")
Back to Archived Journals » Oncolytic Virotherapy » Volume 4
Combining HDAC inhibitors with oncolytic virotherapy for cancer therapy
Authors Nakashima H, Nguyen T, Chiocca EA
Received 30 August 2015
Accepted for publication 15 October 2015
Published 20 November 2015 Volume 2015:4 Pages 183—191
DOI https://doi.org/10.2147/OV.S66081
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Hiroshi Nakashima, Tran Nguyen, Ennio Antonio Chiocca
Department of Neurosurgery, Brigham and Women's Hospital, Boston, MA, USA
Abstract: Histone deacetylase (HDAC) enzymes play a critical role in the epigenetic regulation of cellular functions and signaling pathways in many cancers. HDAC inhibitors (HDACi) have been validated for single use or in combination with other drugs in oncologic therapeutics. An even more novel combination therapy with HDACi is to use them with an oncolytic virus. HDACi may lead to an amplification of tumor-specific lytic effects by facilitating increased cycles of viral replication, but there may also be direct anticancer effects of the drug by itself. Here, we review the molecular mechanisms of anti-cancer effects of the combination of oncolytic viruses with HDACi.
Keywords: epigenetics, glioma, oncolytic virus, HDAC inhibitor, HSV-1, cancer
Introduction
Significant genetic heterogeneity and instability are a hallmark of many cancers, and this contributes to treatment resistance. This is made even more complex by tumor epigenetic changes that also contribute to genomic instability and tumor plasticity.1,2 Epigenetic mechanisms include DNA methylation, post-translational modifications (PTMs) of histones and non-coding RNAs. One of the epigenetic modifiers attracting scientific attention as a potential therapeutic target comprises the histone deacetylases (HDACs). Advanced proteomic analyses with high-resolution mass spectrometry have enabled large-scale acetylome studies and subsequently have identified thousands of acetylation sites on the lysine moieties of more than 1,700 proteins in mammalian cells, including histones. For this reason, HDAC is also referred to as a lysine deacetylase.3,4 Pharmacological inhibitors of HDAC (HDACi) have been promisingly explored in therapeutic applications in cancer. In fact, pharmacological inhibitors, such as suberoylanilide hydroxamic acid (SAHA) and FK228, have been approved for clinical use to treat cancers by the Food and Drug Administration (FDA) in USA.
Oncolytic virotherapy was initially developed to eliminate cancer cells, with clinical treatment applications under development for over a decade.5,6 Oncolytic viruses (OVs) have also been applied to the area of cancer vaccination and immunotherapy. Genetically attenuated viruses are designed to selectively apply lytic effects against tumor cells (oncolysis), while simultaneously sparing non-tumor cells. However, as is the case with many therapeutic medications, OV therapy can be met with tumor resistance. This resistance may be due to less viral susceptibility and permissivity, attenuated apoptotic tumor death, and intrinsic/extrinsic antiviral immune responses. HDACi could address these resistance mechanisms to OVs, and are emerging as enhancers of oncolytic virotherapy. In fact, recent work demonstrates that HDACi could improve OV therapy, reverse antiviral processes in tumor cells, and possibly promote OV-mediated tumor immunotherapy.
Oncolytic viruses
The concept behind OVs is for trying to find strains or mutants that are highly permissive and selective for infection and replication in malignant cells where they can produce their progeny for progressive oncolytic cycles throughout the tumor. In contrast to non-replicating viral vectors, OVs still maintain most of their viral genes intact, particularly those that encode genes required for the viral replicative life cycle, viral evasion mechanisms from host defense, and immune surveillance. OVs are engineered genetically or selected naturally from pathogenic viruses that include herpes simplex virus type 1 (HSV-1), adenovirus serotype 5 (Ad5), vascular stomatitis virus (VSV), measles virus, and many others. Strategies for acquiring and improving tumor selectivity in viruses range widely from genetic mutations and modification of transcriptional regulatory elements to structural modifications that alter infectivity and to arming with additional anticancer genes.7,8 OVs can also be combined with chemical drugs to obtain adjuvant and possibly synergistic anticancer effects both experimentally in animal models and clinically. The host still recognizes the OV as a foreign pathogen and will attempt to fight it via various immune responses that can diminish its oncolysis and lead to premature clearance of the OV. However, there exists a balance and this immune response can also lead to an anticancer effect. Immune activation in response to OV administration can thus also benefit treatment via a local cytokine storm in the tumor and recognition of tumor antigens. Understanding this balance between deleterious immunity that could lead to premature clearance of the OV and anticancer immunity that can lead to an effective anticancer therapy remains a target for scientific research and likely the objective to make OV therapy truly efficacious.9,10
Several clinical trials using OVs have and are being conducted,7,11 but a recent phase III melanoma study using an HSV-based OV (oHSV1) is providing justification for approval by the US FDA as the first therapeutic viral oncolytic bioagent in the US (T-VEC; Amgen Inc. Thousand Oaks, CA, USA).12 HSV-1, a member of alpha-herpes virus family, was the first virus that was genetically engineered to be oncolytic.6 It is one of the largest viruses by size (>150 kb) among OVs. The HSV-1 DNA genome is packed in the virion, which consists of three major layers, lipid envelope, tegument, and capsid. The kinetics of viral gene expression consist of several immediate early (α) genes expressed right after infection, followed by early (β) genes expression. Viral DNA replication then triggers expression of late (γ) genes that encode viral structural components for progeny virions. In the design of oHSV1, appropriately placed mutations in one or several of these genes can attenuate lytic infection in normal tissues, while keeping infection of tumor tissues still to more permissive levels.8 Therapeutic success using OV is still a challenge since responsiveness and efficacy of OV may be different in different cancers and conditions due to different epigenetic states that affect viral permissiveness or susceptibility.
HDACs as modifiers of epigenetic silencing
PTM is an important biochemical process that occurs in both tumorigenesis and virus pathobiology. Acetylation and deacetylation of histones and non-histone substrates at lysine residues have emerged as crucial post-translational mechanisms. They regulate chromatin remodeling and accessibility of transcription factors that bind to specific sites in the regulatory regions of DNA to activate or suppress the transcription of specific genes by complexing with other proteins. Also, histone acetyltransferases (HATs) transfer acetyl groups to the µ-amino group of lysine on target substrates. Acetylation at lysine is a reversible process where HAT-mediated histone acetylation activates transcription, while the removal of the acetyl group causes chromatin condensation and transcriptional repression of genes. This deacetylation process is achieved by HDACs.
HDACs are classified into four groups based on similarity and function. Class I, II, and IV groups are classical zinc-dependent enzymes and are structurally distinct from the nicotinamide adenine dinucleotide (NAD+) dependent class III group, called sirtuins (SIRTs). Inhibitors of zinc-dependent HDACs have shown great potential in the treatment of epigenetic disorders including cancers. Many HDACi are currently in pre-clinical and clinical phases, and SAHA, FK228, and valproic acid (VPA), have been approved for clinical use by the FDA. Although SIRTs are considered to play key roles for physical stresses, metabolism, tumorigenesis, and aging, there is little information available regarding OV therapy with SIRTs and their inhibitors. Therefore, zinc-dependent HDACs and their inhibitors will be the focus of this review.
The class I group (HDAC1-3 and 8) is homologous to the yeast-reduced potassium dependency 3 (Rpd3) protein and is predominantly localized in the nucleus, but they also localize in the cytoplasm to target cytosolic non-histone proteins. Class I HDACs are expressed ubiquitously in human tissues and global defects of this HDAC class in transgenic mice result in embryonic or perinatal lethality.13–16 HDACs often form complexes to deacetylase specific targets. HDAC1 and 2 have undergone little functional divergence from each other and can form homo- and hetero-dimer.17,18 HDAC1 and 2 are found in different transcriptional repressor complexes, including the Sin3 complex, the nucleosome remodeling and deacetylating complex, or the CoREST complex.19–21 HDAC3 is usually found in the complexes containing nuclear receptor corepressor 1 or the silencing mediator of retinoid and thyroid receptors.22
The class II group (HDAC4-7, 9, and 10) is homologous to the yeast Histone deacetylase (Hda1) and localizes to the cytoplasm. This class II group is further subdivided into class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10). The class IIa has a highly conserved deacetylase domain in its carboxyl-terminus and can be shuttled into the nucleus, based on the phosphorylation status of serine residues in the N-terminus, binding sites for the 14-3-3 proteins that regulate nuclear-cytoplasmic localization.23 Since their intrinsic catalytic activities are relatively weak, class IIa HDACs usually interact with HDAC3 and other cofactors in order to increase the kinetics of deacetylation of target substrates.24,25 HDAC7 and 9 play key roles in lineage-determination in adaptive immunity.26–28 HDAC6 is unique among the HDAC family, in that it possesses two tandem catalytically active domains and an ubiquitin (Ub; and Ub-like modifiers) binding domain at its C-terminus. HDAC6 lacks in vivo deacetylase activity for nuclear histone proteins. Instead it is one of the major cytoplasmic deacetylases, targeting non-histone substrates, such as α-tubulin, Hsp90, and β-catenin.29–31 Biologically, HDAC6 functions in selective autophagy and in the aggresome processes of substrates that are conjugated to the moiety of Ub and Ub-like modifiers, and these processes also appear to impact the kinetics of viral infection/replication.32–35
The class IV HDAC11 is the smallest one in the HDAC family. It contains mostly the catalytic domains and shares motifs with both class I and II HDACs. HDAC11 regulates the immunosuppressive cytokine molecule IL-10 in antigen-presenting cells.36,37
HDAC1 and 2 as direct viral silencing factors
The HSV-1 genome is susceptible to host nuclear epigenetic mechanisms during infection, a process that allows the infected cell to regulate viral gene expression. HDAC1 and 2 are major enzymes that deacetylate the histone tails of nucleosomes containing HSV-1 genes and also play an important role in viral latency. Although HDACs themselves do not directly recognize and bind to DNA, HDAC1/2 are incorporated in the CoREST-REST-LSD1 repressor complex that binds repressor element 1 (RE-1) DNA sequences. This HDAC1/2 repressor complex primarily suppresses neuronal genes containing such RE-1s in their regulatory sequences in non-neuronal cells. The HSV-1 genome also contains multiple RE-1 sequences.38,39 HSV-1 infected cells that over-express wild-type REST were shown to preferentially lead to a latent program of viral life cycle, while HSV1 infected cells that express mutated REST (ie, lacking the binding capability to CoREST and HDAC1/2) preferentially lead to a lytic life cycle.40,41 Thus, HDAC1/2 and their repressor complex are critical factors in the decision between the lytic cycle or establishment of latency for HSV-1 in sensory neurons.42
HSV-1 can interfere with HDAC1 and 2 mediated epigenetic silencing: the viral genes ICP0 and Us3 are known viral factors that inhibit HDACs. ICP0 is an immediate early (IE) protein, known as a viral Ub E3 ligase, and transfers Ub-chains to a target substrate for proteasomal degradation. Some intrinsic and innate antiviral factors including components of ND10 bodies (PML, SP100, and SUMO), p53 and the DNA sensor IFI16 are ICP0 substrates for degradation upon infection. ICP0 thus counteracts these antiviral host responses. ICP0’s inhibition of HDAC1/2 seems to be independent of proteasomal degradation.39 ICP0 instead disrupts CoREST-REST-LSD1 complex formation, dissociates HDAC1 from it and then translocates it to the cytoplasm.39 In addition to HDAC1/2, ICP0 is also reported to counteract class II HDACs, implying multiple roles for ICP0 against diverse HDAC functions.43 The Us3 gene products Us3 and Us3.5 kinase (hereafter, referred to as Us3-PKs) are also involved in the cytoplasmic distribution of HDAC1/2.44,45 In contrast to ICP0, Us3-PKs are not essential for viral replication in cultured cells, but are more critical for viral replication and neurotoxicity in mice. Us3-PKs show kinase activity against viral and cellular proteins and counteract apoptotic induction and innate immune responses led by type I interferons (hereafter referred to as IFN-I). To this end, HDAC1 and 2 also are subject to PTM from Us3-PKs.46–48 Although it has not been reported whether this phosphorylation of HDACs affects enzymatic activities, Us3-PKs overexpression changes HDAC1 localization into the cytoplasm.44 Thus, ICP0 and Us3 independently interact with HDAC1/2 to displace them from viral DNA during infection.39
The roles of ICP0 and Us3 in counteracting HDAC1 and 2 functions may not be critical in viral replication in cells that are not able to sustain the viral establishment of latency (ie, non-neuronal cells) and that preferentially sustain the lytic cycle. Viral gene expression of ICP0-null HSV-1 was temporally accelerated in cultured cells pretreated with sodium butyrate (a pan-HDACi), but the expression of viral genes from wild-type HSV-1 was not.49 It is thus uncertain whether HDACi enhances viral replication and yields in these cells. An HSV-1 with a mutation in the Us3 gene is unlikely to exhibit altered replication in cultured cells when pretreated with HDACi (sodium butyrate or HDACi VII).47 In proliferating or non-neuronal cultured cells, HDACs may not play a dominant role in suppression of the viral lytic cycle. Prior to ICP0 and Us3 gene activation, the major tegument protein VP16 regulates IE gene expression, required for the viral lytic cycle. Upon intracellular penetration, VP16 forms a complex with and recruits other histone modifiers, such as lysine-specific demethylase 1 (LSD1) and HAT (CLOCK) in addition to Oct1 and HCF1, to the promoters of IE genes.50,51 Thus, the viral VP16 complex competes with chromatin silencing mediated by the cellular HDACs-repressor complex. On the other hand, HDACs-repressor complex may play a more important role in preventing viral reactivation from the latent state where VP16, ICP0, Us3, and other viral proteins are absent. Experiments using in vitro and in vivo HSV-1 latency models indicate that latent HSV-1 in sensory neurons can be reactivated in response to sodium butyrate.52–54 The use of HDACi to reactivate human immunodeficiency virus (clinical trial in NCT01680094, NCT02513901, and NCT02336074) and Epstein–Barr virus (clinical trial in NCT01341834) from latency is used clinically as a means to flush out these hidden viruses from chronically infected patients.55,56 In summary, viral genes (ICP0 and Us3) associate with HDAC1/2 to suppress host repressor complexes and establish latency but HDACi may be more effective in cells that have already silenced the viral genome.
HDACi and OV in tumor
In cancer, hypoacetylation of histone tails and changing epigenetic signatures are hallmarks of cancer progression, metastasis, and therapeutic resistance, as well as tumor dormancy.57,58 OVs can be targets of genome silencing pathways that diminish oncolytic capability. Otsuki et al addressed the impact of oHSV1 in aggressive brain tumor type when treated with HDACi, mostly using VPA.59 VPA is a widely used antiepileptic drug and is also used in migraine prophylaxis and bipolar disorders. The pharmacological actions of VPA include multiple mechanisms to affect neuroprotection, but the finding that VPA can also act as a HDACi is relatively recent.59–61 Also, several clinical trials using VPA and vorinostat in high-grade glioblastoma patients are currently ongoing in combination with standard chemodrug temozolomide and/or radiation.62 When relatively high doses of VPA (5–30 mM) were administered to human glioma lines prior to oHSV1 infection, the cytotoxic effect of the virus was significantly more pronounced. This was also evidenced by using other pan-HDACi (trichostatin A [TSA], sodium butyrate, and APHA). VPA augmented oHSV1 replication in different glioma cells. This was correlated with higher expression of viral IE genes (ICP0 and ICP4) and late gene at early time points after infection. When VPA was administered to mice with an orthotopic xenograft glioma 1 day before oHSV1 intratumoral injection, there was a significant survival advantage over oHSV1-treated mice that were not treated with VPA.
This significant finding depended on the timing of VPA treatment. While treatment before viral infection (14 hours) significantly improved viral yields, concomitant treatment (8 hours) diminished this augmented OV effect.59 Likely concomitant treatment caused a repression in viral DNA replication and reduction of late genes, whereas IE gene expression was not suppressed but enhanced. Since it is also known that prolonged treatment of HDACi causes cell arrest, pretreatment and cotreatment of HDACi may cause different changes in the cellular environment to affect oHSV1 infection. While VPA pretreatment could induce cell cycle arrest at G1 leading to efficient HSV-1 replication, the administration of HDACi along with the oHSV1 could dampen this benefit to support viral activity.63 A similar negligible effect of TSA in viral replication was observed with oHSV1 infected glioma cells during TSA treatment.64 In this instance, TSA treatment down-regulated cyclin D1 expression that is required for efficient HSV-1 replication. ICP0 facilitates the recruitment of cyclin D to viral DNA replication foci and stabilizes cyclin D proteins through degradation of Cdc34, an E2 Ub-conjugating enzyme.65,66 Cyclin D1 is also a critical driver of the G1-S phase transition and a tumorigenic factor for many cancers. Thus, despite little effect in viral replication, the synergistic cytotoxicity of TSA and oHSV1 was significant in glioma cells in vitro and in vivo.64 In oral squamous cell carcinoma cells, TSA cotreatment led to up-regulation of oHSV1 replication at early time point (24 hours) and enhanced cytotoxicity of oHSV1.67 This correlated with induction of cyclin-dependent kinase inhibitors p21, resulting in G0/G1 arrest in treated cells. In fact, the lack of HDAC1/2 can induce cell cycle arrest at the G1 phase through the up-regulation of cyclin-dependent kinase inhibitor proteins, p21 and p57, which are targets of Cullin 4-RING E3 ligases, an Ub E3 ligase complex.68 CUL4B-containing Cullin 4-RING E3 ligases B and HDAC1/2 containing the Sin3A repressor complex are associated to deacetylase histone H3 and H4 on CDKN1A/p21 promoters, which can be blocked by TSA. Overall, HDACi can suppress tumor proliferation during oHSV1 infection. Whereas oHSV1 replication may be affected negatively or positively depending on the cellular context, HDACi can show enhanced cytotoxic effect with oHSV1 in glioma and squamous cell carcinoma.
The study by Otsuki et al also showed that HDACi can block type-I interferon pathway during oHSV1 infection in glioma cells.59 IFN-I including IFN-α and INF-β are crucial inflammatory cytokines for innate antiviral function in early phases of viral infection.69 Extracellular IFN-Is bind to its receptors on the cellular surface and activates Janus tyrosine kinases followed by phosphorylation of STAT1 and STAT2 proteins.70–72 JAK/STAT pathways through IFN-I binding to its receptors stimulate transcription of genes containing IFN-stimulated gene response elements. While IFN-α is secreted from restricted cell types such as immune cells, IFN-β secretion occurs in nearly any types of cells in response to the stimulation. Administration of IFN-β protein is approved in the USA to treat multiple sclerosis and underway experimentally and clinically in glioma and other cancer therapy.73,74 In OV therapy, however, IFN treatment blocks the oncolytic effect and OV replication is greatly suppressed when IFN-β is added. Under this condition, VPA treatment improved oHSV1 oncolytic effects by neutralizing IFN-induced antiviral effects. At the molecular level, VPA suppressed up-regulation of STAT1 gene expression in response to IFN-β treatment. PKR and PML are involved in intrinsic antiviral functions against HSV-1. PKR is a kinase that phosphorylates translational initiation factor eIF-2α to suppress viral protein synthesis and PML is one of the ND10 nuclear body proteins that associates with viral DNA early during lytic infection and represses viral replication. PKR and PML are ubiquitously expressed and their genes were up-regulated by IFN-β, whereas VPA suppressed them. The mechanism behind these findings implicates a non-canonical role of HDAC1/2 that functions as a transcriptional activator of a subset of genes that are induced by IFN-I.75–77 In addition to oHSV1, HDACi also inhibit IFN-induced antiviral responses that restrict oncolytic activities of VSV and vaccinia viruses in prostate cancer and melanoma cell lines, respectively.78,79 Thus, HDACi would be widely beneficial to support OV replication in tumors where immune responses are intact and interfere with virus activity.
HDACi and immune cells in response to OV
HDACi may change intercellular antivirus immune responses. Natural killer (NK) cells are among the first responders against pathogens and they are recruited to pathogenic sites to attack virus and tumor cells. NK cells require expression of NK-cell specific recognition receptors on the recipient cells. NKG2D is one of the receptor molecules expressed on virus-infected cells and tumors. HDACi is known to induce immunosensitization of tumors by increasing NKG2D ligand expression and sensitize them to NK cytotoxicity that can be applied to cancer immunotherapy.80 HSV-1 infection also increases susceptibility to NK lysis of the infected cells and natural cytotoxicity receptors NKp30 and NKp46 that are upregulated in response to HSV1 infection in gliomas.81 Therefore, early phases of the innate immune response can impact negatively on virotherapy and activation of both NK cells and macrophages in response to oHSV1 infection impedes viral replication and oncolysis. However, it is not clear if HDACi affects the cytotoxic properties and migration of NK cells in the setting of virotherapy of cancer. Thus, Alvarez-Breckenridge et al addressed the impact of HDACi against NK cells early during oHSV1 infection using a human glioma xenograft model.82 The injection of oHSV1 into the orthotopic glioma site of mice led to the infiltration of NK cells and IFN-γ product in that region, while VPA administration temporally suppressed them. When compared to cyclophosphamide that causes robust suppression of innate immune cells (macrophages, lymphocytes, and NK cells), VPA impaired the kinetics and abundance of recruited immune cells (NK and macrophage) after oHSV1 infection. At the cellular level, VPA attenuated IFN-γ production in NK cells in part through reduction in STAT5 phosphorylation and T-BET gene expression, a master regulator of IFN-γ gene expression.83 In summary, HDACi can up-regulate NK ligand expression on tumor cell surface and suppress the inhibitory effect of NK cells against oHSV1 at the tumor site.81 Therefore, VPA pre-treatment may allow the oncolytic effect of oHSV1 in tumor bed to enhance NK-mediated tumor lysis at later time-point when the cytotoxic capability of NK cells recovers after clearance of VPA and viruses.
HDACi can assist OV-mediated immunotherapy. Multiple HDACs regulate the development process and maintain the lineage integrity of lymphocytes: hence, the utility of HDACi is recently expanding to include anti-inflammatory properties for autoimmune disease.26–28,84–87 Furthermore, the application of HDACi in the immune system also boosts vaccinations to treat cancer and chronic infectious diseases as well.88–92 Thus, OVs are potential vaccine and immune-stimulatory agents to apply in tumor immunotherapy in this context.9,93,94 VSV is one of the oncolytic vaccine platforms that stimulates acute immunity along with immunogenic tumor cell death and boosts tumor-specific cytotoxic T lymphocytes when the virus encodes tumor-antigen to express in tumor. Thus, Bridle et al used HDACi to enhance this oncolytic vaccination strategy to target melanoma.95 Oncolytic VSV encoding human dopachrome tautomerase gene (VSV-hDCT) was used to boost vaccination after priming with recombinant Ad expressing hDCT in naive mice bearing B16-F10 melanoma. When coadministered with MS-275, the VSV-reactive CD8+ response was rather attenuated, leading to delayed induction of antibody against the virus until the end of drug administration. However, prolonged VSV-induced lymphopenia was also observed by MS-275, associating with delayed reconstitution of peripheral CD4+, CD8+ T-cells, and B cells. As result, MS-275 increased tumor antigen (DCT)-specific CD8+ numbers with enhanced cytotoxic functionality in the context of TNFα and IFN-γ products at late time point while decreasing Foxp3+ Treg cell number. Of interest, MS-275 coadministration suppressed this prime-boost induced autoimmune response against normal DCT+ melanocytes. This and other studies suggest that HDACi may suppress IFN-I products in diseases associated with autoimmune tissue damage and chronic infection, and thus can restore exhausted adaptive immunity.91,96–98
Conclusion
There is an increasing body of evidence that dysregulated epigenetic factors are associated with chronic diseases such as cancer, inflammation, and infection. Targeting HDACs with pharmacological inhibitors can influence a broad range of transcriptomes. Acetyl-modification of nucleosome components and trancriptional accessories changes the immune and inflammatory signaling cascades, as well as lymphocyte lineages. In oncolytic therapy, HDACi could create a favorable environment for therapeutic viruses and we summarized currently published studies in Table 1 including those where HDACi-enhanced OV effects against tumors. These viruses could then function as self-productive, anti-tumor agents in the tumor bed, and promote tumor immunogenicity. There have not been clinical trials using combinations of HDACi and virotherapy. At this point, it seems that all cancer indications may be worthwhile exploring in a possible clinical trial setting.
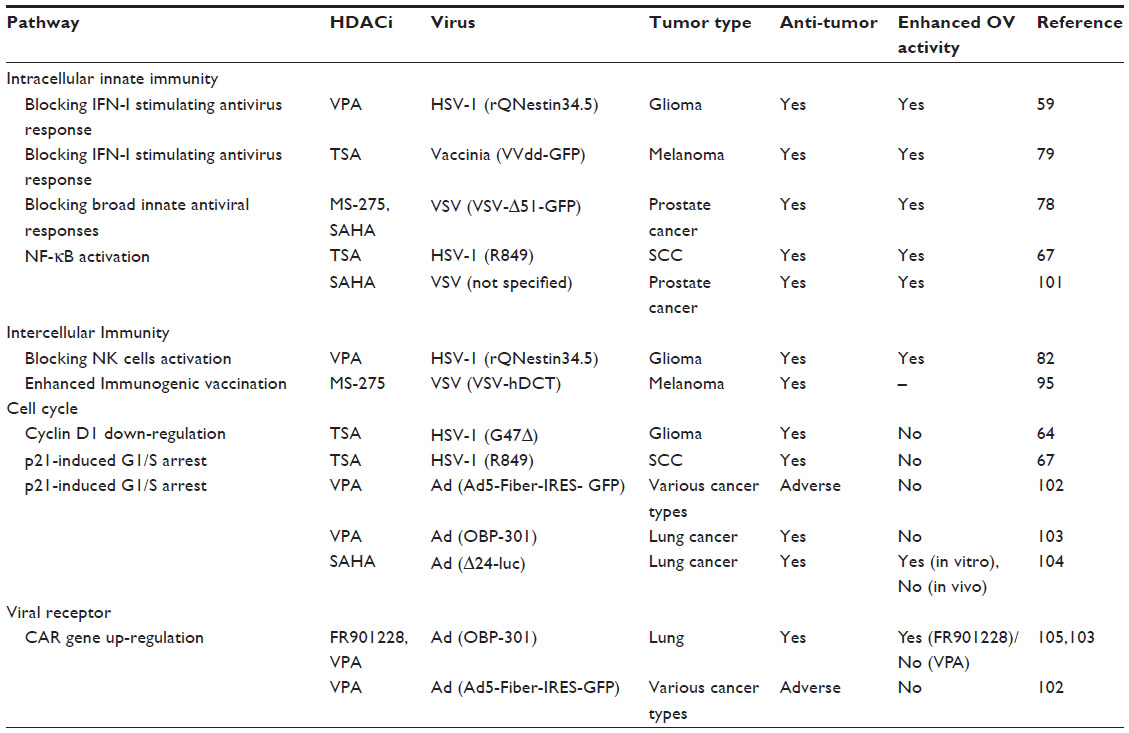 | Table 1 A summary of published effects of combined OV and HDACi |
Although various HDAC inhibitors can be combined experimentally, there are mixed reports of therapeutic efficacy relative to their synergistic effects. This is because HDAC functions are complex, divergent, and redundant among classes. Therefore, the success of combined therapy may require careful evaluation of individual HDACs using a specific HDACi, whereas most studies used pan-HDACi (eg, VPA, TSA, and SAHA) in experimental OV virotherapy. Furthermore, the determination of the ideal time of HDACi administration is necessary for optimal OV therapy synergy with the host immune responses. Since host epigenetic roles in cancers and immune cell lineages are broad and also not restricted in lysine acetylation states, inhibition of other epigenetic modifiers (DNA/protein methylases and non-coding RNAs) are also needed to be evaluated in the setting of OV combination in the future.1,99,100
Acknowledgment
This work was supported by NCI P01CA163205.
Disclosure
The authors report no conflicts of interest in this work.
References
Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014; 54(5):716–727. | |
Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. | |
Lundby A, Lage K, Weinert BT, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2(2):419–431. | |
Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325(5942):834–840. | |
Ganly I, Kirn D, Eckhardt G, et al. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin Cancer Res. 2000;6(3):798–806. | |
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–856. | |
Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev. 2014;12(1):23–34. | |
Grandi P, Peruzzi P, Reinhart B, Cohen JB, Chiocca EA, Glorioso JC. Design and application of oncolytic HSV vectors for glioblastoma therapy. Exp Rev Neurother. 2009;9(4):505–517. | |
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev. 2014;14(8):559–567. | |
Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2(4):295–300. | |
Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol. 2014;16(3):334–351. | |
Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. | |
Haberland M, Mokalled MH, Montgomery RL, Olson EN. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009;23(14):1625–1630. | |
Bhaskara S, Chyla BJ, Amann JM, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30(1):61–72. | |
Montgomery RL, Davis CA, Potthoff MJ, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21(14):1790–1802. | |
Lagger G, O’Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21(11):2672–2681. | |
Moser MA, Hagelkruys A, Seiser C. Transcription and beyond: the role of mammalian class I lysine deacetylases. Chromosoma. 2014; 123(1–2):67–78. | |
Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338(1):17–31. | |
Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437(7057):432–435. | |
Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2(6):851–861. | |
Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell. 1997;89(3):341–347. | |
You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20(2):182–187. | |
Nishino TG, Miyazaki M, Hoshino H, Miwa Y, Horinouchi S, Yoshida M. 14-3-3 regulates the nuclear import of class IIa histone deacetylases. Biochem Biophys Res Commun. 2008;377(3):852–856. | |
Lahm A, Paolini C, Pallaoro M, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104(44):17335–17340. | |
Qian DZ, Kachhap SK, Collis SJ, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66(17):8814–8821. | |
de Zoeten EF, Wang L, Butler K, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. 2011;31(10):2066–2078. | |
Navarro MN, Goebel J, Feijoo-Carnero C, Morrice N, Cantrell DA. Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat Immunol. 2011;12(4):352–361. | |
Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13(11):1299–1307. | |
Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283(19):12686–12690. | |
Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–607. | |
Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. | |
Nakashima H, Nguyen T, Goins WF, Chiocca EA. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J Biol Chem. 2015;290(3):1485–1495. | |
Lee JY, Koga H, Kawaguchi Y, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29(5):969–980. | |
Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447(7146):859–863. | |
Banerjee I, Miyake Y, Nobs SP, et al. Influenza A virus uses the aggresome processing machinery for host cell entry. Science. 2014; 346(6208):473–477. | |
Cheng F, Lienlaf M, Perez-Villarroel P, et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol. 2014;60(1):44–53. | |
Villagra A, Cheng F, Wang HW, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10(1):92–100. | |
Zhou G, Te D, Roizman B. The CoREST/REST repressor is both necessary and inimical for expression of herpes simplex virus genes. mBio. 2011;2(1):e00313–e00310. | |
Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A. 2005;102(21):7571–7576. | |
Zhou G, Du T, Roizman B. HSV carrying WT REST establishes latency but reactivates only if the synthesis of REST is suppressed. Proc Natl Acad Sci U S A. 2013;110(6):E498–E506. | |
Du T, Zhou G, Khan S, Gu H, Roizman B. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc Natl Acad Sci U S A. 2010;107(36):15904–15909. | |
Zhou G, Du T, Roizman B. The role of the CoREST/REST repressor complex in herpes simplex virus 1 productive infection and in latency. Viruses. 2013;5(5):1208–1218. | |
Lomonte P, Thomas J, Texier P, Caron C, Khochbin S, Epstein AL. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J Virol. 2004;78(13):6744–6757. | |
Poon AP, Gu H, Roizman B. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc Natl Acad Sci U S A. 2006;103(26):9993–9998. | |
Bhattacharyya M, Klein JP. A note on testing in Aalen’s additive hazards regression models. Stat Med. 2005;24(14):2235–2240. | |
Kawaguchi Y. Us3, a multifunctional protein kinase encoded by herpes simplex virus 1: how does it function in vivo? Cornea. 2013;32(Suppl 1):S22–S27. | |
Walters MS, Kinchington PR, Banfield BW, Silverstein S. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J Virol. 2010;84(19):9666–9676. | |
Miccheli A, Ricciolini R, Lagana A, Piccolella E, Conti F. Modulation of the free sphingosine levels in Epstein Barr virus transformed human B lymphocytes by phorbol dibutyrate. Biochim Biophys Acta. 1991; 1095(1):90–92. | |
Poon AP, Liang Y, Roizman B. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no 0. J Virol. 2003;77(23):12671–12678. | |
Kalamvoki M, Roizman B. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc Natl Acad Sci U S A. 2010;107(41):17721–17726. | |
Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15(11):1312–1317. | |
Pinnoji RC, Bedadala GR, George B, Holland TC, Hill JM, Hsia SC. Repressor element-1 silencing transcription factor/neuronal restrictive silencer factor (REST/NRSF) can regulate HSV-1 immediate-early transcription via histone modification. Virol J. 2007;4:56. | |
Neumann DM, Bhattacharjee PS, Hill JM. Sodium butyrate: a chemical inducer of in vivo reactivation of herpes simplex virus type 1 in the ocular mouse model. J Virol. 2007;81(11):6106–6110. | |
Frazier DP, Cox D, Godshalk EM, Schaffer PA. The herpes simplex virus type 1 latency-associated transcript promoter is activated through Ras and Raf by nerve growth factor and sodium butyrate in PC12 cells. J Virol. 1996;70(11):7424–7432. | |
Manson McManamy ME, Hakre S, Verdin EM, Margolis DM. Therapy for latent HIV-1 infection: the role of histone deacetylase inhibitors. Antiviral Chem Chemother. 2014;23(4):145–149. | |
Wildes TM, Bartlett NL. Drug development for recurrent and refractory classical Hodgkin lymphoma. Leuk Lymphoma. 2009;50(4):529–540. | |
Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1(1):19–25. | |
Sanna G, Bertolaccini ML, Mathieu A. Central nervous system lupus: a clinical approach to therapy. Lupus. 2003;12(12):935–942. | |
Otsuki A, Patel A, Kasai K, et al. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol Ther. 2008;16(9):1546–1555. | |
Gottlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–6978. | |
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001; 276(39):36734–36741. | |
Bezecny P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol. 2014;31(6):985. | |
Flemington EK. Herpesvirus lytic replication and the cell cycle: arresting new developments. J Virol. 2001;75(10):4475–4481. | |
Liu TC, Castelo-Branco P, Rabkin SD, Martuza RL. Trichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effects. Mol Ther. 2008;16(6):1041–1047. | |
Hagglund R, Roizman B. Herpes simplex virus 1 mutant in which the ICP0 HUL-1 E3 ubiquitin ligase site is disrupted stabilizes cdc34 but degrades D-type cyclins and exhibits diminished neurotoxicity. J Virol. 2003;77(24):13194–13202. | |
Kalamvoki M, Roizman B. ICP0 enables and monitors the function of D cyclins in herpes simplex virus 1 infected cells. Proc Natl Acad Sci U S A. 2009;106(34):14576–14580. | |
Katsura T, Iwai S, Ota Y, Shimizu H, Ikuta K, Yura Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 2009; 16(3):237–245. | |
Ji Q, Hu H, Yang F, et al. CRL4B interacts with and coordinates the SIN3A-HDAC complex to repress CDKN1A and drive cell cycle progression. J Cell Sci. 2014;127(21):4679–4691. | |
Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond Ser B Biol Sci. 1957;147(927):258–267. | |
Silvennoinen O, Ihle JN, Schlessinger J, Levy DE. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature. 1993;366(6455):583–585. | |
Muller M, Briscoe J, Laxton C, et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature. 1993;366(6451):129–135. | |
Watling D, Guschin D, Muller M, et al. Complementation by the protein tyrosine kinase JAK2 of a mutant cell line defective in the interferon-gamma signal transduction pathway. Nature. 1993; 366(6451):166–170. | |
Wakabayashi T, Natsume A, Hashizume Y, Fujii M, Mizuno M, Yoshida J. A phase I clinical trial of interferon-beta gene therapy for high-grade glioma: novel findings from gene expression profiling and autopsy. J Gene Med. 2008;10(4):329–339. | |
Chiocca EA, Smith KM, McKinney B, et al. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol Ther. 2008;16(3):618–626. | |
Icardi L, Mori R, Gesellchen V, et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc Natl Acad Sci U S A. 2012;109(30):12058–12063. | |
Chang HM, Paulson M, Holko M, et al. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc Natl Acad Sci U S A. 2004;101(26):9578–9583. | |
Nusinzon I, Horvath CM. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc Natl Acad Sci U S A. 2003;100(25):14742–14747. | |
MacTavish H, Diallo JS, Huang B, et al. Enhancement of vaccinia virus based oncolysis with histone deacetylase inhibitors. PloS One. 2010;5(12):e14462. | |
Nguyen TL, Abdelbary H, Arguello M, et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A. 2008;105(39):14981–14986. | |
Lopez-Soto A, Huergo-Zapico L, Acebes-Huerta A, Villa-Alvarez M, Gonzalez S. NKG2D signaling in cancer immunosurveillance. Int J Cancer. 2015;136(8):1741–1750. | |
Alvarez-Breckenridge CA, Yu J, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18(12):1827–1834. | |
Alvarez-Breckenridge CA, Yu J, Price R, et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J Virol. 2012; 86(8):4566–4577. | |
Townsend MJ, Weinmann AS, Matsuda JL, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20(4):477–494. | |
Boucheron N, Tschismarov R, Goschl L, et al. CD4(+) T cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC2. Nat Immunol. 2014;15(5):439–448. | |
Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal. 2012;5(229):ra45. | |
Chang S, Collins PL, Aune TM. T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J Immunol. 2008;181(12):8372–8381. | |
Li N, Zhao D, Kirschbaum M, et al. HDAC inhibitor reduces cytokine storm and facilitates induction of chimerism that reverses lupus in anti-CD3 conditioning regimen. Proc Natl Acad Sci U S A. 2008; 105(12):4796–4801. | |
Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood. 2012;119(11):2443–2451. | |
Lee SY, Huang Z, Kang TH, et al. Histone deacetylase inhibitor AR-42 enhances E7-specific CD8(+) T cell-mediated antitumor immunity induced by therapeutic HPV DNA vaccination. J Mol Med. 2013;91(10):1221–1231. | |
Kato Y, Yoshino I, Egusa C, Maeda T, Pili R, Tsuboi R. Combination of HDAC inhibitor MS-275 and IL-2 increased anti-tumor effect in a melanoma model via activated cytotoxic T cells. J Dermatol Sci. 2014;75(2):140–147. | |
Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y, Shen H. Epigenetic manipulation restores functions of defective CD8(+) T cells from chronic viral infection. Mol Ther. 2014;22(9):1698–1706. | |
Woan KV, Lienlaf M, Perez-Villaroel P, et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation. Mol Oncol. 2015; 9(7):1447–1457. | |
Westrom BR, Tagesson C, Leandersson P, Folkesson HG, Svendsen J. Decrease in intestinal permeability to polyethylene glycol 1000 during development in the pig. J Dev Physiol. 1989;11(2):83–87. | |
Bartlett DL, Liu Z, Sathaiah M, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12(1):103. | |
Bridle BW, Chen L, Lemay CG, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013;21(4):887–894. | |
Lang KS, Recher M, Junt T, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11(2):138–145. | |
Teijaro JR, Ng C, Lee AM, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013; 340(6129):207–211. | |
Wilson EB, Yamada DH, Elsaesser H, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340(6129):202–207. | |
Ruiz AJ, Russell SJ. MicroRNAs and oncolytic viruses. Curr Opin Virol. 2015;13:40–48. | |
Okemoto K, Kasai K, Wagner B, et al. DNA demethylating agents synergize with oncolytic HSV1 against malignant gliomas. Clin Cancer Res. 2013;19(21):5952–5959. | |
Shulak L, Beljanski V, Chiang C, et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-kappaB-dependent autophagy. J Virol. 2014;88(5):2927–2940. | |
Hoti N, Chowdhury W, Hsieh JT, Sachs MD, Lupold SE, Rodriguez R. Valproic acid, a histone deacetylase inhibitor, is an antagonist for oncolytic adenoviral gene therapy. Mol Ther. 2006;14(6):768–778. | |
Watanabe Y, Hashimoto Y, Kagawa S, et al. Enhanced antitumor efficacy of telomerase-specific oncolytic adenovirus with valproic acid against human cancer cells. Cancer Gene Ther.2012;19(11):767–772. | |
Kim DR, Park MY, Lim HJ, et al. Combination therapy of conditionally replicating adenovirus and histone deacetylase inhibitors. Int J Mol Med. 2012;29(2):218–224. | |
Watanabe T, Hioki M, Fujiwara T, et al. Histone deacetylase inhibitor FR901228 enhances the antitumor effect of telomerase-specific replication-selective adenoviral agent OBP-301 in human lung cancer cells. Exp Cell Res. 2006;312(3):256–265. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.