for icotinib).
Mechanisms underlying DDIs for EGFR-TKIs used for the treatment of NSCLC
Interactions via effects on gastrointestinal pH
Other than the physicochemical properties of the different TKIs, the most important factor affecting the solubility of/exposure to these agents is gastric pH.1 For TKIs with a pKa of less than 4–5, concomitant administration of agents that increase stomach pH can reduce TKI solubility, absorption, and bioavailability.5,50 In particular, clinically relevant DDIs due to changes in gastric pH have been demonstrated between histamine H2-receptor antagonists such as ranitidine, or proton pump inhibitors (PPIs) such as omeprazole, and a number of TKIs, including crizotinib, dasatinib, erlotinib, gefitinib, lapatinib, and pazopanib.50,51 As patients with cancer often take acid suppressants for symptoms of gastrointestinal reflux, the potential for such interactions is clinically important.5
Interactions via effects on cytochrome P450 (CYP)-dependent metabolism
Phase I metabolism (mostly oxidative) by liver cytochrome P450 (CYP)–dependent enzymes is the most important route of drug metabolism in vivo.1 Many TKIs are metabolized by this family of enzymes, which makes them prone to metabolic DDIs.5,50 Indeed, most pharmacokinetic (PK) interactions that affect the EGFR-TKIs involve effects on metabolism, especially via CYP enzymes. Potent enzyme inhibitors and inducers can modify the exposure (the area under the plasma concentration–time curve [AUC] and the maximum plasma concentration [Cmax]) of specific EGFR-TKIs, while EGFR-TKIs that are CYP enzyme substrates can affect the PK of other drugs.5 Increased or decreased exposure due to alteration of CYP enzyme activity could lead to clinically relevant toxicity or reduced effectiveness of the EGFR-TKIs.1 Cigarette smoking is also known to induce key CYP enzymes52 and has been shown to affect the PK profiles of specific TKIs.5
As shown in Table 1, the extent to which different EGFR-TKIs are metabolized by CYP enzymes varies markedly, as do their effects as inducers or inhibitors of CYP enzyme activity. Nevertheless, there is wide potential for interactions with other drugs used in supportive therapy or for the treatment of comorbidities in patients with NSCLC.
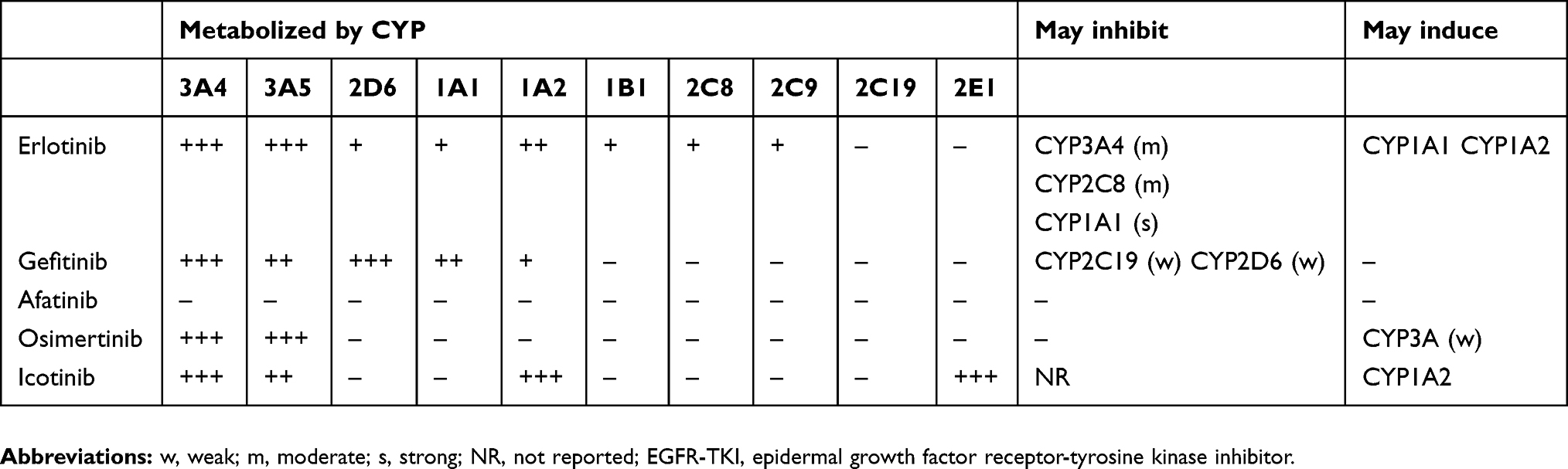 |
Table 1 Cytochrome P450 (CYP450) enzymes involved in the metabolism of EGFR-TKIs approved for the treatment of NSCLC |
CYP2D6, in particular, has been reported to be responsible for the metabolism of up to 25% of commonly prescribed drugs.53,54 Importantly, CYP2D6 is encoded by a highly polymorphic gene, with more than 70 alleles and 130 genetic variations,55 which could have a significant influence on up to a half of the drugs metabolized by this enzyme.56 There is marked inter-ethnic variation in the frequency of the different alleles, leading to substantial variability in the prevalence of the four main phenotypes (poor, intermediate, extensive, and ultra-rapid metabolizers).57 The poor metabolizer phenotype, in particular, has been widely examined in relation to adverse drug reactions.58 Poor metabolizers account for 5–10% of the Caucasian populations, but are rarely found in Asia, and are highly variable among people of African ancestry.59,60 Conversely, about 10–15% of the Caucasians are intermediate metabolizers, compared with up to 50% of the Asians and 30% of the Africans.57
Interactions via effects on UDP-glucuronosyltransferase
Uridine-diphosphate (UDP)-glucuronosyltransferase (UGT) catalyzes the conjugation of glucuronic acid to endogenous substances and exogenous compounds.4 Most UGT isoforms are expressed in the liver.4 Since UGTs are rate-limiting enzymes in the metabolism of various compounds, co-administration of UGT-inhibiting drugs can lead to an increase in the concentration of such compounds in the circulation.61 In particular, UGT1A1 plays a key role in the metabolism and detoxification of many potentially harmful compounds and drugs, and thus inhibition of UGT1A1 can lead to severe DDIs and other undesirable effects.62 UGT1A1 is also involved in the metabolic elimination of endogenous bilirubin, preventing accumulation to toxic levels.62 Several TKIs are potent inhibitors of UGT1A1 (eg, erlotinib, lapatinib, nilotinib, pazopanib, regorafenib, and sorafenib),63–65 and this may underlie some of the adverse events observed with these agents, such as hyperbilirubinemia and hepatotoxicity.62,64,66
Interactions via effects on transporter proteins
In order to reach the portal blood circulation, TKIs need to pass through the gut wall. This involves both passive diffusion and active transport via organic anion-transporting peptide (OATP), organic cation-transporting peptide (OCTP), multidrug-resistance–associated proteins such as ATP-Binding Cassette (ABC) transporter G2 (ABC-G2), efflux transporters such as P-glycoprotein (P-gp, also known as multidrug resistance protein 1 [MDR1], ABC sub-family B, member 1 [ABC-B1] or cluster of differentiation 243 [CD243]), and intestinal metabolic enzymes such as CYP3A4.1 P-gp and ABC-G2 are expressed in the small intestine, liver, kidneys, and blood–brain barrier (BBB); they appear to regulate the oral absorption, biliary and renal elimination, and also the BBB penetration of several anticancer drugs, including TKIs.1,2
The role of P-gp in the absorption of TKIs has been widely studied. Some TKIs (eg, crizotinib) are P-gp substrates, so inhibition or induction of P-gp due to concomitant administration of another drug could lead to clinically relevant DDIs.1 Others such as pazopanib, lapatinib, and gefitinib directly inhibit P-gp activity, so could increase the bioavailability of concomitantly administered P-gp substrates.1 Other ABC drug transporters such as Breast Cancer Resistance Protein (BCRP) and Multidrug Resistance-associated Protein 2 (MRP2 or ABC-C2) are also recognized for their potential for DDIs.5,67–69 The role of the uptake solute carrier transporters (eg, OATP, OCTP) in transporter-mediated drug interactions with EGFR-TKIs is less well defined.5,69,70
Clinically relevant DDIs with selected EGFR-TKIs
Gefitinib
Gefitinib was the first oral quinazoline compound to be referred to as a “selective” EGFR-TKI.71 In the US, gefitinib is indicated for first-line treatment of patients with metastatic NSCLC who harbor EGFR exon 19 deletions or exon 21 (L858R) substitution mutations, as detected by a test approved by the Food and Drug Administration (FDA).72
Acid-reducing agents
Medications that cause significant sustained elevation in gastric pH, such as PPIs and H2-receptor antagonists, may reduce the bioavailability, plasma concentration, and efficacy of gefitinib.72,73 High doses of short-acting antacids may have similar effects if taken regularly close to the time of administration of gefitinib.73,74 In a recent preclinical study, treatment with omeprazole (10–100 mg/kg orally [p.o.]) and vonoprazan (1–5 mg/kg p.o.) produced significant dose-dependent increases in gastric pH, and the AUC0–3h of gefitinib (5 mg/kg, p.o.) declined with increasing pH.75 In healthy male volunteers, use of the rapid-acting H2-receptor antagonist ranitidine (at a dose of 450 mg p.o., that increased gastric pH to ≥5 for ≥4 hrs), taken 1 hr before a 250 mg dose of gefitinib, markedly reduced gefitinib exposure (geometric least-squares mean AUC0–∞ reduced by 47%; Cmax by 71%).74,76 However, inhibitors of gastric secretion had no effect on the efficacy of gefitinib in patients with NSCLC or in those harboring EGFR-activating mutations.77
Concomitant use of gefitinib with PPIs should be avoided but, if deemed essential, gefitinib should be taken 12 hrs before or after the PPI.72 Similarly, gefitinib should be taken 6 hrs before or after an H2-receptor antagonist or an antacid.72
CYP450-dependent metabolism
Gefitinib was metabolized at a similar rate when incubated in vitro with recombinant human CYP3A4 or CYP2D6, less efficiently with CYP3A5 and CYP1A1.78 The range of metabolites produced by recombinant human CYP3A478 are similar to those generated by human liver microsomes, in which gefitinib was found to be rapidly and extensively metabolized.79
O-desmethyl-gefitinib, the major metabolite of gefitinib in human plasma,80 was formed mainly by recombinant human CYP2D6,78 but in liver microsomes, O-desmethyl-gefitinib was only a minor product;79 the metabolism of gefitinib was primarily dependent on CYP3A4 and was not notably reduced in microsomes from CYP2D6 poor metabolizers.79 Gefitinib is excreted either unchanged or after metabolism.73,79,80
Concomitant administration of gefitinib with strong CYP3A4 inhibitors may reduce metabolism and clearance of gefitinib, and may increase its plasma concentration.72,73 This may be clinically important, as adverse reactions with gefitinib are related to dose and exposure.73 In healthy volunteers, pretreatment with the potent CYP3A4 inhibitor itraconazole (200 mg QD for 12 days) prior to gefitinib (a single dose of 250 mg on day 4) led to an increase in gefitinib exposure (mean AUC) by up to 78%.74,81 Patients receiving potent CYP3A4 inhibitors should be closely monitored for adverse reactions to gefitinib.73 Notably, the effect of CYP3A4 inhibitors on gefitinib exposure may be greater in CYP2D6 poor metabolizers.73
CYP3A4 inducers may increase gefitinib metabolism and reduce the plasma concentration and efficacy of gefitinib.72,73 Thus, pretreatment with the strong CYP3A4 inducer rifampicin (600 mg QD for 16 days) prior to gefitinib (a single dose of 500 mg on day 10) led to a reduction in mean gefitinib AUC of up to 83%.81 Administration of gefitinib with the moderate-to-strong CYP3A4 inducer phenytoin led to a 26% reduction in Cmax and a 47% reduction in AUC.82 A potential interaction was also reported between gefitinib and herbal medicines including ginseng (a CYP3A4/5 inducer); the patient was a non-responder but became a partial responder after discontinuation of the herbal medicines.83 A similar interaction might be expected with St. John’s Wort (Hypericum perforatum), another CYP3A4 inducer.1 Concomitant administration of gefitinib with CYP3A4 inducers (phenytoin, carbamazepine, rifampicin, barbiturates, St. John’s Wort, ginseng) should, therefore, be avoided, as treatment efficacy may be reduced.73 If use with a moderate-to-strong CYP3A4 inducer is essential, the dose of gefitinib should be increased to 500 mg/day (provided no severe adverse drug reactions are apparent).72 The standard dose (250 mg/day) should be resumed after discontinuation of the CYP3A4 inducer.72 Patients taking gefitinib with potent CYP3A4 inhibitors should be carefully monitored, due to the potential for toxicity, while those taking CYP3A4 inducers should be monitored for reduced efficacy.5,72
As previously noted, gefitinib is mainly metabolized by CYP3A4, and to a lesser extent by CYP2D6,79 although the impact of CYP2D6 inhibitors on gefitinib PK has not been evaluated.5,73 The role of CYP2D6 in the clearance of gefitinib has been evaluated in healthy volunteers genotyped for CYP2D6 status. In CYP2D6 poor metabolizers, O-desmethyl gefitinib was unmeasurable in plasma (confirming that production of this metabolite is mediated by CYP2D6), and gefitinib exposure was twofold higher than in extensive metabolizers.72,84 The investigators suggested that the absence of metabolite is unlikely to be clinically relevant, as it contributes little to the overall activity of gefitinib, while poor metabolizers have higher exposure to unchanged gefitinib (this is also unlikely to lead to clinically significant changes in the safety and tolerability of gefitinib).84 Consequently, prospective screening for CYP2D6 genotype is not warranted before starting gefitinib, and dose adjustments and changes in clinical management strategy are not required for CYP2D6 poor metabolizers.72,73,84 Giving a potent CYP2D6 inhibitor concomitantly with gefitinib might also increase gefitinib exposure. Consequently, poor CYP2D6 metabolizers and patients who begin taking a CYP2D6 inhibitor together with gefitinib should be closely monitored for adverse reactions to gefitinib.72,73
Gefitinib is also a weak inhibitor of CYP2D6 in vitro.72,81 In patients with solid tumors, co-administration of gefitinib with the CYP2D6 substrate metoprolol led to a 35% increase in metoprolol exposure.81 This effect may be relevant to CYP2D6 substrates with a narrow therapeutic index73 as it may be necessary to modify the dose of such agents when used concurrently with gefitinib.73
Smoking status is not a relevant consideration for gefitinib.5
UDP-glucuronosyltransferases
Gefitinib demonstrated broad inhibition of UGT-mediated glucuronidation in vitro, particularly against UGT1A1, UGT1A7, UGT1A9, and UGT2B7 isotypes.63 The risk of potential DDIs in vivo was predicted by calculating the ratios between the area under the plasma concentration–time curve in the presence and absence of inhibitor (AUCi/AUC). For gefitinib, the AUC ratio at the highest evaluated dose (700 mg/day) was less than 1.3 for the substrates of each inhibited UGT isoform. While the authors acknowledged that in vivo DDIs extrapolated from in vitro data should be interpreted with caution, they concluded that the use of gefitinib is unlikely to lead to clinically significant DDIs via inhibition of glucuronidation.63
Transporter proteins
In vitro evidence indicates that gefitinib is a substrate of P-gp but according to the EMA assessment report for gefitinib, there is no evidence to suggest that this effect has clinical consequences.74 Effects of other agents on P-gp are unlikely to influence gefitinib absorption, as P-gp is saturated at higher concentrations.72 In one study, gefitinib was reported to directly inhibit the function of P-gp in multidrug-resistant lung and breast cancer cells. The authors suggested that gefitinib may inhibit the excretion of P-gp substrate drugs and that potential DDIs should be evaluated,85 although there is no evidence that this effect is clinically relevant. In a preclinical study, simultaneous administration of gefitinib dramatically increased the oral bioavailability of irinotecan,86 and in children with refractory solid tumors, use of gefitinib led to a fourfold increase in the bioavailability of oral irinotecan versus historical controls, and significantly reduced the clearance of irinotecan and its active metabolite, SN-38. The authors suggested that these effects may have occurred via inhibition of ABC-G2 by gefitinib.87 Gefitinib has been shown to inhibit BCRP in vitro,88 but the clinical importance of this effect is unknown.5,73
Other clinically relevant interactions
Increases in the international normalized ratio (INR) and/or the rate of bleeding events have occurred in patients taking warfarin and gefitinib concomitantly.72,73,89 Patients taking this combination should be monitored regularly for changes in prothrombin time or INR.72,73
Concomitant use of sorafenib reduced gefitinib exposure (Cmax by 26%, AUC by 38%) via an unknown mechanism, whereas sorafenib exposure was unaffected.90 In phase II clinical trials, concomitant use of gefitinib and vinorelbine exacerbated the neutropenic effect of vinorelbine,73,91 although in a phase I/II trial of gefitinib plus vinorelbine and gemcitabine in patients with metastatic breast cancer the incidence of febrile neutropenia was not a major limiting factor.92
Erlotinib
Erlotinib is a reversible EGFR-TKI that is approved by the FDA as first-line, maintenance, or second-line or subsequent treatment following progression after at least one prior chemotherapy regimen, in patients with metastatic NSCLC who harbor EGFR exon 19 deletions or exon 21 (L858R) substitution mutations, as detected by an FDA-approved test.93
Acid-reducing agents
The solubility of erlotinib is pH-dependent and decreases above pH 5;1,5,94 therefore, drugs that alter gastrointestinal pH could alter the solubility and absorption of erlotinib, leading to potentially clinically relevant changes in bioavailability.94 In a recent preclinical study of concomitant treatment with omeprazole (10–100 mg/kg p.o.) and vonoprazan (1–5 mg/kg p.o.), both of which induced significant dose-dependent increases in gastric pH, the AUC0–3h of erlotinib (5 mg/kg p.o.) decreased as pH increased.75
Combining erlotinib with PPIs should be avoided.93,94 In healthy volunteers, concomitant use of the PPI omeprazole (40 mg once daily [QD] for 7 days) led to a reduction in erlotinib exposure (46% reduction in AUC; 61% reduction in Cmax).95 Temporal separation of doses may not eliminate the interaction because PPIs affect the pH of the upper gastrointestinal tract for an extended period.93
Co-administration of H2-receptor antagonists can also reduce the efficacy of erlotinib. Concomitant use of the H2-receptor antagonist ranitidine (300 mg QD for 5 days, given 2 hrs before erlotinib) led to a 33% reduction in erlotinib AUC and a 54% reduction in Cmax.95 Increasing the dose of erlotinib is unlikely to compensate for such reductions in exposure,94 but when dosing was staggered (ranitidine was given as a divided dose of 150 mg twice daily [BID] and erlotinib was given 10 hrs after the previous evening dose of ranitidine and 2 hrs before the next morning dose of ranitidine), the reduction in erlotinib exposure was much less marked (15% reduction in AUC; 17% reduction in Cmax).95 Consequently, if ranitidine co-administration is considered, it should be used in a staggered manner; ie, erlotinib must be taken at least 2 hrs before or 10 hrs after the dose of ranitidine.93,94
A retrospective analysis of 190 patients with advanced NSCLC indicated that concomitant use of gastric acid suppressants had no significant effect on plasma concentrations of erlotinib, progression-free survival (PFS) or overall survival (OS).96 However, in another retrospective review of 544 patients with advanced NSCLC treated with erlotinib, both PFS and OS were significantly reduced in patients who took gastric acid suppressants compared with those who did not (median PFS 1.4 vs 2.3 months, p<0.001; median OS 12.9 vs 16.8 months, p=0.003).97 The authors did not speculate about the reasons for the divergent results. According to the product label, if required, antacids should be taken at least 4 hrs before or 2 hrs after erlotinib.94
Subsequently, two retrospective studies in patients with EGFR mutations receiving either of the first-generation EGFR-TKIs (erlotinib or gefitinib) also reported that use of acid suppressants had no adverse effects on median PFS and median OS.98,99 Recently, however, a further retrospective observational study of NSCLC patients taking erlotinib or gefitinib found that median PFS was 84 days in patients taking acid suppressants, compared with 221 days in those not taking acid suppressants (p<0.0001); the type of acid suppressant used did not seem to be important.100 In the earlier studies, the presence of activating EGFR mutations in a proportion of patients may have conferred increased sensitivity to EGFR TKIs. Consequently, when erlotinib and gefitinib were given concomitantly with an acid suppressant, despite the reduction in bioavailability, the concentrations of erlotinib and gefitinib achieved in plasma may have been sufficient to inhibit mutant EGFR.98,100 The discrepancy between the outcomes of the earlier studies of erlotinib96,97 might also be explained by a difference in the proportion of patients with EGFR mutations between the two studies, ie, in the study of Chu et al, the majority of patients did not exhibit EGFR mutations, so the outcomes of erlotinib therapy may have been more sensitive to differences in plasma levels of erlotinib.97
CYP450-dependent metabolism
The metabolism of erlotinib is mediated predominantly by CYP3A4/3A5 in liver and intestine, and to a lesser extent by CYP1A2 and CYP2C8, as well as extra-hepatically by pulmonary CYP1A1 and CYP1B1 in tumor tissue.78,101 The active metabolite, O-desmethyl erlotinib, subsequently undergoes oxidation and glucuronidation.5,78,101 Extrahepatic metabolism by CYP3A4 in the intestine, CYP1A1 in the lungs, and CYP1B1 in tumor tissue may also contribute to the clearance of erlotinib.94
Potent inhibitors of CYP3A4 reduce erlotinib metabolism, leading to an increase in plasma erlotinib concentrations.94 Thus, concomitant use of erlotinib with the potent CYP3A4 inhibitor ketoconazole (200 mg p.o., BID for 5 days) led to an 86% increase in erlotinib AUC.94,102 In an open-label, crossover study in male and female healthy volunteers, co-administration of erlotinib (100 mg p.o. on days 1 and 15) with the combined CYP3A4 and CYP1A2 inhibitor ciprofloxacin (750 mg BID on days 13–18) led to a 39% increase in erlotinib AUC and a (non-significant) 17% increase in Cmax.93,103 The EU label advises caution when combining erlotinib with ciprofloxacin or potent CYP1A2 inhibitors such as fluvoxamine; if severe adverse events occur, the dose of erlotinib should be reduced.94 The US label recommends against concurrent use of erlotinib with strong CYP3A4 inhibitors or CYP3A4/1A2 inhibitors, but if this is unavoidable, and if severe adverse reactions occur, it suggests reducing the dose of erlotinib in 50-mg steps.93
Potent CYP3A4 inducers increase erlotinib metabolism and reduce plasma erlotinib concentrations.104 Thus, pretreatment with rifampicin (600 mg p.o., QD for 7 days) led to a 69% reduction in the median AUC of erlotinib.94 Concomitant use of erlotinib with CYP3A4 inducers should be avoided; alternatively, the use of a higher dose of erlotinib may be considered (300 or 450 mg, compared with the standard dose of 150 mg).93,94 Patient safety should be closely monitored (including renal and liver function and serum electrolytes).94
Other strong and moderate CYP3A4 inducers (eg, enzalutamide, phenytoin, carbamazepine, barbiturates, and herbal preparations containing St. John’s Wort) may also reduce erlotinib exposure.1,94 Caution is advised when using these agents concomitantly with erlotinib, and alternative treatments should be considered when possible.94
Cigarette smoking has been shown to markedly reduce erlotinib exposure via an increase in CYP1A1/1A2 activity. In healthy volunteers, the geometric mean of erlotinib AUC0-∞ following a single 150 mg dose was 2.8-fold lower in smokers than in non-smokers, and was similar to that in non-smokers after a dose of 300 mg. Cmax in smokers was two-thirds of that in non-smokers, and C24h was 8.3-fold lower than in non-smokers.105
In patients with solid tumors, clearance of erlotinib was 24% faster in current smokers than former smokers/never-smokers.106 The increase in clearance seems to be related to induction of CYP1A1/1A2 in smokers.2 In current smokers with NSCLC, the response rate to erlotinib was markedly lower than that in never-smokers (3.9% vs 24.7%; p<0.001).107 In another study, the maximum tolerated dose (MTD) of erlotinib in current smokers with NSCLC was 300 mg/day108 (ie, double the MTD previously established in unselected patients), although the authors did not speculate on the underlying mechanisms.109 However, the efficacy and long-term safety of doses greater than the recommended starting doses have yet to be established in patients who continue to smoke.94 In current smokers with locally advanced or metastatic NSCLC, a dose of erlotinib of 300 mg/day led to higher plasma concentrations than were achieved by the standard dose of 150 mg/day, but no incremental efficacy benefit was demonstrated.110 Patients should be encouraged to stop smoking as soon as possible before initiating erlotinib.94 The US label advises increasing the dose of erlotinib in current smokers to 300 mg (maximum), returning immediately to the recommended dose (150 or 100 mg/day) on cessation of smoking.93 The US label also recommends against the use of erlotinib together with moderate CYP1A2 inducers.93
Erlotinib is itself a potent inhibitor of CYP1A1 and a moderate inhibitor of CYP3A4 and CYP2C8.94 The physiologic relevance of CYP1A1 inhibition by erlotinib is unclear, given the limited expression of CYP1A1 in humans.94 Pretreatment with, or co-administration of, erlotinib did not alter the clearance of the CYP3A4 substrates midazolam and erythromycin, but reduced the oral bioavailability of midazolam.94 In another study, concomitant use of erlotinib did not affect the PK of paclitaxel (a CYP3A4/2C8 substrate).111 While the EU label suggests that clinically relevant effects of erlotinib on the PK profiles of other CYP3A4 substrates are unlikely,94 case reports suggest a need for caution during use of erlotinib together with CYP3A4 or CYP2C8 substrates. These include a case of rhabdomyolysis due to increased simvastatin exposure in a patient receiving concomitant erlotinib.112 In another patient, toxicities that occurred during use of phenytoin were exacerbated following addition of erlotinib.113 Clinicians are advised to be aware of these potential interactions when combining these drugs with erlotinib and to proceed with caution.1
UDP-glucuronosyltransferases
Erlotinib is a selective and potent competitive inhibitor of glucuronidation by UGT1A1 in vitro and exerts potent mixed inhibition of bilirubin glucuronidation in human liver microsomes.63 Based on these findings, co-administration of erlotinib (≥100 mg/day) is predicted to increase the AUC of drugs predominantly cleared by UGT1A1 by ≥30% and to cause clinically significant DDIs when given with such agents.63 Cheng et al (2017) found erlotinib to be a potent noncompetitive inhibitor of UGT1A1.62 Patients with low UGT1A1 expression or genetic glucuronidation disorders (eg, Gilbert’s disease) could develop high serum concentrations of bilirubin and must be treated with caution.94
Transporter proteins
In vitro transport studies demonstrated that erlotinib is a substrate for, and inhibitor of, both P-gp and BCRP.114–116 Concomitant administration of P-gp inhibitors such as cyclosporine and verapamil may lead to altered distribution and/or altered elimination of erlotinib.94 The clinical relevance of this interaction is unclear,5 but clinicians should be aware of the potential for an increase in adverse events when using erlotinib in the presence of P-gp inhibitors.94
Erlotinib and its active metabolite OSI-420 are substrates for human organic anion transporter 3 (OAT3) and, to a lesser extent, organic cation transporter 2 (OCT2)116 but the clinical implications of these properties have not been fully elucidated.2
Other clinically relevant interactions
Erlotinib can increase the INR in patients taking warfarin.117 Bleeding events have been reported (including peptic ulcer bleeding, hematemesis, hematochezia, melena, and hemorrhage from possible colitis),93 some of which were fatal.94 Patients taking erlotinib with coumarin-derived anticoagulants such as warfarin should be monitored regularly for changes in prothrombin time or INR.93,94 Dose modifications are not recommended for erlotinib.93
Concomitant use of capecitabine may increase plasma erlotinib concentrations. When combined with capecitabine, there was a significant increase in erlotinib AUC and a borderline increase in Cmax (compared with concentrations measured in another study of erlotinib monotherapy).94
In patients with advanced solid tumors, carboplatin exposure was reported to increase when administered concomitantly with erlotinib.111 However, in an intensive PK study of patients with advanced NSCLC who had participated in a phase III trial of first-line erlotinib plus chemotherapy, the use of erlotinib did not alter systemic exposure of paclitaxel and carboplatin compared with that in the placebo group.118
Afatinib
Afatinib is an oral, irreversible inhibitor of the ErbB family of tyrosine kinases. Afatinib downregulates ErbB signaling by covalently binding to the kinase domains of EGFR, HER2, and HER4, leading to irreversible inhibition of tyrosine kinase autophosphorylation; afatinib also inhibits transphosphorylation of HER3.119 Afatinib is approved by the FDA for first-line treatment of patients with metastatic NSCLC who harbor nonresistant EGFR mutations, as detected by an FDA-approved test. It is also approved for locally advanced or metastatic NSCLC of squamous histology progressing after platinum-based chemotherapy.120,121
Acid-reducing agents
Afatinib is highly soluble throughout the physiologic pH range (1–7.5).122 Consequently, interactions with acid-reducing drugs are not expected.5
Cytochrome P450-dependent metabolism
Afatinib undergoes minimal biotransformation, and oxidative CYP-mediated metabolism is of negligible importance.5,123 Metabolism is mainly governed by non-enzyme catalyzed formation of adducts to proteins and nucleophilic small molecules.5,123 Consequently, DDIs arising from inhibition or induction of CYP450 enzymes by concomitant medications are unlikely to occur.121 Smoking status has no significant effect on exposure to afatinib.5,124
Transporter proteins
Afatinib is a substrate and inhibitor of P-gp in vitro,5,125 and concomitant use of strong P-gp inhibitors can increase exposure to afatinib.120,121 In healthy subjects, ritonavir (a strong inhibitor of P-gp and BCRP) given simultaneously or 6 hrs after a single 40 mg dose of afatinib led to minimal increases in afatinib AUC0–∞ and Cmax (by 5% and 11% respectively).125 However, in a second study, ritonavir given 1 hr before a single 20 mg dose of afatinib led to a 48% increase in afatinib AUC0–∞ and a 39% increase in Cmax.125 Conversely, strong P-gp inducers can reduce exposure to afatinib.120 In healthy subjects, pretreatment with the potent P-gp inducer rifampicin (600 mg QD for 7 days) before a single 40 mg dose of afatinib led to a reduction in plasma exposure (34% reduction in AUC0–∞ and 22% reduction in Cmax).121,125
For patients taking afatinib who require treatment with a P-gp inhibitor, the EMA label recommends using staggered dosing to maximize the interval between the doses of afatinib and the P-gp inhibitor (preferably 6 hrs for P-gp inhibitors dosed BID and 12 hrs for those given QD).120 According to the US label, if a patient taking a concomitant P-gp inhibitor experiences toxicities while taking afatinib, their clinician may reduce the afatinib dose by 10 mg, and resume the original dose after discontinuation of the P-gp inhibitor, provided tolerability is acceptable.121 For those taking a P-gp inducer, the afatinib dose may be increased by 10 mg, subject to tolerability, and the original dose may be resumed 2–3 days after the P-gp inducer is discontinued.121
Afatinib is a moderate inhibitor of P-gp in vitro,5,125 but clinical data suggest that changes in plasma concentrations of other P-gp substrates are unlikely to occur due to concomitant administration of afatinib.120,126
Afatinib is both a substrate and an inhibitor of BCRP in vitro120,121,126 and may increase the bioavailability of BCRP substrates administered orally, such as rosuvastatin and sulfasalazine.120
Osimertinib
Osimertinib is a third-generation potent irreversible EGFR-TKI that has efficacy in patients with advanced NSCLC with EGFR mutations (both sensitizing/activating mutations (del19/L858R) and T790M resistance mutations).47 In the USA, osimertinib is indicated for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, as detected by an FDA-approved test, who have progressed on or after EGFR TKI therapy, and also for first-line treatment of patients with metastatic NSCLC whose tumors have EGFR exon 19 deletions or exon 21 L858R mutations, as detected by an FDA-approved test.127 Similarly, in the EU, osimertinib is indicated for the treatment of adults with locally advanced or metastatic EGFR T790M mutation-positive NSCLC; also for first-line treatment of adults with locally advanced or metastatic NSCLC with activating EGFR mutations.128
Acid-reducing agents
In a preclinical study, the AUC0–3h of osimertinib (5 mg/kg p.o.) was not significantly affected by concomitant omeprazole (10–100 mg/kg p.o.) or vonoprazan (1–5 mg/kg p.o.), both of which caused significant dose-dependent increases in gastric pH.75 In an open-label study in healthy male volunteers (n=68), co-administration of omeprazole did not significantly alter osimertinib exposure: the geometric least-squares mean ratio [90% CI] for AUC was 107% [100–113%] and for Cmax was 102% [95–109%].129
In patients whose gastric pH may be altered by concomitant agents or medical conditions, dose modifications are not required for osimertinib.129 Gastric pH-modifying agents can be used with osimertinib without restriction.128
CYP450-dependent metabolism
In vitro studies indicate that osimertinib is predominantly metabolized by CYP3A4/5 and is a weak inducer of CYP3A.130 Hence, modulators of CYP3A could impact osimertinib metabolism, while osimertinib may alter the exposure of other CYP3A substrates.130
Drug interaction studies with inhibitors, inducers, or substrates of CYP enzymes and transporters have not been conducted systematically for osimertinib.127 The effect of strong CYP3A4 inhibitors and inducers on the PK of osimertinib in patients with advanced NSCLC was investigated in two open-label studies.131 In the first study of 36 patients, concomitant use of the strong CYP3A4 inhibitor itraconazole (200 mg BID; days 6–18) together with osimertinib (80 mg/day, days 1 and 10) had no clinically significant effect on osimertinib exposure; AUC increased by 24% and Cmax decreased by 20% versus osimertinib given alone128,131 (the upper bounds of the 90% CIs of the geometric mean least square mean treatment ratios [itraconazole + osimertinib/osimertinib alone] for AUC and Cmax were both below the pre-specified “no-effect” limit of 200%). Similarly, there were no clinically relevant changes in exposure parameters for the active metabolite of osimertinib, AZ5104. The authors suggested that preclinical hepatocyte and recombinant CYP studies132 may have overestimated the contribution of cytochrome P450 metabolism to the clearance of osimertinib in the clinic, whereas the availability of multiple elimination pathways for osimertinib might also explain the lack of significant effects of itraconazole.131 The minor reduction in osimertinib Cmax and increase in AUC were interpreted as being due to inhibition of CYP3A by itraconazole, leading to changes in elimination of osimertinib and its metabolites. Osimertinib tmax was 2 hrs longer in patients taking osimertinib plus itraconazole than those taking osimertinib alone (p=0.0002), which suggests that concomitant use of itraconazole may alter the absorption of osimertinib.131
In the second study (n=40), concomitant use of osimertinib (80 mg/day, days 1–77) with the CYP3A4 inducer rifampicin (600 mg/day, days 29–49) led to a 78% reduction in osimertinib AUC as well as an 82% reduction in AUC and 78% reduction in Cmax of AZ5104, the metabolite of osimertinib.128,131 Although the proportion of white patients was slightly higher in the rifampicin study than the itraconazole study, the authors felt that this was unlikely to have affected the results given that osimertinib exposure does not appear to be affected by ethnicity.133
Consistent with the US and EU labels,127,128 the researchers concluded that osimertinib can be given concurrently with CYP3A4 inhibitors but that strong CYP3A inducers should be avoided if possible.131 Concomitant use of St. John’s Wort with osimertinib is specifically contraindicated in the EU.128 The US label advises that if concurrent use of a strong CYP3A4 inducer is unavoidable, the dose of osimertinib should be increased to 160 mg/day; the standard dose (80 mg/day) may be resumed 3 weeks after discontinuation of the CYP3A4 inducer.127 Moderate CYP3A4 inducers may also reduce osimertinib exposure, so should be used with caution or avoided if possible.128 No dose adjustments are required when osimertinib is used with moderate and/or weak CYP3A inducers.127
In patients with EGFR mutation-positive NSCLC following disease progression on a prior EGFR-TKI, daily administration of osimertinib increased rosuvastatin exposure but had minimal effects on exposure of the sensitive CYP3A4 substrate, simvastatin.134 Clinically relevant interactions between osimertinib and CYP3A4 substrates are therefore unlikely.128
In a population PK analysis based on data from 780 patients, Brown et al (2017) found that smoking status had no significant effect on osimertinib PK (dose-normalized AUC at steady state),133 which suggests that CYP1A1 induction does not have a major effect on osimertinib metabolism. Only 3% of the patients were current smokers, which limited the strength of the analysis. Nevertheless, no dosage adjustments are required when treating current smokers with osimertinib.128
UDP-glucuronosyltransferases
Based on in vitro studies, osimertinib is not an inhibitor of UGT1A1 or UGT2B7 at clinically relevant concentrations. Intestinal inhibition of UGT1A1 is possible, but the clinical impact is unknown.128
Transporter proteins
Osimertinib is a substrate of P-gp and BCRP in vitro,135 but this is unlikely to lead to clinically significant DDIs at clinically relevant doses.128 Osimertinib is a competitive inhibitor of BCRP transporters in vitro,128,130 and it may, therefore, increase the exposure of BCRP substrates.128,130 Concomitant administration of osimertinib led to a 35% increase in the AUC and a 72% increase in the Cmax of rosuvastatin (a sensitive BCRP substrate).134 Patients taking osimertinib with medications that have a narrow therapeutic index and BCRP-dependent disposition should be closely monitored for changes in tolerability due to increases in osimertinib exposure.127,128 At clinically relevant concentrations, osimertinib is not a substrate or inhibitor of OATP1B1 or OATP1B3 in vitro.128
Icotinib
Icotinib is a second-generation reversible EGFR-TKI, approved by the China Food and Drug Administration (CFDA) for the treatment of advanced NSCLC following progression on at least one platinum-based chemotherapy.18,29,136,137 To date, relatively few studies have reported on potential DDIs with icotinib.138
CYP450-dependent metabolism
A preclinical pharmacokinetic study and a clinical mass balance study showed that more than 90% of icotinib is eliminated by hepatic metabolism, primarily via CYP450 enzymes; four to six main metabolites were identified.139 The main enzymes responsible for icotinib metabolism are CYP3A4, CYP2C19, CYP3A5, and CYP1A2.138,139 According to Shi et al (2013), the involvement of several enzymes in the metabolism of icotinib means that accumulation of the drug is limited, and explains its relatively short half-life (6 h140), which is one of the main differences between icotinib and the other EGFR-TKIs.40
Zhang et al (2018) recently examined the formation of icotinib metabolites by recombinant CYP isozymes in human liver microsomes, to identify the enzymes responsible for icotinib metabolism.138 The metabolic pathways identified in vitro predominantly involved CYP3A4 (accounting for 77–87% of icotinib metabolism), CYP3A5 (5–15%), and CYP1A2 (3.7–7.5%). Metabolism of icotinib via CYP450 2C8, 2C9, 2C19, and 2D6 was insignificant. The authors recommended that clinicians should consider the risk of DDIs when prescribing icotinib with strong CYP3A inhibitors or inducers.138 Induction of CYP1A2 in lung cancer patients with smoking history may also contribute to the PK and pharmacologic variability of icotinib.138
Chen et al (2015) used a physiologically based PK model (validated using data from a phase I trial of icotinib in healthy Chinese subjects) to simulate DDIs with ketoconazole and rifampin (a potent CYP3A4 inhibitor/inducer, respectively). The model-predicted exposure (AUC) for icotinib was higher when given with ketoconazole (400 mg) and rifampin (600 mg) than when given alone; the AUC ratio for icotinib during concomitant use of ketoconazole and rifampin was 3.22 and 0.55, respectively.139
UDP-glucuronosyltransferases
Cheng et al (2017)62 found both icotinib and erlotinib to be non-competitive inhibitors of UGT1A1, but the effect of icotinib was weaker than that of erlotinib (IC50 for inhibition of UGT1A1-mediated NCHN-O-glucuronidation in human liver microsomes was 5.15 μmol/L for icotinib vs 0.68 μmol/L for erlotinib). The authors concluded that use of icotinib is unlikely to lead to clinically significant DDIs due to inhibition of UGT1A1.62
DDIs: aids for clinical decision-making
The growing awareness of the importance of DDIs in the treatment of cancer patients has been reflected by increases in the number and scope of the sources of information and guidance available to aid clinical decision-making.141 Some useful online resources are now available. “Oncology in Practice” (http://oncologypro.esmo.org/Oncology-in-Practice) from the European Society of Medical Oncology includes an overview of the main DDIs for the most frequently used TKIs, prophylaxis and treatment of these DDIs, and information for patients. “Drugs.com” includes a drug interaction checker at https://www.drugs.com/drug_interactions.html, which allows users to specify the agents in a prescription and to obtain information on potential interactions. “SiteGPR” (http://sitegpr.com/fr/) provides evidence-based advice (in French) on dose adjustments for patients with renal impairment, including dose adjustments due to DDIs.141 “Cancer Drug Interactions” (https://cancer-druginteractions.org/), which is endorsed by the British Oncology Pharmacy Association, allows users to select from a list of anticancer drugs and commonly prescribed concomitant medications and obtain information on whether a DDI is likely, together with the rationale and quality of evidence. Watson for Oncology (https://www.ibm.com/watson/health/oncology-and-genomics/oncology/) is an artificial intelligence system that extracts data from medical records and provides evidence-based treatment options tailored to the individual patient.142,143 Lexicomp Online (http://www.lexi.com) and Micromedex 2.0 (http://micromedex.com) both include interactive tools for evaluation of drug interactions.144 Interestingly, Muhič et al (2017) found that different DDI screening systems may differ significantly in their ability to detect clinically relevant DDI-related adverse drug reactions.145 Notably, pharmacy information experts recommend that to address such questions, multiple sources of information should be consulted.144
Summary and recommendations for clinical practice
The development of the EGFR-TKIs has changed the therapeutic landscape of NSCLC and raised expectations among both patients and physicians. The introduction of these drugs into clinical practice presents challenges for physicians, however, not least due to the risk of DDIs with some agents. When formulating individualized therapeutic strategies for their patients, physicians should be aware of how differences between the PK properties of the different EGFR-TKIs may affect the potential for DDIs and, consequently, the efficacy, optimum dose, and tolerability of the treatment regimen. Moreover, given that cancer patients are often highly polymedicated, physicians must always bear in mind the potential impact of concomitant medications when selecting treatment and addressing the management of side effects. The most important interactions for physicians to be aware of, in terms of their significance to the treatment of patients with NSCLC, are described in Table 2; recommended approaches to managing such interactions are summarized below.
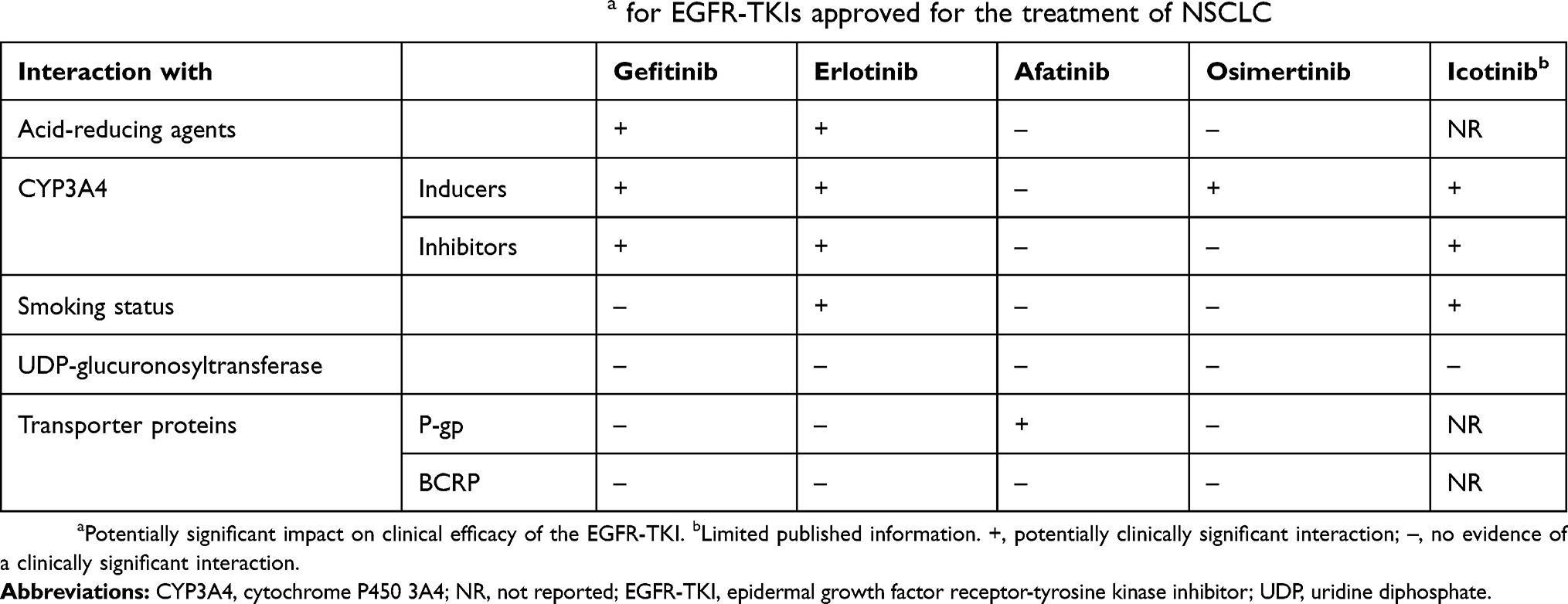 |
Table 2 Potentially clinically significant interactionsa for EGFR-TKIs approved for the treatment of NSCLC |
Clinically significant interactions with acid-suppressive drugs (PPIs, H2-receptor antagonists, and antacids) have been demonstrated for EGFR-TKIs that exhibit pH-dependent solubility (ie, gefitinib and erlotinib). When used concomitantly, bioavailability may be reduced, to such an extent that clinical efficacy may be significantly impaired. As patients taking EGFR-TKIs often experience gastrointestinal side effects and routinely use acid-reducing agents for palliation of gastro-esophageal reflux, dyspepsia, gastritis, and mucositis, these DDIs are clinically relevant. If concomitant use is unavoidable, then staggering the dose of the EGFR-TKI and the acid suppressant by several hours may help to reduce the extent of the interaction. Another option would be to prescribe afatinib, which is not subject to this type of interaction (no information is available for icotinib). To manage a clinically significant DDI, a twice-daily PPI could be replaced by a once-daily regimen. Giving the EGFR-TKI 2 hrs before the PPI (and using an enteric-coated formulation of the PPI) should optimize absorption of the EGFR-TKI.
Physicians should exercise caution when prescribing a known CYP enzyme inhibitor or inducer, as concomitant drugs may need to be substituted or the doses adjusted to account for potential reductions or increases in CYP enzyme-mediated metabolism. In addition to the agents mentioned earlier, commonly used CYP450 inhibitors include: amiodarone, cimetidine (1A2); clarithromycin, diltiazem, grapefruit juice, telithromycin (3A4/3A5); amiodarone, fluconazole, fluoxetine, metronidazole, trimethoprim, sulfamethoxazole (2C9); isoniazid (2C19); and amiodarone, cimetidine, diphenhydramine, fluoxetine, paroxetine, quinidine, terbinafine (2D6). Furthermore, phenobarbital is an inducer of CYP1A2, 3A4/5, and 2C9. A table of important substrates, inhibitors, and inducers (with direct links to PubMed citations) is continually updated by Indiana University School of Medicine and can be accessed at https://drug-interactions.medicine.iu.edu/Home.aspx.146
Gefitinib, erlotinib, osimertinib, and icotinib are predominantly metabolized by CYP3A4; consequently, concomitant administration with a potent CYP3A4 inhibitor may substantially increase plasma concentrations of the EGFR-TKIs. Conversely, co-administration with a strong CYP3A4 inducer may increase EGFR-TKI metabolism, reduce plasma concentrations, and consequently reduce efficacy. Clinicians should take care when treating patients with CYP3A4 inducers, and if an interaction is anticipated, concomitant administration should be avoided if possible.
Plasma concentrations of erlotinib are markedly reduced in smokers; therefore, while receiving erlotinib, current smokers should be advised to stop smoking. For those who continue to smoke, it may be necessary to increase the dose of erlotinib (to a maximum of 300 mg/day). Induction of CYP1A2 in smokers may also influence the metabolism of icotinib.
Gefitinib, erlotinib, osimertinib, and afatinib are substrates for the drug transporter P-gp in vitro (no information is available for icotinib), but clinical findings indicate that clinically relevant DDIs may occur with afatinib only (not with gefitinib, erlotinib or osimertinib). Clinicians should consider staggering or adjusting the dose of afatinib when used in combination with a P-gp inhibitor or inducer.
Abbreviation list
ABC, ATP-binding cassette; AUC, area under the plasma concentration–time curve; BBB, blood–brain barrier; BID, twice daily; Cmax, maximum plasma concentration; CYP, cytochrome P450; DDI, drug–drug interaction; EGFR, epidermal growth factor receptor; EMA, European Medicines Agency; FDA, Food and Drug Administration; INR, international normalized ratio; MTD, maximum tolerated dose; NR, not reported; NSCLC, non-small-cell lung cancer; OAT3, organic anion transporter 3; OATP, organic anion-transporting peptide; OCT2, organic cation transporter 2; OCTP, organic cation-transporting peptide; OS, overall survival; PFS, progression-free survival; P-gp, P-glycoprotein; PK, pharmacokinetic[s]; p.o., orally; PPI, proton pump inhibitor; QD, once daily; TKI, tyrosine kinase inhibitor; UDP, uridine diphosphate; UGT, UDP-glucuronosyltransferase.
Acknowledgments
Medical writing assistance during the preparation of this article was financially supported by Boehringer Ingelheim and was provided by Michael Simpson, PhD, CMPP of GeoMed, an Ashfield company, part of UDG Healthcare plc. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and approved the final version.
Disclosure
Dr Junling Li reports grants from Boehringer Ingelheim for medical writing assistance during the preparation of this article. Dr Ziyi Xu reports grants from Boehringer Ingelheim for medical writing assistance during the preparation of this article. The authors report no other conflicts of interest in this work.
References
1. van Leeuwen RWF, van Gelder T, Mathijssen RHJ, Jansman FGA. Drug-drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol. 2014;15(8):e315–e326. doi:10.1016/S1470-2045(13)70579-5
2. Terada T, Noda S, Inui K. Management of dose variability and side effects for individualized cancer pharmacotherapy with tyrosine kinase inhibitors. Pharmacol Ther. 2015;152:125–134. doi:10.1016/j.pharmthera.2015.05.009
3. Relling MV, Pui CH, Sandlund JT, et al. Adverse effect of anticonvulsants on efficacy of chemotherapy for acute lymphoblastic leukaemia. Lancet. 2000;356(9226):285–290. doi:10.1016/S0140-6736(00)02503-4
4. Zhang N, Liu Y, Jeong H. Drug-drug interaction potentials of tyrosine kinase inhibitors via inhibition of UDP-glucuronosyltransferases. Sci Rep. 2015;5:17778. doi:10.1038/srep17778
5. Peters S, Zimmermann S, Adjei AA. Oral epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: comparative pharmacokinetics and drug-drug interactions. Cancer Treat Rev. 2014;40(8):917–926. doi:10.1016/j.ctrv.2014.06.010
6. Roskoski R
7. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12(8):553–563. doi:10.1038/nrc3309
8. Zer A, Leighl N. Promising targets and current clinical trials in metastatic non-squamous NSCLC. Front Oncol. 2014;4:329. doi:10.3389/fonc.2014.00329
9. Solomon B, Wilner KD, Shaw AT. Current status of targeted therapy for anaplastic lymphoma kinase-rearranged non-small cell lung cancer. Clin Pharmacol Ther. 2014;95(1):15–23. doi:10.1038/clpt.2013.200
10. Yap TA, Popat S. Toward precision medicine with next-generation EGFR inhibitors in non-small-cell lung cancer. Pharmgenomics Pers Med. 2014;7:285–295. doi:10.2147/PGPM.S55339
11. Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4(1):36–54. doi:10.3978/j.issn.2218-6751.2014.05.01
12. Reguart N, Remon J. Common EGFR-mutated subgroups (Del19/L858R) in advanced non-small-cell lung cancer: chasing better outcomes with tyrosine kinase inhibitors. Future Oncol. 2015;11(8):1245–1257. doi:10.2217/fon.15.15
13. Carrera S, Buque A, Azkona E, et al. Epidermal growth factor receptor tyrosine-kinase inhibitor treatment resistance in non-small cell lung cancer: biological basis and therapeutic strategies. Clin Transl Oncol. 2014;16(4):339–350. doi:10.1007/s12094-013-1143-9
14. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–S31. doi:10.1038/onc.2009.198
15. Mok T, Lee K, Tang M, Leung L. Dacomitinib for the treatment of advanced or metastatic non-small-cell lung cancer. Future Oncol. 2014;10(5):813–822. doi:10.2217/fon.14.22
16. Hirsh V. Next-generation covalent irreversible kinase inhibitors in NSCLC: focus on afatinib. BioDrugs. 2015;29(3):167–183. doi:10.1007/s40259-015-0130-9
17. Sullivan I, Planchard D. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. Front Med (Lausanne). 2016;3:76.
18. Tan F, Shi Y, Wang Y, Ding L, Yuan X, Sun Y. Icotinib, a selective EGF receptor tyrosine kinase inhibitor, for the treatment of non-small-cell lung cancer. Future Oncol. 2015;11(3):385–397. doi:10.2217/fon.14.249
19. Hanna N, Johnson D, Temin S, et al. Systemic therapy for stage IV non-small-cell lung cancer: American Society of Clinical Oncology Clinical Practice Guideline update. J Clin Oncol. 2017;35(30):3484–3515. doi:10.1200/JCO.2017.74.6065
20. Planchard D, Popat S, Kerr K, et al. Metastatic non-small cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Supplement_4):iv192–iv237. doi:10.1093/annonc/mdy275
21. Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883–1889. doi:10.1093/annonc/mdv270
22. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi:10.1016/S1470-2045(11)70393-X
23. Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. doi:10.1016/S1470-2045(11)70184-X
24. Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. doi:10.1016/S1470-2045(09)70364-X
25. Park K, Tan EH, O‘Byrne K, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016;17(5):577–589. doi:10.1016/S1470-2045(16)30033-X
26. Wu YL, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18(11):1454–1466. doi:10.1016/S1470-2045(17)30608-3
27. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi:10.1056/NEJMoa0909530
28. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi:10.1056/NEJMoa0810699
29. Biaoxue R, Hua L, Wenlong G, Shuanying Y. Efficacy and safety of icotinib in treating non-small cell lung cancer: a systematic evaluation and meta-analysis based on 15 studies. Oncotarget. 2016;7(52):86902–86913. doi:10.18632/oncotarget.13509
30. Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. doi:10.1200/JCO.2012.44.2806
31. Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–222. doi:10.1016/S1470-2045(13)70604-1
32. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi:10.1056/NEJMoa1713137
33. Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res. 2006;12(24):7232–7241. doi:10.1158/1078-0432.CCR-06-0658
34. Han JY, Park K, Kim SW, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol. 2012;30(10):1122–1128. doi:10.1200/JCO.2011.36.8456
35. Cheng X, Chen H. Tumor heterogeneity and resistance to EGFR-targeted therapy in advanced nonsmall cell lung cancer: challenges and perspectives. Onco Targets Ther. 2014;7:1689–1704.
36. Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17(5):1169–1180. doi:10.1158/1078-0432.CCR-10-2277
37. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi:10.1371/journal.pmed.0020073
38. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi:10.1126/scitranslmed.3002003
39. Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66(16):7854–7858. doi:10.1158/0008-5472.CAN-06-1951
40. Shi Y, Zhang L, Liu X, et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013;14(10):953–961. doi:10.1016/S1470-2045(13)70355-3
41. Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342–350. doi:10.1124/jpet.112.197756
42. Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. doi:10.1038/onc.2008.109
43. Hirsh V. New developments in the treatment of advanced squamous cell lung cancer: focus on afatinib. Onco Targets Ther. 2017;10:2513–2526. doi:10.2147/OTT.S104177
44. Yang JC, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141–151. doi:10.1016/S1470-2045(14)71173-8
45. Paz-Ares L, Tan EH, O‘Byrne K, et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial. Ann Oncol. 2017;28(2):270–277. doi:10.1093/annonc/mdw611
46. Ward RA, Anderton MJ, Ashton S, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J Med Chem. 2013;56(17):7025–7048. doi:10.1021/jm400822z
47. Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. doi:10.1158/2159-8290.CD-14-0337
48. Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. doi:10.1056/NEJMoa1411817
49. Tan CS, Cho BC, Soo RA. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor -mutant non-small cell lung cancer. Lung Cancer. 2016;93:59–68. doi:10.1016/j.lungcan.2016.01.003
50. van Leeuwen RWF, Jansman FGA, Hunfeld NG, et al. Tyrosine kinase inhibitors and proton pump inhibitors: an evaluation of treatment options. Clin Pharmacokinet. 2017;56(7):683–688. doi:10.1007/s40262-016-0503-3
51. Budha NR, Frymoyer A, Smelick GS, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92(2):203–213. doi:10.1038/clpt.2012.73
52. Petros WP, Younis IR, Ford JN, et al. Effects of tobacco smoking and nicotine on cancer treatment. Pharmacotherapy. 2012;32(10):920–931. doi:10.1002/j.1875-9114.2012.01117
53. Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286(5439):487–491.
54. Eichelbaum M, Ingelman-Sundberg M, Evans WE. Pharmacogenomics and individualized drug therapy. Annu Rev Med. 2006;57:119–137. doi:10.1146/annurev.med.56.082103.104724
55. Pharmacogene Variation Consortium (PharmaVar). CYP2D6: cytochrome P450 family 2 subfamily D member 6. Verison 3.3 2018. Available from: https://www.pharmvar.org/gene/CYP2D6
56. Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5(1):6–13. doi:10.1038/sj.tpj.6500285
57. Samer CF, Lorenzini KI, Rollason V, et al. Applications of CYP450 testing in the clinical setting. Mol Diagn Ther. 2013;17(3):165–184. doi:10.1007/s40291-013-0028-5
58. Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007;116(3):496–526. doi:10.1016/j.pharmthera.2007.09.004
59. McGraw J, Waller D. Cytochrome P450 variations in different ethnic populations. Expert Opin Drug Metab Toxicol. 2012;8(3):371–382. doi:10.1517/17425255.2012.657626
60. Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics. 2002;3(2):229–243. doi:10.1517/14622416.3.2.229
61. Fujiwara R, Yokoi T, Nakajima M. Structure and protein-protein interactions of human UDP-glucuronosyltransferases. Front Pharmacol. 2016;7(1):388. doi:10.3389/fphar.2016.00323
62. Cheng X, Lv X, Qu H, et al. Comparison of the inhibition potentials of icotinib and erlotinib against human UDP-glucuronosyltransferase 1A1. Acta Pharm Sin B. 2017;7(6):657–664. doi:10.1016/j.apsb.2017.07.004
63. Liu Y, Ramirez J, House L, et al. Comparison of the drug-drug interactions potential of erlotinib and gefitinib via inhibition of UDP-glucuronosyltransferases. Drug Metab Dispos. 2010;38(1):32–39. doi:10.1124/dmd.109.029660
64. Miners JO, Chau N, Rowland A, et al. Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: implications for hyperbilirubinemia. Biochem Pharmacol. 2017;129(1):85–95. doi:10.1016/j.bcp.2017.01.002
65. Ai L, Zhu L, Yang L, et al. Selectivity for inhibition of nilotinib on the catalytic activity of human UDP-glucuronosyltransferases. Xenobiotica. 2014;44(4):320–325. doi:10.3109/00498254.2013.840750
66. Shah RR, Morganroth J, Shah DR. Hepatotoxicity of tyrosine kinase inhibitors: clinical and regulatory perspectives. Drug Saf. 2013;36(7):491–503. doi:10.1007/s40264-013-0048-4
67. Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y. Impact of drug transporter studies on drug discovery and development. Pharmacol Rev. 2003;55(3):425–461. doi:10.1124/pr.55.3.1
68. Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75(1):13–33. doi:10.1016/j.clpt.2003.09.012
69. Oostendorp RL, Beijnen JH, Schellens JH. The biological and clinical role of drug transporters at the intestinal barrier. Cancer Treat Rev. 2009;35(2):137–147. doi:10.1016/j.ctrv.2008.09.004
70. van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35(8):692–706. doi:10.1016/j.ctrv.2009.08.004
71. Cohen MH, Williams GA, Sridhara R, et al. United States Food and Drug Administration drug approval summary: gefitinib (ZD1839; iressa) tablets. Clin Cancer Res. 2004;10(4):1212–1218.
72. Food and Drug Administration. Iressa (gefitinib) Prescribing Information. 2015.
73. European Medicines Agency. Iressa (gefitinib) Summary of Product Characteristics. 2017.
74. European Medicines Agency. Iressa (gefitinib) assessment report. 2009. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001016/WC500036361.pdf. Accessed November 14, 2018.
75. Yasumuro O, Uchida S, Kashiwagura Y, et al. Changes in gefitinib, erlotinib and osimertinib pharmacokinetics under various gastric pH levels following oral administration of omeprazole and vonoprazan in rats. Xenobiotica. 2017;1:1–7.
76. Tang W, Tomkinson H, Masson E. Effect of sustained elevated gastric pH levels on gefitinib exposure. Clin Pharmacol Drug Dev. 2017;6(5):517–523. doi:10.1002/cpdd.337
77. Saeki S, Sasaki J, Morioka J, et al. The influence of gastric secretion inhibitors on gefitinib therapy in patients with non-small cell lung cancer harboring epidermal growth factor receptor activating mutations. J Clin Oncol. 2012;30(15_suppl):e18059.
78. Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13(12):3731–3737. doi:10.1158/1078-0432.CCR-07-0088
79. McKillop D, McCormick AD, Millar A, Miles GS, Phillips PJ, Hutchison M. Cytochrome P450-dependent metabolism of gefitinib. Xenobiotica. 2005;35(1):39–50. doi:10.1080/00498250400026464
80. McKillop D, Hutchison M, Partridge EA, et al. Metabolic disposition of gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, in rat, dog and man. Xenobiotica. 2004;34(10):917–934. doi:10.1080/00498250400009171
81. Swaisland HC, Ranson M, Smith RP, et al. Pharmacokinetic drug interactions of gefitinib with rifampicin, itraconazole and metoprolol. Clin Pharmacokinet. 2005;44(10):1067–1081. doi:10.2165/00003088-200544100-00005
82. Chhun S, Verstuyft C, Rizzo-Padoin N, et al. Gefitinib-phenytoin interaction is not correlated with the C-erythromycin breath test in healthy male volunteers. Br J Clin Pharmacol. 2009;68(2):226–237. doi:10.1111/j.1365-2125.2009.03438.x
83. Hwang SW, Han HS, Lim KY, Han J-Y. Drug interaction between complementary herbal medicines and gefitinib. J Thorac Oncol. 2008;3(8):942–943. doi:10.1097/JTO.0b013e3181803f1e
84. Swaisland HC, Cantarini MV, Fuhr R, Holt A. Exploring the relationship between expression of cytochrome P450 enzymes and gefitinib pharmacokinetics. Clin Pharmacokinet. 2006;45(6):633–644. doi:10.2165/00003088-200645060-00006
85. Kitazaki T, Oka M, Nakamura Y, et al. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer. 2005;49(3):337–343. doi:10.1016/j.lungcan.2005.03.035
86. Stewart CF, Leggas M, Schuetz JD, et al. Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Res. 2004;64(20):7491–7499. doi:10.1158/0008-5472.CAN-04-0096
87. Furman WL, Navid F, Daw NC, et al. Tyrosine kinase inhibitor enhances the bioavailability of oral irinotecan in pediatric patients with refractory solid tumors. J Clin Oncol. 2009;27(27):4599–4604. doi:10.1200/JCO.2008.19.6642
88. Li J, Cusatis G, Brahmer J, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6(3):432–438.
89. Arai S, Mitsufuji H, Nishii Y, et al. Effect of gefitinib on warfarin antithrombotic activity. Int J Clin Oncol. 2009;14(4):332–336. doi:10.1007/s10147-008-0871-2
90. Adjei AA, Molina JR, Mandrekar SJ, et al. Phase I trial of sorafenib in combination with gefitinib in patients with refractory or recurrent non-small cell lung cancer. Clin Cancer Res. 2007;13(9):2684–2691. doi:10.1158/1078-0432.CCR-06-2889
91. Pujol JL, Viens P, Rebattu P, et al. Gefitinib (Iressa) with vinorelbine or vinorelbine/cisplatin for chemotherapy-naive non-small cell lung cancer patients. J Thorac Oncol. 2006;1(5):417–424.
92. Gioulbasanis I, Saridaki Z, Kalykaki A, et al. Gefitinib in combination with gemcitabine and vinorelbine in patients with metastatic breast cancer pre-treated with taxane and anthracycline chemotherapy: a phase I/II trial. Anticancer Res. 2008;28(5b):3019–3025.
93. Food and Drug Administration. Tarceva (erlotinib) Prescribing Information. 2016.
94. European Medicines Agency. Tarceva (erlotinib) Summary of Product Characteristics. 2018.
95. Kletzl H, Giraudon M, Ducray PS, Abt M, Hamilton M, Lum BL. Effect of gastric pH on erlotinib pharmacokinetics in healthy individuals: omeprazole and ranitidine. Anticancer Drugs. 2015;26(5):565–572. doi:10.1097/CAD.0000000000000212
96. Hilton JF, Tu D, Seymour L, Shepherd FA, Bradbury PA. An evaluation of the possible interaction of gastric acid suppressing medication and the EGFR tyrosine kinase inhibitor erlotinib. Lung Cancer. 2013;82(1):136–142. doi:10.1016/j.lungcan.2013.06.008
97. Chu MP, Ghosh S, Chambers CR, et al. Gastric acid suppression is associated with decreased erlotinib efficacy in non-small-cell lung cancer. Clin Lung Cancer. 2015;16(1):33–39. doi:10.1016/j.cllc.2014.07.005
98. Zenke Y, Yoh K, Matsumoto S, et al. Clinical impact of gastric acid-suppressing medication use on the efficacy of erlotinib and gefitinib in patients with advanced non-small-cell lung cancer harboring EGFR mutations. Clin Lung Cancer. 2016;17(5):412–418. doi:10.1016/j.cllc.2016.01.006
99. Kumarakulasinghe NB, Syn N, Soon YY, et al. EGFR kinase inhibitors and gastric acid suppressants in EGFR-mutant NSCLC: a retrospective database analysis of potential drug interaction. Oncotarget. 2016;7(51):85542–85550. doi:10.18632/oncotarget.13458
100. Nieves Sedano M, Manuel Caro Teller J, Garcia MC, et al. Clinical impact of gastric acid suppressing medication on the effectiveness of tyrosine kinase inhibitors in lung cancer patients. J BUON. 2018;23(3):647–653.
101. Ling J, Johnson KA, Miao Z, et al. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab Dispos. 2006;34(3):420–426. doi:10.1124/dmd.105.007765
102. Johnson JR, Cohen M, Sridhara R, et al. Approval summary for erlotinib for treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen. Clin Cancer Res. 2005;11(18):6414–6421. doi:10.1158/1078-0432.CCR-05-0790
103. Kletzl H, Zwanziger E, Kirkpatrick C, et al. Effect of ciprofloxacin on the systemic exposure to erlotinib. J Clin Oncol. 2008;26(15_suppl):19047. doi:10.1200/JCO.2007.15.2777
104. Hamilton M, Wolf JL, Drolet DW, et al. The effect of rifampicin, a prototypical CYP3A4 inducer, on erlotinib pharmacokinetics in healthy subjects. Cancer Chemother Pharmacol. 2014;73(3):613–621. doi:10.1007/s00280-014-2390-3
105. Hamilton M, Wolf JL, Rusk J, et al. Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res. 2006;12(7 Pt 1):2166–2171. doi:10.1158/1078-0432.CCR-05-2235
106. Lu JF, Eppler SM, Wolf J, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80(2):136–145. doi:10.1016/j.clpt.2006.04.007
107. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi:10.1056/NEJMoa050753
108. Hughes AN, O‘Brien ME, Petty WJ, et al. Overcoming CYP1A1/1A2 mediated induction of metabolism by escalating erlotinib dose in current smokers. J Clin Oncol. 2009;27(8):1220–1226. doi:10.1200/JCO.2008.19.3995
109. Hidalgo M, Siu LL, Nemunaitis J, et al. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol. 2001;19(13):3267–3279. doi:10.1200/JCO.2001.19.13.3267
110. Smit EF, Wu YL, Gervais R, et al. A randomized, double-blind, phase III study comparing two doses of erlotinib for second-line treatment of current smokers with advanced non-small-cell lung cancer (CurrentS). Lung Cancer. 2016;99:94–101. doi:10.1016/j.lungcan.2016.06.019
111. Patnaik A, Wood D, Tolcher AW, et al. Phase I, pharmacokinetic, and biological study of erlotinib in combination with paclitaxel and carboplatin in patients with advanced solid tumors. Clin Cancer Res. 2006;12(24):7406–7413. doi:10.1158/1078-0432.CCR-06-1886
112. Veeraputhiran M, Sundermeyer M. Rhabdomyolysis resulting from pharmacologic interaction between erlotinib and simvastatin. Clin Lung Cancer. 2008;9(4):232–234. doi:10.3816/CLC.2008.n.036
113. Grenader T, Gipps M, Shavit L, Gabizon A. Significant drug interaction: phenytoin toxicity due to erlotinib. Lung Cancer. 2007;57(3):404–406. doi:10.1016/j.lungcan.2007.02.011
114. Shi Z, Peng XX, Kim IW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67(22):11012–11020. doi:10.1158/0008-5472.CAN-07-2686
115. Marchetti S, de Vries NA, Buckle T, et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther. 2008;7(8):2280–2287. doi:10.1158/1535-7163.MCT-07-2250
116. Elmeliegy MA, Carcaboso AM, Tagen M, et al. Role of ATP-binding cassette and solute carrier transporters in erlotinib CNS penetration and intracellular accumulation. Clin Cancer Res. 2011;17(1):89–99. doi:10.1158/1078-0432.CCR-10-1934
117. Thomas KS, Billingsley A, Amarshi N, Nair BA. Elevated international normalized ratio associated with concomitant warfarin and erlotinib. Am J Health Syst Pharm. 2010;67(17):1426–1429. doi:10.2146/ajhp090202
118. Tran HT, Zinner RG, Blumenschein GR
119. Keating GM. Afatinib: a review of its use in the treatment of advanced non-small cell lung cancer. Drugs. 2014;74(2):207–221. doi:10.1007/s40265-013-0170-8
120. European Medicines Agency. Giotrif (afatinib) Summary of Product Characteristics. 2018.
121. Food and Drug Administration. Gilotrif (afatinib) Prescribing Information. 2018.
122. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP) assessment report for giotrif (afatinib). 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002280/WC500152394.pdf. Accessed 14 November, 2018.
123. Stopfer P, Marzin K, Narjes H, et al. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother Pharmacol. 2012;69(4):1051–1061. doi:10.1007/s00280-011-1803-9
124. Freiwald M, Schmid U, Fleury A, Wind S, Stopfer P, Staab A. Population pharmacokinetics of afatinib, an irreversible ErbB family blocker, in patients with various solid tumors. Cancer Chemother Pharmacol. 2014;73(4):759–770. doi:10.1007/s00280-014-2403-2
125. Wind S, Giessmann T, Jungnik A, et al. Pharmacokinetic drug interactions of afatinib with rifampicin and ritonavir. Clin Drug Investig. 2014;34(3):173–182. doi:10.1007/s40261-013-0161-2
126. Wind S, Schnell D, Ebner T, Freiwald M, Stopfer P. Clinical pharmacokinetics and pharmacodynamics of afatinib. Clin Pharmacokinet. 2017;56(3):235–250. doi:10.1007/s40262-016-0440-1
127. Food and Drug Administration. Tagrisso (osimertinib) Prescribing Information. 2018.
128. European Medicines Agency. Tagrisso (osimertinib) Summary of Product Characteristics. 2017.
129. Vishwanathan K, Dickinson PA, Bui K, et al. The effect of food or omeprazole on the pharmacokinetics of osimertinib in patients with non-small-cell lung cancer and in healthy volunteers. J Clin Pharmacol. 2018;58(4):474–484. doi:10.1002/jcph.1035
130. Pilla Reddy V, Walker M, Sharma P, Ballard P, Vishwanathan K. Development, verification, and prediction of osimertinib drug-drug interactions using PBPK modeling approach to inform drug label. CPT Pharmacometrics Syst Pharmacol. 2018;7(5):321–330. doi:10.1002/psp4.12289
131. Vishwanathan K, Dickinson PA, So K, et al. The effect of itraconazole and rifampicin on the pharmacokinetics of osimertinib. Br J Clin Pharmacol. 2018;84(6):1156–1169. doi:10.1111/bcp.13534
132. Dickinson PA, Cantarini MV, Collier J, et al. Metabolic disposition of osimertinib in rats, dogs, and humans: insights into a drug designed to bind covalently to a cysteine residue of epidermal growth factor receptor. Drug Metab Dispos. 2016;44(8):1201–1212. doi:10.1124/dmd.115.069203
133. Brown K, Comisar C, Witjes H, et al. Population pharmacokinetics and exposure-response of osimertinib in patients with non-small cell lung cancer. Br J Clin Pharmacol. 2017;83(6):1216–1226. doi:10.1111/bcp.13223
134. Harvey RD, Isambert N, Rafii S, et al. Effect of multiple-dose osimertinib (AZD9291) on the pharmacokinetics (PK) of simvastatin and rosuvastatin. J Clin Oncol. 2016;34(15_suppl):e14098. doi:10.1200/JCO.2016.34.15_suppl.e14098
135. Ballard P, Yates JW, Yang Z, et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res. 2016;22(20):5130–5140. doi:10.1158/1078-0432.CCR-16-0399
136. Liang W, Wu X, Fang W, et al. Network meta-analysis of erlotinib, gefitinib, afatinib and icotinib in patients with advanced non-small-cell lung cancer harboring EGFR mutations. PLoS One. 2014;9(2):e85245. doi:10.1371/journal.pone.0085245
137. Shi Y, Sun Y, Ding C, et al. [China experts consensus on icotinib for non-small cell lung cancer treatment(2016 version)]. Zhongguo Fei Ai Za Zhi. 2016;19(7):489–494. doi:10.3779/j.issn.1009-3419.2016.07.12
138. Zhang T, Zhang K, Ma L, et al. Metabolic pathway of icotinib in vitro: the differential roles of CYP3A4, CYP3A5, and CYP1A2 on potential pharmacokinetic drug-drug interaction. J Pharm Sci. 2018;107(4):979–983. doi:10.1016/j.xphs.2017.12.007
139. Chen J, Liu D, Zheng X, Zhao Q, Jiang J, Hu P. Relative contributions of the major human CYP450 to the metabolism of icotinib and its implication in prediction of drug-drug interaction between icotinib and CYP3A4 inhibitors/inducers using physiologically based pharmacokinetic modeling. Expert Opin Drug Metab Toxicol. 2015;11(6):857–868. doi:10.1517/17425255.2015.1034688
140. Zhao Q, Shentu J, Xu N, et al. Phase I study of icotinib hydrochloride (BPI-2009H), an oral EGFR tyrosine kinase inhibitor, in patients with advanced NSCLC and other solid tumors. Lung Cancer. 2011;73(2):195–202. doi:10.1016/j.lungcan.2010.11.007
141. Stepney R, Lichtman SM, Danesi R Drug-drug interactions in older patients with cancer: a report from the 15th conference of the international society of geriatric oncology, Prague, Czech Republic.
142. Somashekhar S, Sepúlveda M-J, Norden A, et al. Early experience with IBM Watson for Oncology (WFO) cognitive computing system for lung and colorectal cancer treatment. J Clin Oncol. 2017;35(15_suppl):8527. doi:10.1200/JCO.2017.35.15_suppl.8527
143. Sarre-Lazcano C, Armengol Alonso A, Huitzil Melendez F, et al. Cognitive computing in oncology: A qualitative assessment of IBM Watson for Oncology in Mexico. J Clin Oncol. 2017;35(15_suppl):e18166. doi:10.1200/JCO.2017.35.15_suppl.e18166
144. Chatfield AJ. Lexicomp online and micromedex 2.0. J Med Libr Assoc. 2015;103(2):112–113. doi:10.3163/1536-5050.103.2.016
145. Muhič N, Mrhar A, Brvar M. Comparative analysis of three drug-drug interaction screening systems against probable clinically relevant drug-drug interactions: a prospective cohort study. Eur J Clin Pharmacol. 2017;73(7):875–882. doi:10.1007/s00228-017-2232-4
146. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. Available from: https://drug-interactions.medicine.iu.edu
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.