")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Combined Pulmonary Fibrosis Emphysema: Role of Cigarette Smoking and Pulmonary Hypertension in a Rural Cohort
Authors Sangani R , Ghio A, Culp S, Patel Z, Sharma S
Received 5 March 2021
Accepted for publication 20 May 2021
Published 21 June 2021 Volume 2021:16 Pages 1873—1885
DOI https://doi.org/10.2147/COPD.S307192
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Rahul Sangani,1 Andrew Ghio,2 Stacey Culp,3 Zalak Patel,4 Sunil Sharma1
1Section of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, West Virginia University, Morgantown, WV, USA; 2Human Studies Facility, US Environmental Protection Agency, Chapel Hill, NC, USA; 3Department of Biostatistics, West Virginia University, Morgantown, WV, USA; 4Department of Radiology, West Virginia University, Morgantown, WV, USA
Correspondence: Andrew Ghio
Human Studies Facility, US Environmental Protection Agency, Chapel Hill, NC, USA
Tel +1 919 966-0670
Fax +1 919 966-6271
Email [email protected]
Background: Disease heterogeneity in idiopathic pulmonary fibrosis (IPF) often complicates the systematic study of disease, management of patients and clinical investigations.
Objective: To describe combined pulmonary fibrosis emphysema (CPFE) phenotype in a rural Appalachian IPF cohort with the highest smoking rates in the United States.
Methods: CPFE patients (n = 60) in a developed IPF cohort (n = 153) were characterized. Groups (CPFE vs IPF without emphysema) were categorized based on the predominant HRCT patterns of UIP (n = 109). Demographics, clinical variables, and treatment details were recorded. Kaplan–Meier survival and multivariate logistic regression analysis were performed.
Results: The prevalence of CPFE in our IPF cohort was 45% (n = 49). The CPFE group was younger (73.9 vs 78.2), had a more extensive smoking history (93.9% vs 53.3%) with greater mean smoking pack years (49.09 vs 15.39) and had lower percentage predicted DLCO on presentation (38.35 vs 51.09) compared to IPF without emphysema group. Both groups shared equivalent higher burden of comorbidities, including pulmonary hypertension (PH) (46.9% vs 33.3%). One-fifth of patients were prescribed antifibrotics and only a subset (5%) of patients underwent lung transplantation. There was a non-significant trend towards reduced survival in CPFE (p = 0.076). Smoking status and DLCO predicted CPFE in our cohort. Body mass index (BMI), PH, and pirfenidone use were significant predictors of mortality.
Conclusion: CPFE was highly prevalent in our rural IPF cohort. In contrast to previous studies, CPFE group was older and had higher female (approx. 30%) occurrence. A greater exposure to cigarette smoke and reduced DLCO at diagnosis predicted CPFE. Lower BMI and PH predicted higher mortality whereas use of pirfenidone improved survival in our cohort. This study highlights a complex interaction of cigarette smoking, advanced fibrosis of UIP, PH and potential utility of antifibrotic agents in CPFE phenotype. Substantial burden of comorbidities, older age, and the limited utilization of advanced therapeutics in the cohort emphasize the challenges faced by rural Appalachian patients.
Keywords: combined pulmonary fibrosis emphysema; CPFE, cigarette smoking, pulmonary hypertension; PH, pirfenidone, body mass index; BMI, rural population
Introduction
The clinically accepted dichotomy between emphysema and pulmonary fibrosis can reflect the constraints of the current diagnostic approach resulting in an imperfect recognition of the coexistence of these entities in the same individuals.1 However, there is increasing appreciation that emphysema and pulmonary fibrosis can be documented in the same patient, producing a clinical syndrome known as combined pulmonary fibrosis and emphysema (CPFE).2–4 The diagnosis of the CPFE syndrome is established using high resolution CT (HRCT) imaging with pathology when available. The radiographic appearance includes the presence of upper-lobe emphysema and lower-lobe pulmonary fibrosis.5–7 Clinical characteristics can include male gender, a heavy smoking history, the presence of significant dyspnea, relatively preserved lung volumes but a disproportionate impairment of gas exchange, and hypoxemia.8 Pathology frequently reveals both usual interstitial pneumonia and emphysema. The effect of emphysema on pulmonary function tests (PFT) likely contributes to alter or mask the estimation of baseline disease severity and its progression over time.9 Early identification of this phenotype potentially has important clinical implications as CPFE is often complicated by pulmonary hypertension (PH), lung cancer, acute exacerbation of underlying disease, and postoperative cardiopulmonary complications.8,10,11
It has been proposed that a combined syndrome of pulmonary fibrosis and emphysema can be described among individuals with idiopathic pulmonary fibrosis (IPF). Reports suggest that the incidence of such CPFE among IPF cohorts may include up to 35% of patients.8 The characterization of CPFE as a distinct clinical entity has identified several inherent limitations including a lack of an international consensus definition of CPFE, variable interpretation of honeycombing on radiographs particularly when emphysema is admixed with reticulation, an impact of coincidental findings of smoking related ILDs (e.g., respiratory bronchiolitis-interstitial lung disease [RB-ILD], desquamative interstitial pneumonia [DIP], and unclassifiable smoking-related interstitial fibrosis [SRIF]), and an underestimation of disease severity with employment of FVC for clinical monitoring.12–14 The recently recognized pattern of airspace enlargement with fibrosis (AEF) can represent an additional complicating feature of CPFE with predominant thick-walled cysts in lower lung zones in association with reticulation.15 Reflecting this uncertainty, a variable impact of emphysema on survival of IPF patients has been reported.16
At our rural central Appalachian academic center, we are uniquely challenged with the highest rates of smoking (26.7%) in the United States.17 We present a detailed analysis of a locally identified CPFE phenotype and compare it to an IPF cohort without emphysema. Considering the recommended approach of avoiding variable disease severity between the groups, we performed our analysis of outcome measures (CPFE vs IPF without emphysema) in a cohort with radiologic evidence supporting an advanced fibrosis pattern of UIP.18 While evaluating the complex interactions of smoking with injury patterns of fibrosis, emphysema, and PH, this study also examines the potential role of antifibrotic agents in this challenging population.
Materials and Methods
Subjects
A single-center, retrospective, observational cohort study was conducted at West Virginia University Hospital. The study protocol was reviewed and approved by our institutional review board (ID #1904548975), which waived the requirement for informed consent. During the study period of January 1, 2015 to December 31, 2019, the target patients were identified with International Classification of Diseases (ICD) coding for the idiopathic ILD (ICD 9-CM: 515–516.9; ICD-10-CM J84-84.9) within the electronic medical records (EMR) of West Virginia University Hospital (WVUH) system.
Case Definitions
Charts were reviewed systematically and IPF was diagnosed in accordance to the latest ATS/ERS/JRS/ALAT diagnostic guidelines.19 These diagnostic criteria require exclusion of other known causes of ILD (domestic and occupational environmental exposures, connective tissue disease and, drug toxicity), and either the presence of the HRCT pattern of UIP or specific combinations of HRCT patterns and histopathology patterns in patients subjected to lung tissue sampling. CPFE was defined as basilar UIP pattern on HRCT with coexisting upper lobe predominant emphysematous changes (centrilobular and/or paraseptal).8 Patients were further categorized in subgroups of: 1) CPFE; or 2) IPF without emphysema. Figure 1 describes the development of the study groups.
 |
Figure 1 Development of the study groups. |
Data Collection
We collected demographic and historical data including age, gender, co-morbid conditions, smoking status, family history, symptom(s) duration prior to diagnosis, years since diagnosis, and whether the patient was evaluated at the tertiary or ILD center. In addition, clinical variables of interest including PFT (maximum of 3 which included that testing at the time of diagnosis, intermediate and the latest), six-minute walk distance (6MWD), use of supplemental oxygen, use of antifibrotic agents (pirfenidone and/or nintedanib), and lung transplant status were also recorded. Data was aggregated using REDCap, which is a HIPAA compliant data aggregation tool.20
Statistical Analysis
Descriptive statistics were generated, using means, medians, and standard deviations to summarize the continuous variables and frequency distributions to describe the categorical variables. Chi-square tests were used to detect differences in categorical variables by group, while means of continuous variables were compared using two-sided independent-samples t-tests. The Mann–Whitney U-test was used when normality could not be assumed. Logistic regression analysis was used to determine significant predictors of group status (CPFE vs IPF without emphysema) and to predict mortality at the end of the study. Kaplan-Meier curves were generated, and the Log rank test was used to compare survival since diagnosis between the two groups. Level of significance α = 0.05 was used for all analyses. All analyses were conducted using statistical software SPSS version 26.0.
Results
From 2015–2019, there was a total of 453 patients who had a diagnostic code of idiopathic ILD within the EMR of WVUH system (Figure 1). Applying the most recent ATS/ERS/JRS/ALAT guidelines, 153 of the 453 patients (33.8%) were found to meet the diagnostic criteria for IPF (Figure 1). Two hundred twenty-seven patients (50.1%) were determined to have non-IPF ILD diagnosis and 73 patients (16.1%) did not have HRCT in the system to allow an independent review (Figure 1). UIP was the most common HRCT pattern (n = 109, 71.2%), followed by probable UIP (n = 29, 19%). A minority of patients had a CT scan which was considered either indeterminate for UIP (n = 10, 6.5%) or supportive of an alternate diagnosis (n = 5, 3.3%). However, with multidisciplinary discussion and availability of pathology reports, we were able to include these patients in the final IPF cohort.
Table 1 describes the baseline demographic, comorbidity, diagnostic and management details of the cohort with UIP pattern in CPFE and IPF without emphysema groups. Of total 109 subjects, there were 49 patients in the CPFE group (45%) and 60 patients in the IPF without emphysema (55%). The CPFE group was significantly younger than the non-CPFE IPF group (73.90±10.42 vs 78.20±9.46 years, p = 0.026) and they had both a higher percentage of ever-smokers (93.9% vs 53.3%m, p < 0.001) and a greater smoking history (49.09±29.58 vs 15.39±14.84 pack years, p < 0.001). Ever smoking history of the CPFE and IPF without emphysema groups consisted of 76.7% (n = 46/60) and 51.6% (n = 48/93) former smokers and 18.3% (n = 11/60) and 1.1% (n = 1/93) current smokers, respectively. Approximately 60% of patients in each group were male and were overweight with a mean body mass index (BMI) of 27.12±4.77 vs 27.17±5.29 kg/m2, respectively (p = 0.958). A family history of IPF was provided by a minority of patients (10.6% vs 8.8%). An excess of comorbidities was observed among the patients of both groups and these were not significantly different in prevalence. The comorbidities included hypertension (83.7% vs 70%), hyperlipidemia (75.5% vs 73.3%), gastro-esophageal reflux disease (GERD; 73.5% vs 66.7%), coronary artery disease (CAD; 61.2% vs 53.3%), PH (46.9% vs 33.3%), heart failure (HF; 44.9% vs 31.7%), atrial arrhythmia (34.7% vs 33.3%), anemia (32.7% vs 33.3%) sleep disordered breathing (30.6% vs 38.3%), diabetes (30.6% vs 30.6%), anxiety/mood disorders (28.6% vs 28.3%) and lung cancer (10.2% vs 3.3%). Approximately half of patients in each group were seen at the tertiary or specific ILD center. Two thirds of patients in the cohort carried a prevalent diagnosis whereas one third had an incident (within 6 months of presentation) diagnosis and this was not significantly different between the CPFE and non-CPFE groups. The average duration of symptoms prior to diagnosis was 20 months, however it ranged from 1 month to 10 years. A minority of patients (12.2% vs 15%, p=0.678) underwent surgical lung biopsy.
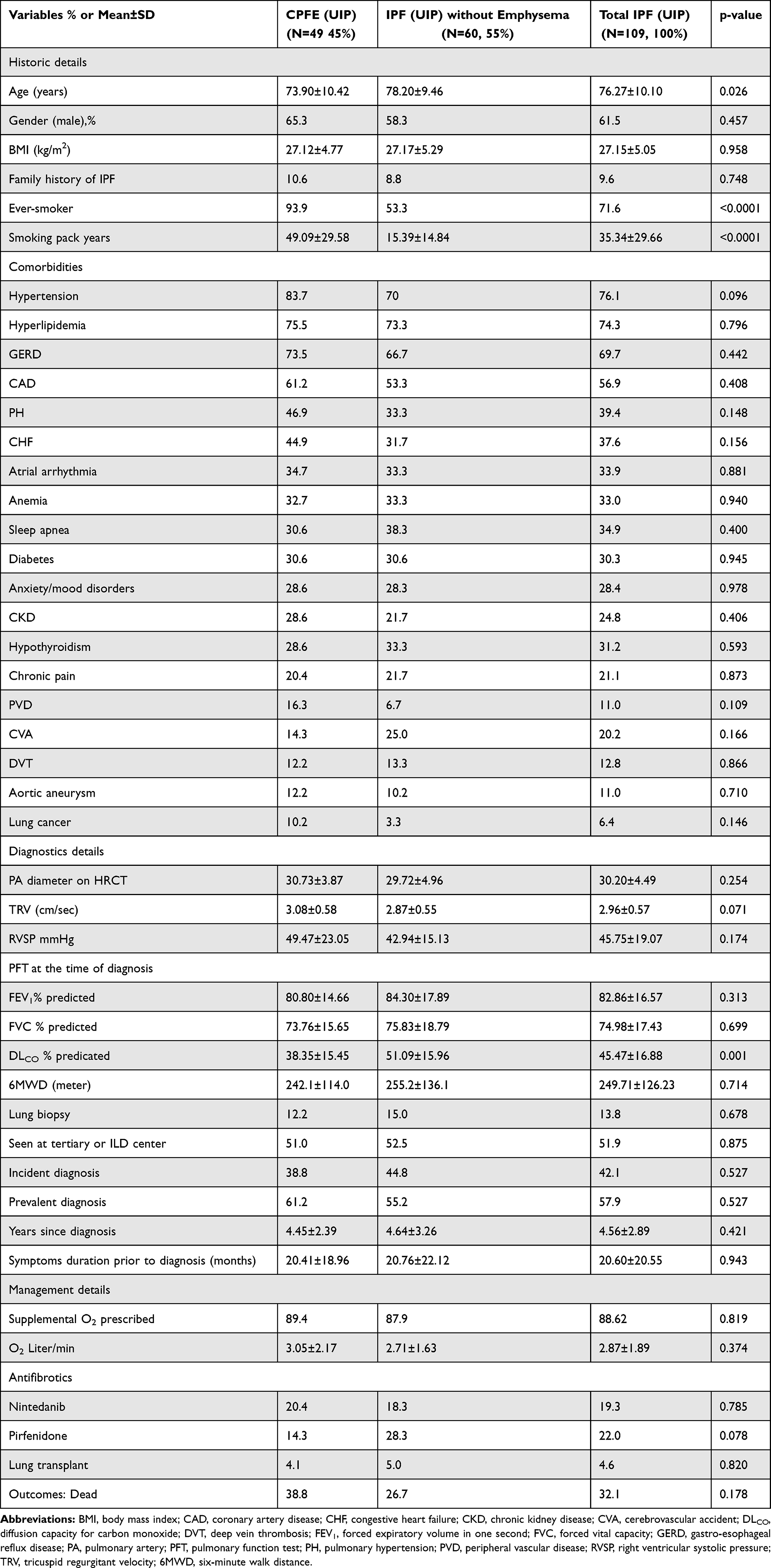 |
Table 1 Clinical Characteristics and Outcomes in the CPFE and IPF Without Emphysema Groups |
Diagnostic parameters of main pulmonary artery (PA) diameter on HRCT (30.73±3.87 vs 29.72±4.96 in mm, p = 0.254) and transthoracic echocardiography (TTE) measurement of right ventricular systolic pressure (RVSP) (49.47±23.05 vs 42.94±15.13 in mmHg, p = 0.174) were not different between the CPFE vs IPF without emphysema groups, respectively. Interestingly, with inclusion of all HRCT patterns in the analysis, the CPFE group showed higher comorbidity burden of PH (45% vs 29%, p = 0.044), significant dilatation of the main PA on HRCT (30.77±4.08 vs 28.60±4.80 in mm, p = 0.005) and elevated RVSP on TTE (50.52±22.40 vs 42.21±14.11 in mmHg respectively, p=0.044). In our cohort, dilatation of the main PA was noted to be an excellent indicator for PH. Main PA diameter has been shown as an useful non-invasive tool for diagnosing PH.21 Mean PA diameter was enlarged to 32.41±3.56 mm in patients with PH vs 27.75±4.32 mm in patients without PH (p = 0.0001).
The CPFE group had a significantly reduced percent predicted diffusion capacity for carbon monoxide (DLCO) at diagnosis compared to IPF without emphysema group (38.35±15.45 vs 51.09±15.96 mL/min/mm Hg respectively, p = 0.001). Percent predicted values for forced vital capacity (FVC) and forced expiratory volume at one second (FEV1) were not statistically different between the groups. In the CPFE and IPF without emphysema groups, mean FEV1 and FVC values at diagnosis were 2.15±0.57 vs 2.04±0.70 liters and 2.68±0.73 vs 2.49±0.95 liters, respectively. During the study, there were decrements in both FEV1 and FVC with latest values of 2.09±0.5 vs 2.01±0.6 liters and 2.59±0.65 vs.2.42±0.73 liters but these were not significant. Similarly, both groups had reduced six-minute walk distance (6MWD; 242.1±114.0 vs 255.2±136.1 meters respectively, p = 0.714). There was progressive decline of pulmonary function over time. Figure 2 demonstrates the trends of FEV1, FVC and DLCO during the study duration in the two groups.
 |
Figure 2 Trend of pulmonary function tests between CPFE vs IPF without emphysema groups. Time 1: at the time of diagnosis; Time 2: latest during the disease course. There was no significant difference in the differences of decline in PFT data when measured at two different time frames of disease course (at time of diagnosis and the latest). (A) FEV1 and FVC % predicted at time 1 and 2 between the groups (non-significant difference). (B) DLCO % predicated at time 1 and 2 between the groups. CPFE group had significantly lower DLCO % predicted at the time 1 (38.35±15.45 vs 51.09±15.96, p = 0.001). |
A majority of the patients included in the study were oxygen dependent at the time of diagnosis. The mean requirement for supplemental O2 was not dissimilar between the groups (3.05±2.17 vs 2.71±1.63 L/minute respectively, p = 0.374). Approximately 20% of patients were prescribed antifibrotic agents (nintedanib or pirfenidone) and the prescription practice was not different between the groups. Only a subset of patients underwent lung transplantation (4.1% vs 5% respectively, p = 0.820).
Table 2 describes the predictors for CPFE in a multivariate logistic model. Ever smoking status and reduced percentage predicted DLCO at the time of diagnosis were found to be significant predictors of CPFE within our cohort (OR = 29.83, 95% CI 3.56–249.82, p = 0.002 and OR = 0.93, 95% CI of 0.89–0.97, respectively; p = 0.001).
 |
Table 2 Multivariate Logistic Regression Analysis: Predictors of CPFE |
Figure 3 shows the Kaplan–Meier survival curve for the two groups. Approximately, one third of patients from our cohort died and there was no significant difference in survival between the subjects in the CPFE vs IPF without emphysema groups (38.8% vs 26.7% respectively, p = 0.178) (Table 1). There was a trend towards a decreased survival for CPFE patients, but this was not statistically significant (Log rank test, p = 0.076). The mean years alive since diagnosis was 3.61±2.62 vs 4.31±3.06 years (p = 0.215), respectively in the groups. The median survival for both groups was 3.00 years. Multivariate logistic regression analysis (Table 3) showed predictors of survival in the total IPF cohort to include BMI (OR = 1.10, 1.005–1.207, 95% CI, p = 0.038), PH (OR = 0.215, 0.085–0.544, 95% CI, p = 0.001), and pirfenidone (OR = 3.698, 1.040–13.153, 95% CI, p = 0.043), indicating that those with a higher BMI, without PH, and taking pirfenidone had an improved survival. With inclusion of percent predicted DLCO into the logistic regression model, significant predictors of survival in the IPF cohort could not be identified due to missing values at the time of diagnosis.
 |
Table 3 Multivariate Logistic Regression Analysis: Predictors of Survival for Total IPF Cohort |
 |
Figure 3 Kaplan–Meier survival analysis of CPFE and IPF without emphysema groups. There was a trend towards reduced survival between the groups over time since their diagnosis, however not statistically different (log-rank p = 0.076). X-axis signifies years alive since diagnosis during the study time frame and Y-axis shows survival rate. The mean years since diagnosis between the groups were 4.45±2.39 vs 4.64±3.26 (p = 0.740), respectively. |
Discussion
Since the introduction of CPFE as a clinical syndrome by Cottin et al in 2005, there has been an ongoing evaluation of the impact of emphysema in the natural history of pulmonary fibrosis.7,8 The need for future studies to determine the etiology, morbidity, mortality and novel therapeutics for CPFE either with or without PH, has been emphasized.22 Accordingly, we identified a cohort with the CPFE phenotype in a rural Appalachian population, demonstrating one of the highest smoking rates in the United States, to delineate associations between cigarette smoking, fibrosis pattern of usual interstitial pneumonia, PH, and potential utility of antifibrotic agent.
In the locally identified IPF cohort included in this investigation, the group demonstrating CPFE manifested several distinctive features. CPFE was highly prevalent (45%) comparable to previous studies.23 While two thirds of our CPFE patients were male, this is lower compared to a previously noted remarkable (9:1) male predominance.24 Our cohort demonstrated that females shared a significant smoking habit with males in this rural Appalachian region of WV (25.6% vs 27.8%).25 Diagnostic delay (approximately 20 months) was common with only half patients were seen at specific ILD center in our rural cohort. Barriers to early diagnosis in IPF were noted to be multifactorial including older age, male gender, referral pattern from general practitioners and impact of comorbid conditions and alternate diagnoses.26 While our CPFE patients were significantly younger than those in the IPF without emphysema group, they were older than other comparable cohorts included in previous reports.24 The CPFE group shared an equivalent burden of comorbidities, including obstructive sleep apnea, which were associated with the clinical course of disease.27,28 Comorbid PH in CPFE has been shown to cause severe dyspnea (functional class III or IV), exercise limitation (reduced 6MWD) and earlier onset hemodynamic changes on right heart catheterization.29 Similar phenomenon of exercise limitation and relatively severe PH observed in our CPFE group may have contributed to younger age presentation for diagnostic evaluation. Well known physiologic consequences of CPFE were evident with relatively preserved spirometry but severely reduced DLCO compared to IPF without emphysema, both of which decline progressively during the disease course.23,30 This gas exchange abnormality contributed to the need for supplemental oxygen in the majority of patients.1 DLCO has been demonstrated to reflect effects of both emphysema and fibrosis and is considered a measure of disease severity for CPFE.31 For the entire cohort, use of antifibrotics was limited to one fifth of patients whereas only a subset (5%) of patients underwent lung transplantation. Despite the recommendation of prompt initiation of antifibrotics, multiple barriers have been identified for the early adoption of antifibrotics.32 Rural health disparity in terms of known socio-economic challenges, community hospitals having limited access to an ILD referral or lung transplant center, older age, and a higher burden of comorbidities may have restricted our patients’ eligibility for advance therapeutics.
The pathogenesis of CPFE remains poorly understood. Cigarette smoking has been described as a major risk factor for development of both emphysema and IPF.33,34 A meta-analysis demonstrated that smokers had 1.38 times higher risk for developing IPF.35 A majority of smokers with IPF do not demonstrate radiographic evidence of emphysema. Likewise, most patients with emphysema/COPD are not reported to manifest interstitial fibrosis. Recent HRCT data indicated that interstitial lung abnormality was inversely associated with the presence of emphysema in smokers,36 suggesting that these patterns may represent distinct outcomes of smoking that reflect unique individual susceptibilities. Fast metabolizers with greater CYP2A6 activity have been shown to have a higher occurrence of COPD suggesting a production of toxic molecules with smoking.37 Despite these observations, evidence suggests that these two diseases can assume concomitant courses in smokers. Our CPFE cohort had a higher mean smoking (>40 pack years) exposure and ever smoking status was found to be one of the major determinants of CPFE. Animal experiments have confirmed that tobacco smoke can lead to the occurrence of emphysema and pulmonary fibrosis simultaneously.38 Histologically, the co-existence of emphysema and fibrosis without craniocaudal segregation has been recognized.39 In addition, over half of lobectomy specimens from a cohort of smokers with lung cancer have subclinical interstitial fibrosis and have only radiographic evidence of emphysema.3 Although CPFE has been recently classified in the group of smoking-induced chronic lung diseases,40 analysis of pathogenic mechanisms of smoke-induced lung diseases can provide insight into the homeostatic pathways critical to preserve lung health.41
In addition to airway and parenchymal damage in the lung, there is increasing evidence that cigarette smoke directly impacts the pulmonary vasculature suggesting that PH and cor pulmonale are not products of hypoxia alone.42 Thickening of the arterial intima and vessel narrowing are noted as early changes in smokers’ lungs which correlate with the severity of emphysema and bronchiolitis during disease progression.43,44 Cigarette smoke exposure produces mediators that control vasoreactivity and vascular cell proliferation ultimately leading to remodeling.45 In addition, it causes direct oxidative damage, inflammation and hypercoagulability, all potentially contributing to the development of PH.46 Autopsy series of CPFE, IPF and emphysema patients suggested that pulmonary vasculopathy developed in whole lung tissues including fibrosed, emphysematous and preserved lung areas. Notably, these changes were seen significantly higher in unaffected CPFE lung tissue compared to other groups.47 Comparing the groups with all HRCT fibrotic patterns, we observed a higher proportion of comorbid PH and its non-invasive markers (main PA diameter on HRCT and TTE defined RVSP) however, such an association was not evident when limiting the groups to the advanced fibrosis of UIP. Comorbid PH has been shown to impact the survival of CPFE patients but the association of PH with survival was no longer significant in these patients after controlling for baseline CT extent of fibrosis and emphysema.8,10,29,48 This suggests the co-existence of fibrosis, emphysema, and PH as analogous tissue injuries resulting from exposure to cigarette smoke. Prior systemic study of HRCT has observed excess radiologic severity of emphysema and fibrosis in smokers with IPF relative to smokers with non-IPF UIP pattern, lending credence to the hypothesis of prevailing pathobiologic synergy between IPF, emphysema and smoking.49 Smoking cessation has been effective in improving several clinical and physiologic parameters in COPD.50 It may be prudent to design studies which can determine the true order of progression of changes and develop optimal smoking cessation strategy and medications (e.g., antifibrotics) for overlapping therapeutic effects across this spectrum of pathologies.45,51
After more than a decade of research in CPFE, the prognostic implication of comorbid emphysema in IPF remains unclear. One third of patients died during our study duration with a non-significant trend toward reduced survival in CPFE compared to IPF without emphysema (p = 0.076). A recent meta analysis of 13 studies showed comparable poor survival rates at one, three and five years among CPFE and IPF alone.52 Most studies evaluating the impact of emphysema on IPF prognosis have failed to adjust for baseline severity and hence yielded mixed results. CPFE patients are likely to have less extensive fibrosis at the time of diagnosis and this is associated with a comparable poor outcome as IPF alone while CPFE portends a worse prognosis when these patients and those with IPF present with similar ILD burden.18,53 In addition to the visual review of HRCT, the computer-based analytical system of CALIPER has been shown to provide an improved adjustment for baseline severity.31 Given the inherent limitation of FVC in CPFE, a composite physiologic index (CPI) has similarly been developed to predict mortality in IPF patients with or without emphysema.54 Longitudinal analysis of an IPF registry has shown an association of disease severity, based on physiological impairment and phenotypic clustering, to the outcomes of patients.55 While adjusting for a similar UIP pattern between the groups, our multivariate analysis suggested BMI and pirfenidone as predictors of survival in addition to PH. It’s not surprising to observe lower BMI reduced survival in CPFE given its known association for poor prognosis in IPF and emphysema individual disease processes.56,57 Since spirometric endpoints can function as determinants in initiating antifibrotic therapy, the presence of emphysema potentially is problematic since it can affect FVC in a manner dissimilar to fibrosis.58 A single center retrospective study of 45 CPFE patients detected no significant difference in the efficacy of either pirfenidone or nintedanib on measured variables including mortality.59 Randomized control trials, meta-analysis, and an observation study of pirfenidone in IPF have observed improved progression-free survival and reduced all-cause mortality.60–62 However, such an impact has not yet been established in CPFE. Despite a primary end-point focused on FVC, a post-hoc analysis of the INPULSIS trial suggested that the treatment effect of nintedanib was not affected by baseline emphysema.63 Overall, there’s no randomized trial evidence of any impact of antifibrotics on mortality. Notably, given a minority of patients prescribed antifibrotics in our retrospective cohort (22% pirfenidone vs 19.3% nintedanib), such an evaluation for a differential efficacy on mortality is not feasible. Lastly, more than 10% emphysema in IPF provided good discrimination for CPFE where reduced DLCO was the most reliable prognostic marker.64 Considering lack of quantification of emphysema in our CPFE cohort and missing DLCO values for patients who died during the study, we did not identify DLCO as a significant prognostic marker. In addition, over the course of study duration, DLCO reduction was equivalent between the groups with no significant difference noted in the value at the end of study. Akagi et al have recorded similar slower annual decrease of percentage predicted DLCO in CPFE patients as compared to IPF-alone patients.12
Our study notes few limitations. There are several inherent diagnostic challenges described in developing IPF cohorts including evolving diagnostic criteria, data collection practices, identifying an accurate time of diagnosis, and impact of missing values. Certainly, the last was evident in our cohort as 16% patients had missing data precluding their inclusion in the study. HRCT characterization of IPF cohort required exclusion of CTD-associated ILD which can also be associated with CPFE, but such an exclusion may not be totally effective. In addition, we did not quantify the extent of emphysema on HRCT. Survival analysis could have been underestimated as some patients may had shorter follow-up given recent diagnosis, and some patients may have been transferred to other referral centers. Lastly, the unmeasured confounding factors may have contributed to the findings of the study.
Conclusion
Our efforts to define CPFE in a rural Appalachian population with the highest smoking rates in the USA provided key epidemiological, clinical, radiographic, and pathologic insights. CPFE was highly prevalent (45%) in locally identified IPF cohort. Relative to previously defined cohorts, CPFE patients were older, had a higher prevalence of females, carried a major burden of comorbidities, and had limited implementation of advanced therapeutics. PH was a prevalent (approximately 40%) finding between the groups, however, inclusive of all HRCT patterns, PH was noted to be significantly severe in CPFE group. The results of this investigation support the hypothesis that PH presents as a co-existing smoke-induced pathology rather than a comorbid manifestation of chronic hypoxemia. Smoking status and gas exchange impairment with reduced DLCO were significant predictors of CPFE. Both CPFE and IPF without emphysema were progressively fatal conditions but a non-significant trend towards reduced survival in CPFE was observed. Higher BMI, lack of PH and use of pirfenidone improved survival in our cohort which could propose a potential utility of antifibrotics for CPFE. Our study results suggest that fibrosis, PH, and emphysema likely represent a spectrum of smoke induced injury. Prospective studies are required to confirm these findings, to identify the true order of smoke lung pathology so that preventive measures can be undertaken in timely manner, and to better recognize novel therapeutic targets.
Statement of Ethics
The study protocol was approved by the West Virginia University Institutional Review Board (#1904548975, date 01-10-2020).
Consent to participate not applicable given retrospective nature of the study and was waived by IRB.
All relevant ethical safeguards have been met in relation to patient protection.
The study was conducted in accordance with the Declaration of Helsinki.
Transplant patients: all organs were donated voluntarily with written informed consent, and that this was conducted in accordance with the Declaration of Istanbul.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no relevant conflict of interests in relation to this work.
References
1. Jankowich MD, Rounds S. Combined pulmonary fibrosis and emphysema alters physiology but has similar mortality to pulmonary fibrosis without emphysema. Lung. 2010;188(5):365–373. doi:10.1007/s00408-010-9251-6
2. Auerbach O, Garfinkel L, Hammond EC. Relation of smoking and age to findings in lung parenchyma: a Microscopic Study. Chest. 1974;65(1):29–35. doi:10.1378/chest.65.1.29
3. Katzenstein AL, Mukhopadhyay S, Zanardi C, Dexter E. Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Pathol. 2010;41(3):316–325. doi:10.1016/j.humpath.2009.09.003
4. Keller CA, Naunheim KS, Osterloh J, Espiritu J, McDonald JW, Ramos RR. Histopathologic diagnosis made in lung tissue resected from patients with severe emphysema undergoing lung volume reduction surgery. Chest. 1997;111(4):941–947. doi:10.1378/chest.111.4.941
5. Wiggins J, Strickland B, Turner-Warwick M. Combined cryptogenic fibrosing alveolitis and emphysema: the value of high resolution computed tomography in assessment. Respir Med. 1990;84(5):365–369. doi:10.1016/S0954-6111(08)80070-4
6. Lim TK. Respiratory failure from combined emphysema and pulmonary fibrosis. Singapore Med J. 1993;34(2):169–171.
7. Cottin V, Cordier JF. The syndrome of combined pulmonary fibrosis and emphysema. Chest. 2009;136(1):1–2. doi:10.1378/chest.09-0538
8. Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26(4):586–593. doi:10.1183/09031936.05.00021005
9. Ley B, Collard HR, King TE
10. Mejia M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136(1):10–15. doi:10.1378/chest.08-2306
11. Sato S, Koike T, Hashimoto T, et al. Surgical outcomes of lung cancer patients with combined pulmonary fibrosis and emphysema and those with idiopathic pulmonary fibrosis without emphysema. Ann Thorac Cardiovasc Surg. 2016;22(4):216–223. doi:10.5761/atcs.oa.15-00315
12. Akagi T, Matsumoto T, Harada T, et al. Coexistent emphysema delays the decrease of vital capacity in idiopathic pulmonary fibrosis. Respir Med. 2009;103(8):1209–1215. doi:10.1016/j.rmed.2009.02.001
13. Ciccarese F, Attina D, Zompatori M. Combined pulmonary fibrosis and emphysema (CPFE): what radiologist should know. Radiol Med. 2016;121(7):564–572. doi:10.1007/s11547-016-0627-4
14. Watadani T, Sakai F, Johkoh T, et al. Interobserver variability in the CT assessment of honeycombing in the lungs. Radiology. 2013;266(3):936–944. doi:10.1148/radiol.12112516
15. Inomata M, Ikushima S, Awano N, et al. An autopsy study of combined pulmonary fibrosis and emphysema: correlations among clinical, radiological, and pathological features. BMC Pulm Med. 2014;14(1):104. doi:10.1186/1471-2466-14-104
16. Lai RS, Chen CF, Chu KA, Lin MH. The effect of emphysema on survival in patients with idiopathic pulmonary fibrosis: a retrospective study in Taiwan. J Chin Med Assoc. 2019;82(12):922–928. doi:10.1097/JCMA.0000000000000201
17. Prevention WVDoT. Addressing Tobacco Use and Its Associated Health Conditions in West Virginia. Promotion OoCHSaH, editor. 2016.
18. Cottin V. Combined pulmonary fibrosis and emphysema: bad and ugly all the same? Eur Respir J. 2017;50(1):1700846. doi:10.1183/13993003.00846-2017
19. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68. doi:10.1164/rccm.201807-1255ST
20. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi:10.1016/j.jbi.2008.08.010
21. Shen Y, Wan C, Tian P, et al. CT-base pulmonary artery measurement in the detection of pulmonary hypertension: a meta-analysis and systematic review. Medicine (Baltimore). 2014;93(27):e256. doi:10.1097/MD.0000000000000256
22. Cottin V. The impact of emphysema in pulmonary fibrosis. Eur Respir Rev. 2013;22(128):153–157. doi:10.1183/09059180.00000813
23. Lin H, Jiang S. Combined pulmonary fibrosis and emphysema (CPFE): an entity different from emphysema or pulmonary fibrosis alone. J Thorac Dis. 2015;7(4):767–779. doi:10.3978/j.issn.2072-1439.2015.04.17
24. Jankowich MD, Rounds SIS. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141(1):222–231. doi:10.1378/chest.11-1062
25. Nguyen KH, Marshall L, Brown S, Neff L. State-specific prevalence of current cigarette smoking and smokeless tobacco use among adults - United States, 2014. MMWR Morb Mortal Wkly Rep. 2016;65(39):1045–1051. doi:10.15585/mmwr.mm6539a1
26. Hoyer N, Prior TS, Bendstrup E, Wilcke T, Shaker SB. Risk factors for diagnostic delay in idiopathic pulmonary fibrosis. Respir Res. 2019;20(1):103. doi:10.1186/s12931-019-1076-0
27. Buendia-Roldan I, Mejia M, Navarro C, Selman M. Idiopathic pulmonary fibrosis: clinical behavior and aging associated comorbidities. Respir Med. 2017;129:46–52. doi:10.1016/j.rmed.2017.06.001
28. Caminati A, Lonati C, Cassandro R, et al. Comorbidities in idiopathic pulmonary fibrosis: an underestimated issue. Eur Respir Rev. 2019;28:153. doi:10.1183/16000617.0044-2019
29. Cottin V, Le Pavec J, Prevot G, et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35(1):105–111. doi:10.1183/09031936.00038709
30. Amariei DE, Dodia N, Deepak J, et al. Combined pulmonary fibrosis and emphysema: pulmonary function testing and a pathophysiology perspective. Medicina (Kaunas). 2019;55(9). doi:10.3390/medicina55090580
31. Jacob J, Bartholmai BJ, Rajagopalan S, et al. Functional and prognostic effects when emphysema complicates idiopathic pulmonary fibrosis. Eur Respir J. 2017;50(1). doi:10.1183/13993003.00379-2017
32. Maher TM, Strek ME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir Res. 2019;20(1):205. doi:10.1186/s12931-019-1161-4
33. Stockley RA, Mannino D, Barnes PJ. Burden and pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6(6):524–526. doi:10.1513/pats.200904-016DS
34. Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
35. Park Y, Ahn C, Kim T-Y. Occupational and environmental risk factors of idiopathic pulmonary fibrosis: a systematic review and meta-analyses. Sci Rep. 2021;11(1):4318. doi:10.1038/s41598-021-81591-z
36. Washko GR, Hunninghake GM, Fernandez IE, et al. Lung volumes and emphysema in smokers with interstitial lung abnormalities. N Engl J Med. 2011;364(10):897–906. doi:10.1056/NEJMoa1007285
37. Pezzuto A, Lionetto L, Ricci A, Simmaco M, Borro M. Inter-individual variation in CYP2A6 activity and chronic obstructive pulmonary disease in smokers: perspectives for an early predictive marker. Biochim Biophys Acta Mol Basis Dis. 2021;1867(1):165990. doi:10.1016/j.bbadis.2020.165990
38. Lucattelli M, Bartalesi B, Cavarra E, et al. Is neutrophil elastase the missing link between emphysema and fibrosis? Evidence from two mouse models. Respir Res. 2005;6(1):83. doi:10.1186/1465-9921-6-83
39. Kinoshita Y, Watanabe K, Ishii H, Kushima H, Fujita M, Nabeshima K. Distribution of emphysema and fibrosis in idiopathic pulmonary fibrosis with coexisting emphysema. Histopathology. 2019;74(7):1103–1108. doi:10.1111/his.13831
40. Clinical Assembly contribution to the celebration of 20 years of the ERS, Balbi B, Cottin V, Singh S, et al. Smoking-related lung diseases: a clinical perspective. Eur Respir J. 2010;35(2):231–233. doi:10.1183/09031936.00189309
41. Morse D, Rosas IO. Tobacco smoke-induced lung fibrosis and emphysema. Annu Rev Physiol. 2014;76(1):493–513. doi:10.1146/annurev-physiol-021113-170411
42. Weissmann N, Grimminger F, Seeger W. Smoking: is it a risk factor for pulmonary vascular diseases? Pulm Circ. 2012;2(4):395–396. doi:10.4103/2045-8932.105027
43. Higenbottam T. Pulmonary hypertension and chronic obstructive pulmonary disease: a case for treatment. Proc Am Thorac Soc. 2005;2(1):12–19. doi:10.1513/pats.200411-053SF
44. Santos S, Peinado VI, Ramirez J, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. 2002;19(4):632–638. doi:10.1183/09031936.02.00245902
45. Wright JL, Levy RD, Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax. 2005;60(7):605–609. doi:10.1136/thx.2005.042994
46. Lee JH, Lee DS, Kim EK, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172(8):987–993. doi:10.1164/rccm.200501-041OC
47. Awano N, Inomata M, Ikushima S, et al. Histological analysis of vasculopathy associated with pulmonary hypertension in combined pulmonary fibrosis and emphysema: comparison with idiopathic pulmonary fibrosis or emphysema alone. Histopathology. 2017;70(6):896–905. doi:10.1111/his.13153
48. Jacob J, Bartholmai BJ, Rajagopalan S, et al. Likelihood of pulmonary hypertension in patients with idiopathic pulmonary fibrosis and emphysema. Respirology. 2018;23(6):593–599. doi:10.1111/resp.13231
49. Mitchell PD, Das JP, Murphy DJ, et al. Idiopathic pulmonary fibrosis with emphysema: evidence of synergy among emphysema and idiopathic pulmonary fibrosis in smokers. Respir Care. 2015;60(2):259–268. doi:10.4187/respcare.03389
50. Pezzuto A, Carico E. Effectiveness of smoking cessation in smokers with COPD and nocturnal oxygen desaturation: functional analysis. Clin Respir J. 2020;14(1):29–34. doi:10.1111/crj.13096
51. Behr J, Nathan SD, Harari S, et al. Sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: a Phase IIb, Randomised, Double-Blind, Placebo-Controlled Study - rationale and study design. Respir Med. 2018;138:13–20. doi:10.1016/j.rmed.2018.03.019
52. Jiang CG, Fu Q, Zheng CM. Prognosis of combined pulmonary fibrosis and emphysema: comparison with idiopathic pulmonary fibrosis alone. Ther Adv Respir Dis. 2019;13:1753466619888119. doi:10.1177/1753466619888119
53. Ryerson CJ, Hartman T, Elicker BM, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. 2013;144(1):234–240. doi:10.1378/chest.12-2403
54. Wells AU, Desai SR, Rubens MB, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167(7):962–969. doi:10.1164/rccm.2111053
55. Gao J, Kalafatis D, Carlson L, et al. Baseline characteristics and survival of patients of idiopathic pulmonary fibrosis: a longitudinal analysis of the Swedish IPF registry. Respir Res. 2021;22(1):40. doi:10.1186/s12931-021-01634-x
56. Alakhras M, Decker PA, Nadrous HF, Collazo-Clavell M, Ryu JH. Body mass index and mortality in patients with idiopathic pulmonary fibrosis. Chest. 2007;131(5):1448–1453. doi:10.1378/chest.06-2784
57. Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(10):1005–1012. doi:10.1056/NEJMoa021322
58. Moua T, Ryu JH. Obstacles to early treatment of idiopathic pulmonary fibrosis: current perspectives. Ther Clin Risk Manag. 2019;15:73–81. doi:10.2147/TCRM.S160248
59. Romero F, Marchetti N, Hu A, et al. Efficacy of oral antifibrotic agents in the management of combined pulmonary fibrosis and emphysema. Am J Respir Crit Care Med. 2018;197:A1646.
60. King TE
61. Margaritopoulos GA, Trachalaki A, Wells AU, et al. Pirfenidone improves survival in IPF: results from a real-life study. BMC Pulm Med. 2018;18(1):177. doi:10.1186/s12890-018-0736-z
62. Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
63. Cottin V, Azuma A, Raghu G, et al. Therapeutic effects of nintedanib are not influenced by emphysema in the INPULSIS trials. Eur Respir J. 2019;53(4). doi:10.1183/13993003.01655-2018
64. Yoon HY, Kim TH, Seo JB, et al. Effects of emphysema on physiological and prognostic characteristics of lung function in idiopathic pulmonary fibrosis. Respirology. 2019;24(1):55–62. doi:10.1111/resp.13387
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.