")
Back to Journals » International Journal of General Medicine » Volume 14
Combined Identification of Novel Markers for Diagnosis and Prognostic of Classic Hodgkin Lymphoma
Received 6 October 2021
Accepted for publication 19 November 2021
Published 18 December 2021 Volume 2021:14 Pages 9951—9963
DOI https://doi.org/10.2147/IJGM.S341557
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Zhixing Kuang,1,* Jiannan Tu,2,* Xun Li3
1Department of Radiation Oncology, Nanping First Hospital Affiliated to Fujian Medical University, Nanping, People’s Republic of China; 2Department of Oncology, Nanping First Hospital Affiliated to Fujian Medical University, Nanping, People’s Republic of China; 3Department of Oncology, Changzhou Tumor Hospital Affiliated to Soochow University, Changzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xun Li
Department of Oncology, Changzhou Tumor Hospital Affiliated to Soochow University, Changzhou, 213002, People’s Republic of China
Email [email protected]
Background: An effective diagnostic and prognostic marker based on the gene expression profile of classic Hodgkin lymphoma (cHL) has not yet been developed. The aim of the present study was to investigate potential markers for the diagnosis and prediction of cHL prognosis.
Methods: The gene expression profiles with all available clinical features were downloaded from the Gene Expression Omnibus (GEO) database. Then, multiple machine learning algorithms were applied to develop and validate a diagnostic signature by comparing cHL with normal control. In addition, we identified prognostic genes and built a prognostic model with them to predict the prognosis for 130 patients with cHL which were treated with first-line treatment (ABVD chemotherapy or an ABVD-like regimen).
Results: A diagnostic prediction signature was constructed and showed high specificity and sensitivity (training cohort: AUC=0.981,95% CI 0.933– 0.998, P< 0.001, validation cohort: AUC=0.955,95% CI 0.895– 0.986, P< 0.001). Additionally, nine prognostic genes (LAMP1, STAT1, MMP9, C1QB, ICAM1, CD274, CCL19, HCK and LILRB2) were screened and a prognostic prediction model was constructed with them, which had been confirmed effectively predicting prognosis (P< 0.001). Furthermore, the results of the immune infiltration assessment indicated that the high scale of the fraction of CD8 + T cells, M1 macrophages, resting mast cells associated with an adverse outcome in cHL, and naive B cells related to prolonged survival. In addition, a nomogram that combined the prognostic prediction model and clinical characteristics is also suggested to have a good predictive value for the prognosis of patients.
Conclusion: The new markers found in this study may be helpful for the diagnosis and prediction of the prognosis of cHL.
Keywords: novel markers, diagnosis, prognosis, classic Hodgkin lymphoma
Introduction
Classic Hodgkin lymphoma (cHL) is a kind of B-cell neoplasm which characterized by the paucity of Hodgkin/Reed-Sternberg (HRS) cells in conjunction with a microenvironment which contains abundant infiltrating reactive cells. cHL is one type of HL, which constitutes more than 90% of HL.1 The incidence of HL is 2.8 per 100,000 people, it is estimated that 8,830 people in the United States will be diagnosed with HL and 960 people will die from this disease in 2021.2 Currently, the main treatments of cHL include multi-agent chemotherapy and involved field radiation therapy, which can significantly reduce the rate of death and the long-term remission of classic Hodgkin lymphoma can achieve 85%–95%, however, approximately 20%–30% of patients will be seen with disease progression or death within 5 years.3 Until now, many efforts have been made in the area of new drug development to improve treatment efficacy and reduce toxicity; the best known drugs for us are anti-CD30 inhibitors and PD-1 antibodies, which have been reported to show promising results for patients with relapsed and refractory cHL.4 Anti-CD30 inhibitors, called rentuximab vedotin, have been approved for the treatment of classic Hodgkin lymphoma patients after failure with autologous stem cell transplantation or multi-agent chemotherapy regimens,5 but only approximately 17% to 29% of those patients who experienced failure or relapsed with both brentuximab vedotin (CD30-targeted therapy) and autologous stem cell transplantation (ASCT) can expect complete remission (CR) after accepting the treatment of PD-1 antibodies.6 To meet the patient ‘s needs, our ability to develop newly approaches for patients with cHL should keep pace with the development of those suffering from cHL. Expanding knowledge of the changes in cHL gene expression levels may be helpful for the discovery of potential therapeutic targets.
On the other hand, in light of the tremendous developments in the treatment of cHL, an appropriate attention should be paid to novel markers for diagnostic and prognostic factors.
Novel diagnostic markers will be helpful for accurate diagnosis and the development of new therapeutic targets. Excessive medication or radiation therapy may compromise survival due to adverse effects, so a suitable and accurate risk stratification is essential for the treatment of all patients. The most widely used clinical indicator to evaluate the risk of cHL is the international prognostic score (IPS) and is used primarily to evaluate advanced cHL.7 In recent years, the value of interim fluorodeoxyglucose positron emission tomography (PET)/computer tomography (CT) was gradually established to differentiate the risk of cHL.8 However, given the heterogeneity among individual patients with cHL, all these tools that we mentioned use for risk stratification provide limited information underlying the HL biology.9 In light of the pathogenesis of cHL is multifactorial and complicated, therefore, a better understanding of the molecular mechanism in the pathogenesis of cHL is essential to us to find a new prognostic factor.
In recent years, the development of medical technologies that allow gene expression profiling from archival paraffin-embedded tissues pushed forward the recognition of pathogenetic and contributed to the creation of new markers for predicting diagnosis and prognosis based on gene expression. Here, multiple statistical methods were utilized to identify novel markers for diagnostic and prognostic prediction (Figure 1). The discovery of the present research may be helpful in establishing new diagnostic and prognostic markers and to put forward new therapeutic targets.
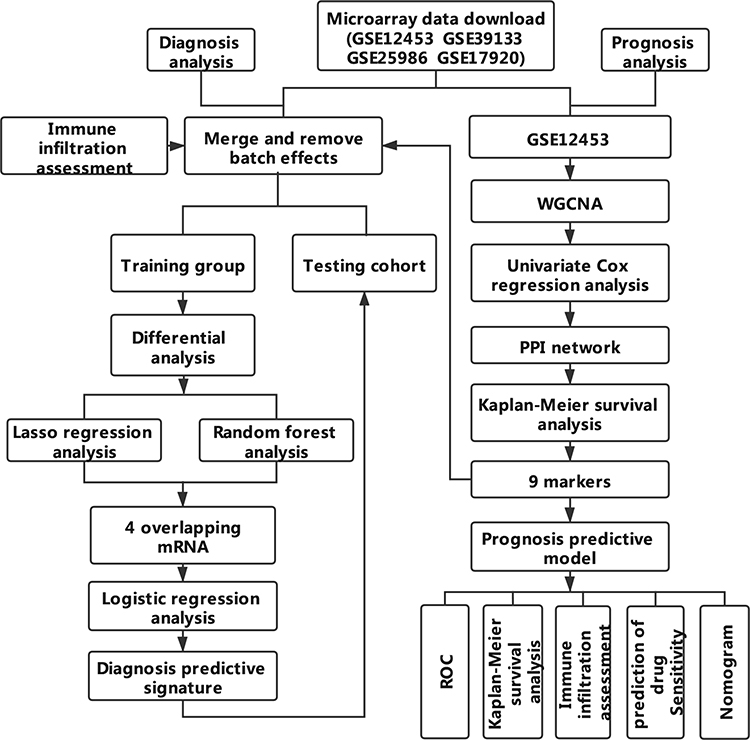 |
Figure 1 Workflow chart of data generation and analysis. |
Materials and Methods
Data Sources and Data Processing
The raw “CEL” data of GSE12453 (12 samples of cHL and 25 samples of normal B cells), GSE39133 (29 samples of cHL and 5 cases of normal B cells), GSE25986 (5 cHL and 5 normal B cell) and GSE17920 (130 case of cHL), which all based on the platform of GPL570 [HG-U133_Plus_2] and were downloaded from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) database. All raw chip data undergoes quality assessment and quality control to identify unqualified samples, and then background correction and normalization were performed. The process which mentioned above were completed by “simpleaffy”,10 “affyPLM”, ‘arrayQualityMetrics’11 packages. After accomplishment of preprocessing and analysis, one disqualified sample in GSE12453 was excluded from our following analysis.
Identification of a mRNA Signature Discriminating Between cHL and Normal Control
To enhance the reliability of the research, four data sets were merged to a big one with 210 case samples and the package of “sva” was used to get rid of batch effects. Subsequently, all 210 cases of cHL patients were randomly divided into a training group and a testing group with a ratio of 1:1. The package of “limma” was utilized to identify differentially expressed genes (DEGs) in the training cohort, while P< 0.05 and |log2 FC| > 1 were set as cutoff values. Then, the random forest (RF) algorithm and least absolute shrinkage and selection operator (LASSO) analysis were used to reduce the dimensionality of DEGs. The optimal values of the penalty parameter λ were determined by the results of 10 times cross-validations. Random forest is a collection of decision trees; these decision trees are generated by recursive binary division of different random sub-samples of training data. In the random forest analysis, the optimal genes were selected according to the mean decrease in accuracy to select optimal genes. Finally, the overlapping markers from the two algorithms were obtained and were used to build a diagnostic signature and get the diagnostic score by using the mRNA coefficients that obtained in logistic regression. Diagnostic score was constructed as follows diagnostic score . Where β represents the regression coefficient of mRNA which derived from the analysis of logistic regression, and i represents the expression level of mRNA. LASSO analysis was performed by the “glmnet” package, random forest analysis was performed by “randomForest” package.
. Where β represents the regression coefficient of mRNA which derived from the analysis of logistic regression, and i represents the expression level of mRNA. LASSO analysis was performed by the “glmnet” package, random forest analysis was performed by “randomForest” package.
Co-Expression Network Construction and Identity Candidate Genes
The top 6000 variant of expression profiles of 24 samples of normal B cells and 12 samples of cHL in GSE12453 were selected and utilized to construct a co-expression network by using “WGCNA” package. The best soft thresholding parameter was selected to make our gene distribution conform to the scale-free network. To counteract the effects of missing or spurious connections between the nodes in the network, the topological overlap matrix (TOM) was transformed by adjacent matrices. According to the measure of TOM-based dissimilarity measure, the genes that highly correlated were clustered into the same module. The Gene Significance (GS) values represented the relationship of each gene with cHL, and the module membership (MM) was represented the correlation of genes with module eigengenes (MEs). Finally, only the module most relevant to cHL was used for further analysis. In our study, to screen the hub genes in the module and ensure the reliability of the results, the criterion of genes of GS > 0.2 and MM > 0.8 were seated.
Construction of a PPI Network and Detection of Prognosis-Related Genes
To reduce the number of genes, the univariate Cox proportional hazards regression analysis was performed by assessing the relationship between overall survival of patients with cHL (OS) and the expression of genes retained with WGCNA, only genes with p< 0.05 in the analysis results were sorted out and used to build a protein-protein interactions (PPI) network. The Search Tool for the Retrieval of Interacting Genes (STRING 10.0) is a database that was designed to predict the protein–protein interactions of genes, and only combination scores > 0.4 were chosen to be significant. Only candidate genes with degree > 10 will be considered hub genes. To investigate real hub mRNAs, Kaplan-Meier survival analysis was performed using the median expression value as the cut-off point in GSE17920. In addition, the Wilcoxon Signed-Rank test was used to calculate the expression difference between cHL and normal B cells for real hub genes in the combinational dataset with 210 case samples.
Multivariate Cox Regression Conduction and Identification of a Risk Model
Multivariate Cox regression analysis was performed to construct a prognosis model using the nine genes related to the prognosis of cHL. The risk score of the prognostic risk model for each patient with cHL was calculated as follows: risk score = β1e1 +β2e2 +β3e3 + … … +βnen, e represents the expression level of gene n, where β represents the expression level derived from the multivariate Cox regression analysis. All patients with cHL were divided into high- and low-risk groups according to the median value of the risk score as the cutoff value. Survival curves were calculated and visualized by the Kaplan-Meier analysis, and the package of “survivalROC” was utilized to illustrate the accuracy of the survival prediction based on the risk score. Apart from this, we also studied the correlation between risk score and clinical characteristics. Furthermore, the IC50 of six commonly used cytotoxicity drugs was predicted for all high and low-risk groups using the “pRRophetic” package. Finally, the disagreement of immune landscape between the patients with different risk was analysed by using the tool of CIBERSORT.
Integrated Analysis by Combining the Clinical Characteristic and the Risk Model
The analyses of univariate and multivariate Cox regression were performed to investigate the effect of the risk model on the prognosis of cHL patients. The risk stratification which obtained from the risk model and other clinicopathological characteristics including age, gender, albumin, hemoglobin, stage, white cell count and lymphocyte rate were used as covariates. In addition to this, the risk model with clinical characteristics was used to build a nomogram. Calibration curves were used to estimate the conformity between the real outcomes and the predicted outcomes for the nomogram, and the discrimination for the nomogram was evaluated by C-index. The package of ‘rms’ was applied to construct the nomogram and calibration curves. Statistical analyzes in our study were performed using R software.
Results
Pre-Processing of the Data Sets and Identification of DEGs in cHL
In total, 34 samples of normal B cells and 176 cases of classical Hodgkin lymphoma(cHL) in four cohorts were chosen for subsequent analysis after excluding a disqualified sample. After completion from the preprocessing, all data of microarray were converted into an expression matrix. To reduce the differences between batches, we regroup the data from four batches after the effects of the removed batch, so that the downstream analysis can only consider biological differences (Figure S1). After that, all 210 case samples were randomly divided into training data set and validation data set. The training group was utilized to identify DEGs, and a total of 61 upregulated and 114 downregulated genes were filtered out (Figure S2).
Building and Validation of an mRNA Signature for Diagnosis and Prediction of cHL
We obtained the top 11 markers with the maximum value of the mean decrease in accuracy by using the analysis of RandomForest, and 27 markers were obtained by using analysis of LASSO (Figure S3). There were eight overlapping markers between these two methods. We obtained four markers by using the method of logistic regression variable selection and constructed a diagnostic prediction signature with the four markers (Table 1). Applying the signature in the training cohort yielded a sensitivity of 98.86% and a specificity of 82.35% for cHL in the training dataset (Figure 2A) and a specificity of 88.32% and sensitivity of 89.77% in the testing dataset (Figure 2B). We also found that this signature also was demonstrated could effectively distinguish cHL from normal controls both in the training cohort (AUC=0.981) and the testing dataset (AUC=0.955) (Figure 2C and D). Unsupervised hierarchical clustering analysis of ADD3, FOXC1, LAPTM5 and MAL showed high specificity and sensitivity to differentiate cHL from normal controls (Figure 2E and F).
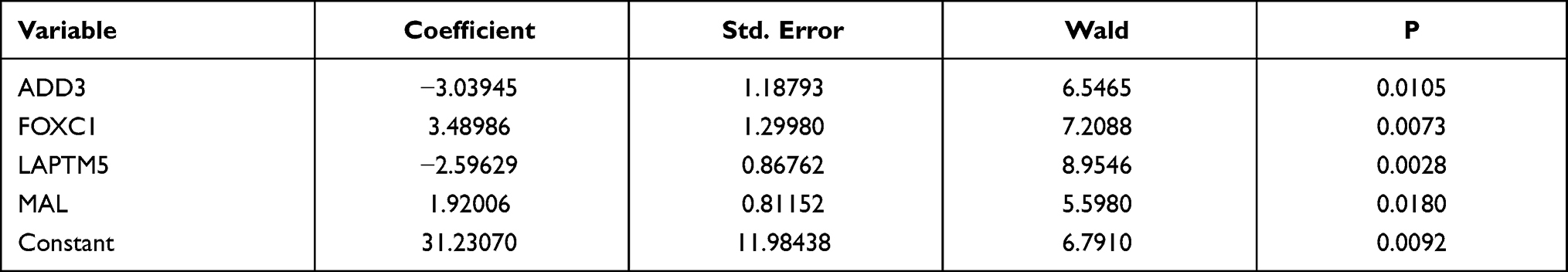 |
Table 1 Characteristics of Four Gene Markers and Their Coefficients in the Diagnosis of cHL |
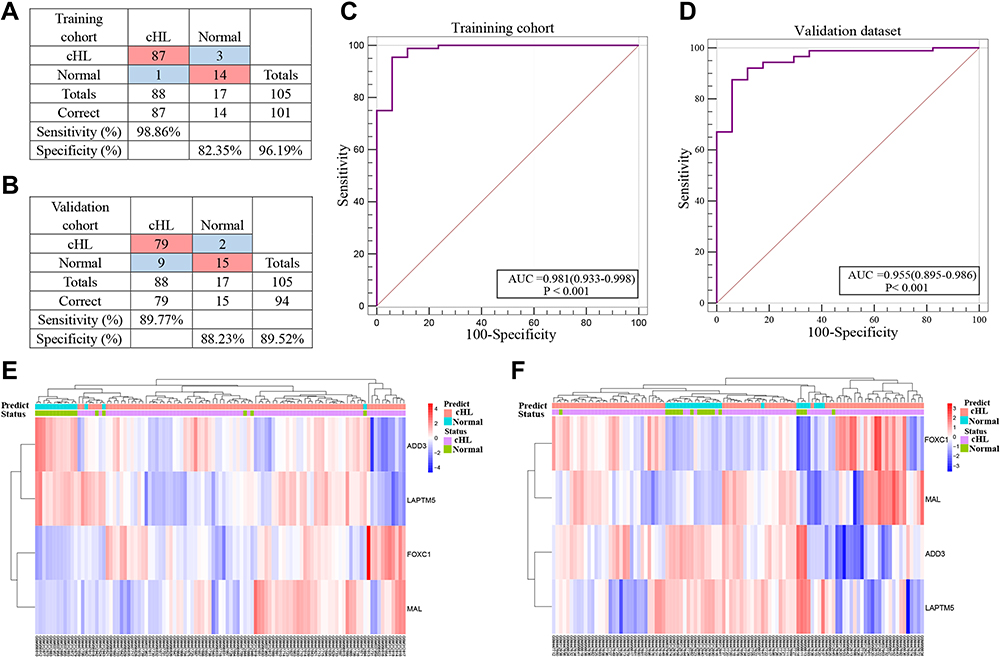 |
Figure 2 mRNA expression analysis of cHL diagnosis. (A and B) Confusion tables of binary results of the diagnostic prediction signature in the training and validation cohorts. (C and D) ROC of the diagnostic prediction signature with four mRNA markers in the training and validation cohorts. (E and F) Unsupervised hierarchical clustering of four mRNA markers in the diagnostic prediction model in the training and validation cohorts. |
Construction of Weighted Co-Expression Network and Identification of Hub Genes
Six thousand genes in GSE12453 were obtained and utilized to construct the co‐expression network (Figure 3A). The power of β = 28 was chosen as the best soft-thresholding parameter (Figure 3B and C). Additionally, the closely correlative modules were merged into a bigger one by setting the MEDissThresas parameter as 0.25 (Figure 3D). Ultimately, 10 modules were generated and the yellow modules showed significantly positive correlation with cHL patients (weighted correlation = 0.95, P=2e −19, Figure 3E). Five hundred and seventy candidate genes selected by WGCNA were used to screen survival‐related mRNA by conducting the analysis of univariable Cox survival. One hundred and seventy survival‐related mRNA were screened out and used to construct the PPI, only the 18 genes ‘degree in the PPI network > 10 were figure out (Supplement Table 1). We evaluated the relationship between the expression of 18 genes and the survival of the patient, and the results showed that 9 genes including LAMP1, STAT1, MMP9, C1QB, ICAM1, CD274, CCL19, HCK, and LILRB2 were significantly negatively correlated with the prognosis of the patient with cHL in GSE17920 (Figure 4). The 9 genes had significantly high expression in cHL compared to normal B cells within the combinational data set with 210 case samples except HCK (Figure S4A).
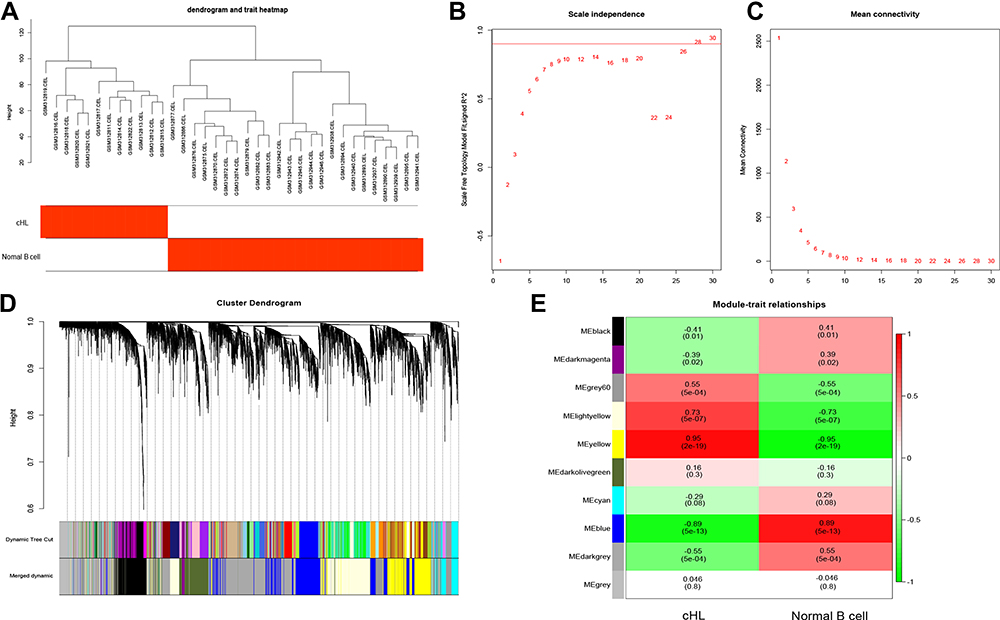 |
Figure 3 Identification of candidate genes in cHL. (A) Clustering dendrogram of cHL and normal B cell. (B and C) analysis of scale-free fit for soft thresholding powers and 28 was selected as the best value. (D) Dendrogram of all 6000 genes clustered on a dissimilarity measure. (E) Heatmap of the relationships between modules and cHL by Pearson correlation. |
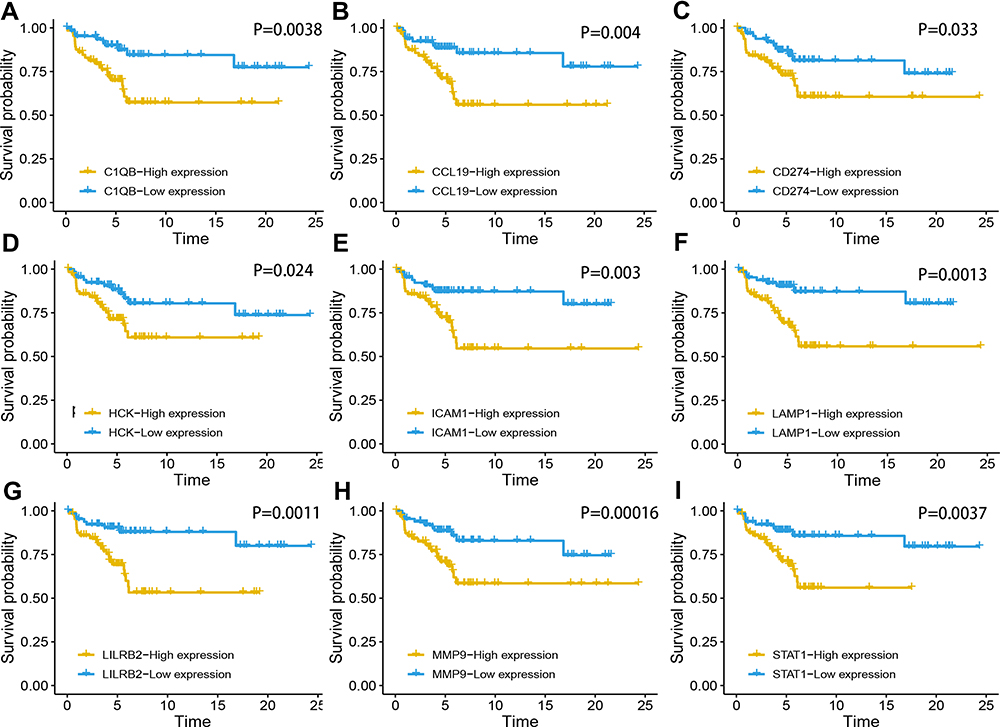 |
Figure 4 Survival analysis of nine genes in cHL. Kaplan–Meier survival curves were generated for (A) C1QB, (B) CCL19, (B) CCL19, (D) HCK, (E) ICAM1, (F) LAMP1, (G) LILRB2, (H) MMP9 and (I) STAT1. |
Identification of the Nine-Gene Risk Prognostic Model for Survival
The nine hub genes were used to perform multivariate regression analysis and build a risk prognostic model in the GSE17920 cohort. The risk score of prognostic model was calculated with the following formula: risk score = (0.6891×STAT1 expression) + (5.1633× LAMP1 expression) + (0.1953× GBP2 expression) + (0.4689 × ICAM1 expression) + (1.8556×C1QB expression) + (0.7998× MMP9 expression) + (0.6767× LILRB2 expression) + (0.9942× HCK expression) + (1.5728× CCL19 expression) + (1.5587× 1.5587 expression). All patients diagnosed with cHL were categorized into a high- and low-risk groups according to the median value of the risk score, which was illustrated by Kaplan-Meier curves (Figure 5A). The Kaplan-Meier curves also showed a significantly different prognosis in patients separated by staging (p<0.001, Figure 5B). The 1-year, 3-year, and 5-year areas under the curve were 0.735, 0.768 and 0.785, respectively (Figure 5C). Among these, nine genes, all significantly elevated in the group with high-risk (Figure S4B). We also found that the risk score is positively correlated with staging, and the risk score significantly increased in the group of hemoglobin <10.5 g/dl, serum albumin <4 g/dl and age ≥45 Years (Figure 5D, E, H, J). There is no statistical difference in the distribution of the risk score in the groups with different lymphocyte rate, white cell, and sex (Figure 5F, G, I). The distribution of patients with different clinical characteristics between high risk and low risk patients was calculated, and found that patients who younger than 45 years old, with stage I–II and albumin greater than 40mg/DL were obviously concentrated on low-risk group. And there is no statistical difference between different gender, hemoglobin, white cell count and lymphocyte rate in different risk groups (Figure S5A-G). Furthermore, cytarabine and vinblastine in our study had a lower IC50 in low-risk patients, suggesting that low-risk patients are more sensitive to cytotoxic drugs (Figure 6D and E), and there is no difference in the drug sensitivity of bleomycin doxorubicin, etoposide, and methotrexate between the high-risk category and the low-risk category (Figure 6A, B, C, F).
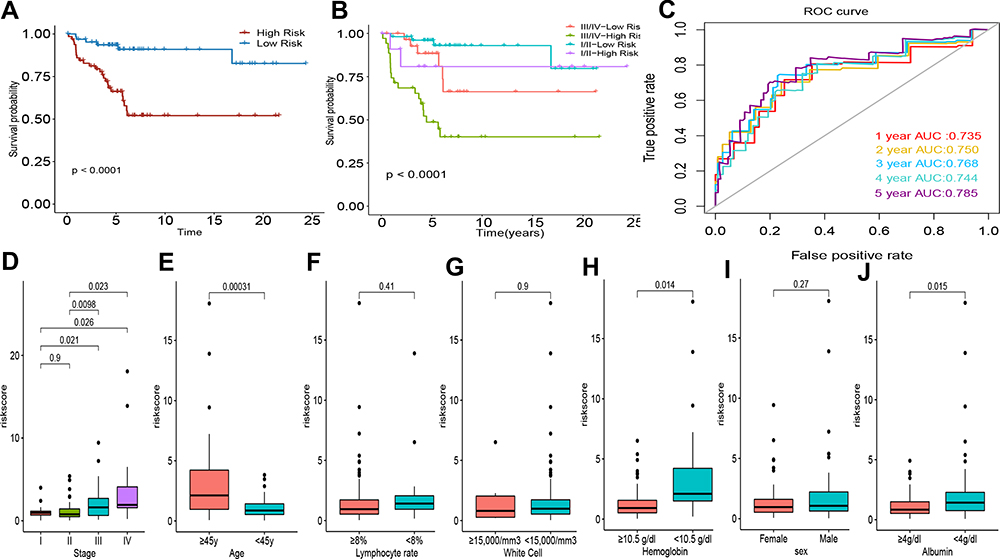 |
Figure 5 Analysis of the prognostic risk model in cHL. (A) Kaplan-Meier survival curves for all cHL patients; (B) Survival curves of cHL patients with combinations of risk core and stage. (C) time-dependent ROC curves for the nine-mRNA risk model in the cohorts of GSE17920. (D) Risk score in patients with different stage. (E) Risk score in patients with age of ≥45 and <45y. (F) Risk score in patients with lymphocyte rate <8% and ≥8%. (G) Risk score in patients with white Cell of ≥15,000 and <15,000/mm3. (H) Risk score in patients with hemoglobin <105 g/L and ≥105 g/L. (I) Risk score in female and male patients. (J) Risk score in in patients with Albumin <40 g/L and ≥40 g/L. |
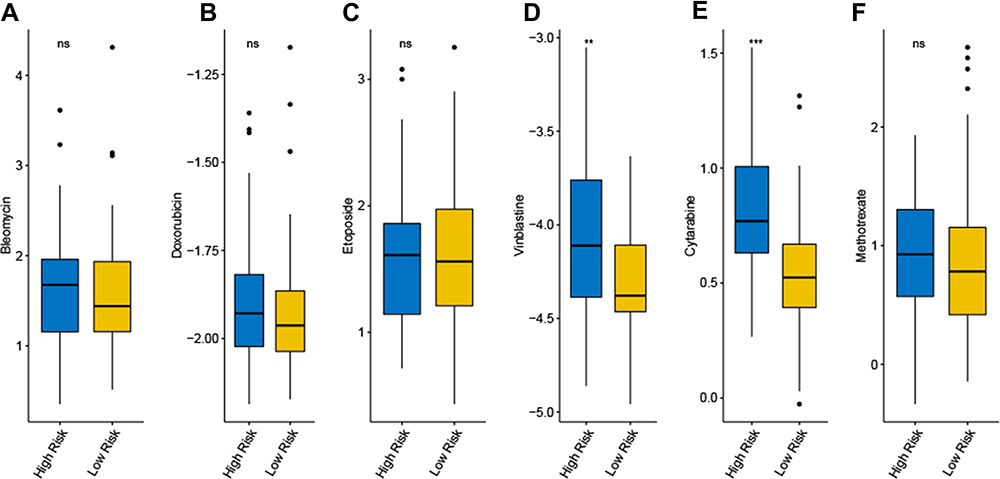 |
Figure 6 Prediction of chemotherapy of six common therapeutic drugs in patients of high risk and low risk. (A) Bleomycin; (B) Doxorubicin; (C) Etoposide; (D) Vinblastine; (E) Cytarabine (F) Methotrexate.(ns not significant, *P < 0.05; **P < 0.01 and ***P < 0.001). |
Immune Infiltration Assessment Based on Sample Type and Risk Model
After the analysis of CIBERSORT immune was completed, we found that the distribution of immune cells in cHL compared to normal tissues is similar to the distribution of immune cells in the high-risk group compared to the low-risk group. In the combinational cohort incorporated by all cHL and normal samples, 176 cases of cHL and 34 normal samples had shown a substantial divergence in the proportion of nine types of immune cells (M1 macrophages, activated CD4+ memory T cells, CD8+T cell, activated dendritic cells, naive B cells, resting mast cells, memory B cells, resting NK cells and activated NK cells, Figure 7A). The presence of nine immune cells types (activated dendritic cells, naive B cells, M1 macrophages, CD8+T cells, resting mast cells, M0 macrophages, helper follicular T cells, resting memory CD4+ T cells, eosinophils, Figure 7B) in the low-risk category and the high-risk category also showed a considerable discrepancy. Among the 22 types of immune cell, the content of CD8+T cells, resting mast cells and M1 macrophages significantly high in cHL and high-risk category contrast to normal controls and low-risk category, which contrary to the distribution of naive B cells. The results indicating that the high scale of the fraction of the CD8+T cells, M1 macrophages and resting mast cells led to adverse outcome in cHL and naive B cells related to prolonged survival.
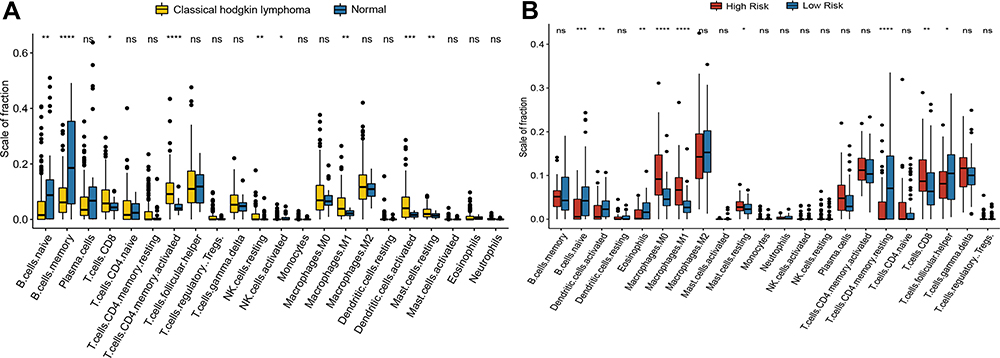 |
Figure 7 Immune infiltration assessment based on sample type and risk model. (A) Differential distribution of immune cells between the cHL and low normal control in the merged cohort. (B) Distribution of immune cells between the high risk and low risk group.(ns not significant, *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001). |
Independent Role of the Risk Model in Patients with cHL
In order to evaluate whether the risk model has independent role in evaluating the prognosis of cHL patients, univariate and multivariate Cox regression analysis were performed in GSE17920 dataset by including the risk stratification and all available clinical characteristics as explanatory variables. According to the international prognostic score (IPS) criterion, all clinical characteristics parameters were grouped by hemoglobin <10.5 g/dl, age ≥45 years, male sex, lymphocyte count <8% of white cell count, white cell count (WCC) ≥15,000/mm3, serum albumin <4 g/dL, stage III/ IV disease by Ann Arbor classification. In univariate Cox regression, age, stage, hemoglobin, WCC and risk stratification were significantly related to OS. After multivariate adjustment was completed, risk stratification, age, stage, and WCC also showed an independent role in affecting the prognosis (risk stratification: HR = 4.4, 95% CI = 1.57–12.35, P=0.0048, Table 2). A nomogram which including all clinical characteristic parameters and risk stratification was constructed and the calibration curve also exhibited high consistency between predictive outcome and observation survival time in the cHL cohort, and C-index for OS were 0.852 (Figure S6A and B).
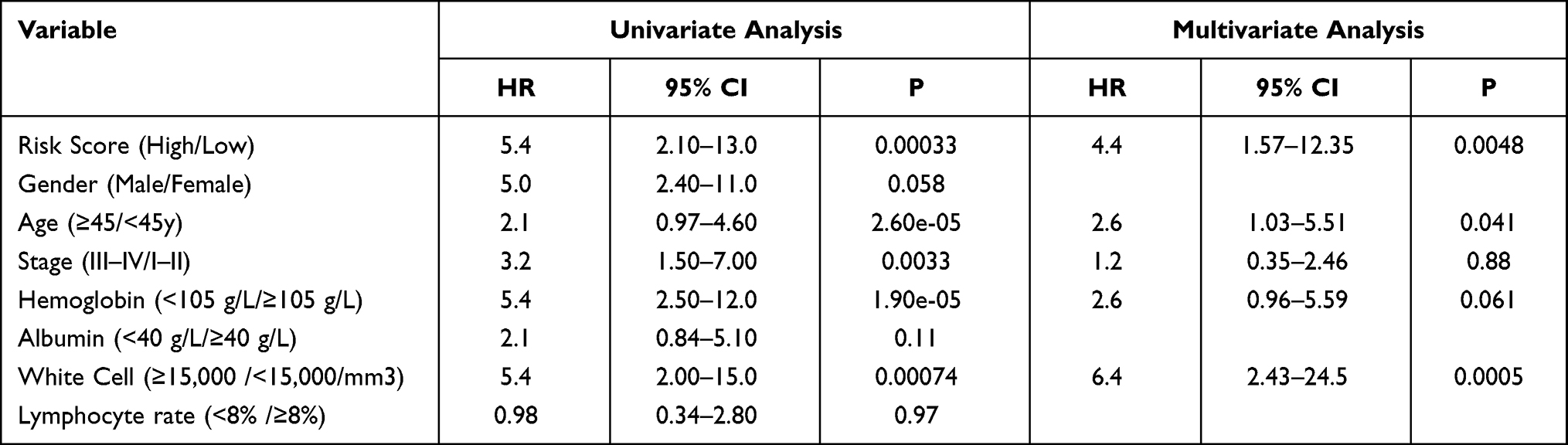 |
Table 2 Univariate and Multivariate Cox Regression Analyses of the Nine-Gene Risk Model in Patients with cHL |
Discussion
cHL is a kind of malignancy that originates from B cells and typically involved lymphonodus, sometimes other organs. The pathogenic mechanisms of cHL are still not very clear. In the present research, we investigate the molecular mechanism of cHL by using bioinformatics methods and try to provide a new clue to the development of diagnosis and risk stratification. In the present study, we built and validated a mRNA signature with four genes (MAL, FOXC1, LAPTM5, and ADD3) for the diagnosis and prediction of cHL, with a sensitivity of 98.86%, a specificity of 82.35%, and an AUC of 0.981 (95% CI,0.933 to 0.998) in the training dataset, and with a sensitivity of 89.77%, a specificity of 88.23% in the validation dataset. In addition, we identified nine genes that included LAMP1, STAT1, MMP9, C1QB, ICAM1, CD274, CCL19, HCK and LILRB2 with a significant prognosis of cHL, and a nine-mRNA-based risk model was developed to better assess the risk of the patient with cHL.
Several candidate genes for gene expression or genome methylation-based markers have been proposed to predict cancer diagnosis, such as the one study conducted by Xu et al in which a diagnosis signature was used to discriminate hepatocellular carcinoma and healthy controls, and achieved AUC values of 0.966 and 0.944, respectively.12 Another research to assess the accuracy of a signature for detecting colorectal cancer patients in the training dataset and validation dataset, the results showed the AUC of 0.851 and 0.923, respectively.13 In the present study, our diagnostic prediction signature showed better performance in differentiating cHL from normal controls and yielded new insights into the diagnosis of cHL. FOCX1 in our signature is an essential component of FOX family members which involved in carcinogenesis and the growth tumor cell and express a higher level in a variety of carcinomas.14 Overexpression of FOXC1 has also been observed in the HL.15 Furthermore, FOXC1 has been demonstrated to activate the NOTCH and NF-kB signaling pathway, and these pathways are the two most important pathways in cHL to promote the survival of HRS cells.16 cHL and mediastinal large B-cell lymphoma (MLBL) have similar molecular genetics, histopathology and clinical manifestations. Myelin and lymphocytes (MAL) has been reported to overexpressed in the majority of MLBL and a minority of cHL, but the role of MAL in those diseases have not reach definitive agreement. In this context, elevated expressions of FOXC1 and MAL were found in the cHL group compared to the normal group, consistent with the observation mentioned in the above research. Widespread loss of classic B lineage phenotype markers in HRS cells is the most important features of cHL,9 which in line with our results that the proportion of memory B cells and naïve B cells in cHL is significantly lower than which in the control group. There is evidence that lysosomal-associated protein transmembrane 5 (LAPTM5) is abundantly expressed in mature B cells.17 In our study, LAPTM5 was significantly down-regulated in cHL compared to the normal control; this may be attributed to the decrease in the number of normal mature B cells and leads to decreased expression of LAPTM5. Adducin 3 (ADD3) is a crucial membrane skeleton protein and has been confirmed to be aberrantly low expressed in multitype cancers, and loss of ADD3 can promote tumor cell growth and angiogenesis that occurs in glioblastoma multiforme.18 Moreover, ADD3 deletions were significantly negatively correlated with prognosis.19
Epidemiological and serologic studies have implicated that approximately 40% of Hodgkin lymphoma cases are associated with Epstein-Barr virus (EBV). Epstein-Barr virus infection and genetic alterations at 9p24.1 contribute to overexpression of CD274/PD-L1,6 which in turn contributes to HRS cells evading T cell immune surveillance.20 Overexpression of PD-L1 expression has been detected and associated with poor prognosis in diffuse large B-cell lymphoma, nasopharyngeal carcinoma and colorectal cancer,21–23 similar conclusions have also been reported in cHL.9 However, the interesting thing is that was a trend with higher HRS cell CD274/PD-L1 expression, patients exhibit more favorable responses to anti–programmed death-1 monoclonal antibody and had superior outcome.3,24 In our study, CD274 has significantly high expression in patients with cHL and high-risk group, which means that patients in the high-risk group will benefit more from immunotherapy. STAT1, ICAM1 and CCL19 which also have been found to be significantly elevated in patients with cHL and correlated with a short period of survival of the cHL patient. High levels of STAT1 contribute to proliferation and bad outcomes of breast cancer,25,26 however, it has also been demonstrated that high levels of STAT1 has tumor suppressive functions and are beneficial to prognosis in some selected cancer.27,28 The divergent role of STAT1 in tumorigenesis and development can be due to heterogeneity in different type of cancer and Different tumor microenvironment.29 Accumulating evidences suggest that Intercellular cell adhesion molecule-1 (ICAM-1) upregulated in multitype types of cancer and plays an important role in tumor invasion and metastasis,30,31 but it also has been proposed that elevated ICAM1 expression in colorectal cancers, gastric cancer and breast cancer is correlated with more favorable prognosis.32 CCL19 is one of the chemokines and has been found to be overexpressed in many tumors, it can not only modulate the immune response and take part in the proliferation of cancer,33 but also recruit tumor cells to the T cell zone, leading to lymph node metastasis,34,35 in addition, it was discovered that CCL19 in signaling pathway of NF-kB can promoting central nervous system lymphoma (CNSL) formation.36
Chemokines and cytokines which produced by the HRS cells as and inflammatory cells in the microenvironment of cHL work in some kind manner to promote the survival of HRS cells.37 Based on current research it is evident that chemokines secreted by HRS cells and inflammatory cells contribute to mediate the expression of MMPs that needed for malignant cell invasion and metastasis via degradate the extracellular matrix (ECM).38 MMP9 is a major constituent of the MMPs and has been identified as a prognostic factor in glioblastoma and clear cell renal carcinoma.39,40 Lysosome-associated membrane protein-1 (LAMP1) is a member of the lysosomal membrane protein that protects the lysosomal membrane from hydrolysis, growing evidence has shown LAMP1 participate in process of tumor cell and overexpression of it correlates with adverse outcome of diffuse Large B-cell Lymphoma and breast cancer,41,42 but the exact role involved in cancer is still unknown. Leukocyte immunoglobulin-like receptor subfamily B member 2 (LILRB2) has also been reported to be associated with promoted tumor cell growth and correlates with poor prognosis,43–45 which is consistent with our study. LILRB2 antibodies can not only inhibit AKT and STAT6 activation but also increase the efficacy of anti-PD-L1 and enhance antitumor immunity,46 suggesting that LILRB2 may act as a novel potential immune-targeted therapeutic checkpoint.
Hematopoietic cell kinase (HCK) is a kind of protein tyrosine that belongs to SRC kinase family, which originally found in the cells of hematopoietic and now it also been studied in other solid tumors and lymphoma.47 Blocking HCK can reduce the activation of MAPK/ERK and PI3K/AKT pathways which has been widely recognized to promote the formation of various cancers.48 Highly expressed HCK is negatively correlated with colorectal cancer patients’ prognosis,49 which in line with our results in cHL. Complement component 1, Q subcomponent B chain (C1QB) is a submember of the complement classical pathway; it has been demonstrated to enhance chemokine secretion that are expected to promote immunosuppression and play a role in tumor growth and progression.50 To date, C1QB overexpression in gliomas has been associated with a reduced prognosis,51 however, recent research revealed that high C1QB in breast cancer is related to a prolonged prognosis.52 Implementing a further study on where C1QB can have a protective or deleterious effect on cancer progression is necessary. In this context, the expression of C1QB increased significantly in cHL patients compared to the normal group and was associated with poor outcomes.
Advances in bioinformatics technology contribute to the construction of multigene risk signature by using public data sets that have been used,53 however, the gene-based risk signatures and the nomogram based on it that as a prognostic tool for cHL has not yet been investigated. In this report, a risk model was constructed and showed powerful performance to stratify all cHL patients into low-risk and high-risk groups, and exhibit an independent impact on the prognostic value of cHL. To further investigate the prognostic value of the risk model, a nomogram was built to predict the personalized risk of every cHL patient. The c index used to evaluate the effectiveness of the nomogram in cHL patients was 0.852, significantly better than previous research used to predict the outcome of other cancer patients.54 Having the ability to accurately assess individual risk and prognosis is the basis of personalized treatment for patients, and the risk model may add complementary value to clinicians in making treatment decisions.
However, the limitations of the present study should also not be ignored. First, the sample size to identify the marker for diagnosis and prognosis needs to be further expanded to avoid selection bias. Secondly, the lack of available validation groups to further confirm the reliability of the risk model in the study. Finally, further experimental research is needed in our future study to elucidate the mechanism of these markers in the carcinogenesis, proliferation and progression of cHL.
Collectively, this is the first study that has been used to investigate the ability of the signature based on mRNA as a novel biomarker for the diagnosis and prediction of the prognosis for cHL. In present study, we identified a diagnostic prediction signature, nine prognostic genes, and a prognostic prediction model, which has been confirmed the usefulness in diagnosis and prognosis prediction of cHL. The novel genes may be helpful for researchers to find new therapeutic targets.
Data Sharing Statement
All data generated or analyzed during this study are included in this article.
Ethics Approval and Consent to Participate
The need for ethics approval was waived by the Department of Scientific Research Management, Changzhou Tumor Hospital Affiliated to Soochow University.
Acknowledgments
This work was supported by a grant from Startup Fund for scientific research, Fujian Medical University (2018QH1171). And all authors would like to thank Christian Steidl, MD, for sharing survival information of dataset GSE17920.
Disclosure
The authors declare that they have no competing interests.
References
1. Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2018;68(2):116–132. doi:10.3322/caac.21438
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi:10.3322/caac.21654
3. Ramchandren R, Domingo-Domenech E, Rueda A, et al. Nivolumab for newly diagnosed advanced-stage classic Hodgkin lymphoma: safety and efficacy in the Phase II CheckMate 205 STUDY. J Clin Oncol. 2019;37(23):1997–2007. doi:10.1200/JCO.19.00315
4. Ansell SM. Immunotherapy in Hodgkin lymphoma: the road ahead. Trends Immunol. 2019;40(5):380–386. doi:10.1016/j.it.2019.03.003
5. Connors JM, Jurczak W, Straus DJ, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N Engl J Med. 2018;378(4):331–344. doi:10.1056/NEJMoa1708984
6. Nie J, Wang C, Liu Y, et al. Addition of low-dose decitabine to anti-PD-1 antibody camrelizumab in relapsed/refractory classical Hodgkin lymphoma. J Clin Oncol. 2019;37(17):1479–1489. doi:10.1200/JCO.18.02151
7. Diefenbach CS, Li H, Hong F, et al. Evaluation of the International Prognostic Score (IPS-7) and a Simpler Prognostic Score (IPS-3) for advanced Hodgkin lymphoma in the modern era. Br J Haematol. 2015;171(4):530–538. doi:10.1111/bjh.13634
8. Zaucha JM, Chauvie S, Zaucha R, Biggii A, Gallamini A. The role of PET/CT in the modern treatment of Hodgkin lymphoma. Cancer Treat Rev. 2019;77:44–56. doi:10.1016/j.ctrv.2019.06.002
9. Mottok A, Steidl C. Biology of classical Hodgkin lymphoma: implications for prognosis and novel therapies. Blood. 2018;131(15):1654–1665. doi:10.1182/blood-2017-09-772632
10. Wilson CL, Miller CJ. Simpleaffy: a BioConductor package for affymetrix quality control and data analysis. Bioinformatics. 2005;21(18):3683–3685. doi:10.1093/bioinformatics/bti605
11. Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics–a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009;25(3):415–416. doi:10.1093/bioinformatics/btn647
12. Xu R, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16(11):1155–1161. doi:10.1038/nmat4997
13. Onwuka J, Li D, Liu Y, et al. A panel of DNA methylation signature from peripheral blood may predict colorectal cancer susceptibility. BMC Cancer. 2020;20(1):692. doi:10.1186/s12885-020-07194-5
14. Han B, Bhowmick N, Qu Y, Chung S, Giuliano A, Cui X. FOXC1: an emerging marker and therapeutic target for cancer. Oncogene. 2017;36(28):3957–3963. doi:10.1038/onc.2017.48
15. Elian F, Yan E, Walter M. FOXC1, the new player in the cancer sandbox. Oncotarget. 2018;9(8):8165–8178. doi:10.18632/oncotarget.22742
16. Nagel S, Meyer C, Kaufmann M, Drexler H, MacLeod R. Deregulated FOX genes in Hodgkin lymphoma. Genes Chromosomes Cancer. 2014;53(11):917–933. doi:10.1002/gcc.22204
17. Kawano Y, Ouchida R, Wang J, et al. A novel mechanism for the autonomous termination of pre-B cell receptor expression via induction of lysosome-associated protein transmembrane 5. Mol Cell Biol. 2012;32(21):4462–4471. doi:10.1128/MCB.00531-12
18. Kiang K, Zhang P, Li N, Zhu Z, Jin L, Leung G. Loss of cytoskeleton protein ADD3 promotes tumor growth and angiogenesis in glioblastoma multiforme. Cancer Lett. 2020;474:118–126. doi:10.1016/j.canlet.2020.01.007
19. Olsson L, Castor A, Behrendtz M, et al. Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia. 2014;28(2):302–310. doi:10.1038/leu.2013.206
20. Song Y, Wu J, Chen X, et al. A single-arm, multicenter, Phase II Study of camrelizumab in relapsed or refractory classical Hodgkin lymphoma. Clin Cancer Res. 2019;25(24):7363–7369. doi:10.1158/1078-0432.CCR-19-1680
21. Liu X, Shan C, Song Y, Du J. Prognostic value of programmed cell death Ligand-1 expression in nasopharyngeal carcinoma: a meta-analysis of 1,315 patients. Front Oncol. 2019;9: p. 1111. doi:10.3389/fonc.2019.01111
22. Qiu L, Zheng H, Zhao X. The prognostic and clinicopathological significance of PD-L1 expression in patients with diffuse large B-cell lymphoma: a meta-analysis. BMC Cancer. 2019;19(1):273. doi:10.1186/s12885-019-5466-y
23. Wu Z, Yang L, Shi L, et al. Prognostic impact of Adenosine Receptor 2 (A2aR) and programmed Cell Death Ligand 1 (PD-L1) expression in colorectal cancer. Biomed Res Int. 2019;2019:8014627. doi:10.1155/2019/8014627
24. Roemer MGM, Redd RA, Cader FZ, et al. Major histocompatibility complex Class II and programmed death Ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J Clin Oncol. 2018;36(10):942–950. doi:10.1200/JCO.2017.77.3994
25. Hou Y, Li X, Li Q, et al. STAT1 facilitates oestrogen receptor alpha transcription and stimulates breast cancer cell proliferation. J Cell Mol Med. 2018;22(12):6077–6086. doi:10.1111/jcmm.13882
26. Goodman ML, Trinca GM, Walter KR, et al. Progesterone receptor attenuates STAT1-mediated IFN signaling in breast cancer. J Immunol. 2019;202(10):3076–3086. doi:10.4049/jimmunol.1801152
27. Leon-Cabrera S, Vazquez-Sandoval A, Molina-Guzman E, et al. Deficiency in STAT1 signaling predisposes gut inflammation and prompts colorectal cancer development. Cancers (Basel). 2018;10:9. doi:10.3390/cancers10090341
28. Crncec I, Modak M, Gordziel C, et al. STAT1 is a sex-specific tumor suppressor in colitis-associated colorectal cancer. Mol Oncol. 2018;12(4):514–528. doi:10.1002/1878-0261.12178
29. Meissl K, Macho-Maschler S, Muller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine. 2017;89:12–20. doi:10.1016/j.cyto.2015.11.011
30. Schellerer VS, Langheinrich MC, Zver V, et al. Soluble intercellular adhesion molecule-1 is a prognostic marker in colorectal carcinoma. Int J Colorectal Dis. 2019;34(2):309–317. doi:10.1007/s00384-018-3198-0
31. Shimura T, Shibata M, Gonda K, et al. Clinical significance of soluble intercellular adhesion Molecule-1 and Interleukin-6 in patients with extrahepatic cholangiocarcinoma. J Invest Surg. 2018;31(6):475–482. doi:10.1080/08941939.2017.1358310
32. Figenschau SL, Knutsen E, Urbarova I, et al. ICAM1 expression is induced by proinflammatory cytokines and associated with TLS formation in aggressive breast cancer subtypes. Sci Rep. 2018;8(1):11720. doi:10.1038/s41598-018-29604-2
33. Zhang X, Wang Y, Cao Y, Zhang X, Zhao H. Increased CCL19 expression is associated with progression in cervical cancer. Oncotarget. 2017;8(43):73817–73825. doi:10.18632/oncotarget.17982
34. Tokunaga R, Naseem M, Lo JH, et al. B cell and B cell-related pathways for novel cancer treatments. Cancer Treat Rev. 2019;73:10–19. doi:10.1016/j.ctrv.2018.12.001
35. Saxena V, Li L, Paluskievicz C, et al. Role of lymph node stroma and microenvironment in T cell tolerance. Immunol Rev. 2019;292(1):9–23. doi:10.1111/imr.12799
36. O’Connor T, Zhou X, Kosla J, et al. Age-related gliosis promotes central nervous system lymphoma through CCL19-mediated tumor cell retention. Cancer Cell. 2019;36(3):250–267 e259. doi:10.1016/j.ccell.2019.08.001
37. Matsuki E, Younes A. Lymphomagenesis in Hodgkin lymphoma. Semin Cancer Biol. 2015;34:14–21. doi:10.1016/j.semcancer.2015.02.002
38. Ren Z, Liang S, Yang J, et al. Coexpression of CXCR4 and MMP9 predicts lung metastasis and poor prognosis in resected osteosarcoma. Tumour Biol. 2016;37(4):5089–5096. doi:10.1007/s13277-015-4352-8
39. Li Q, Chen B, Cai J, et al. Comparative analysis of matrix metalloproteinase family members reveals that MMP9 predicts survival and response to temozolomide in patients with primary glioblastoma. PLoS One. 2016;11(3):e0151815. doi:10.1371/journal.pone.0151815
40. Yang XZ, Cui SZ, Zeng LS, et al. Overexpression of Rab1B and MMP9 predicts poor survival and good response to chemotherapy in patients with colorectal cancer. Aging (Albany NY). 2017;9(3):914–931. doi:10.18632/aging.101200
41. Wang Q, Yao J, Jin Q, et al. LAMP1 expression is associated with poor prognosis in breast cancer. Oncol Lett. 2017;14(4):4729–4735. doi:10.3892/ol.2017.6757
42. Dang Q, Zhou H, Qian J, et al. LAMP1 overexpression predicts for poor prognosis in diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18(11):749–754. doi:10.1016/j.clml.2018.07.288
43. Shao H, Ma L, Jin F, Zhou Y, Tao M, Teng Y. Immune inhibitory receptor LILRB2 is critical for the endometrial cancer progression. Biochem Biophys Res Commun. 2018;506(1):243–250. doi:10.1016/j.bbrc.2018.09.114
44. Gao A, Sun Y, Peng G. ILT4 functions as a potential checkpoint molecule for tumor immunotherapy. Biochim Biophys Acta Rev Cancer. 2018;1869(2):278–285. doi:10.1016/j.bbcan.2018.04.001
45. Cai Z, Wang L, Han Y, et al. Immunoglobulinlike transcript 4 and human leukocyte antigenG interaction promotes the progression of human colorectal cancer. Int J Oncol. 2019;54(6):1943–1954. doi:10.3892/ijo.2019.4761
46. Chen HM, van der Touw W, Wang YS, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest. 2018;128(12):5647–5662. doi:10.1172/JCI97570
47. Poh AR, Love CG, Masson F, et al. Inhibition of hematopoietic cell kinase activity suppresses myeloid cell-mediated colon cancer progression. Cancer Cell. 2017;31(4):563–575 e565. doi:10.1016/j.ccell.2017.03.006
48. Roversi FM, Pericole FV, Machado-Neto JA, et al. Hematopoietic cell kinase (HCK) is a potential therapeutic target for dysplastic and leukemic cells due to integration of erythropoietin/PI3K pathway and regulation of erythropoiesis: HCK in erythropoietin/PI3K pathway. Biochim Biophys Acta Mol Basis Dis. 2017;1863(2):450–461. doi:10.1016/j.bbadis.2016.11.013
49. Roseweir AK, Powell A, Horstman SL, et al. Src family kinases, HCK and FGR, associate with local inflammation and tumour progression in colorectal cancer. Cell Signal. 2019;56:15–22. doi:10.1016/j.cellsig.2019.01.007
50. Mangogna A, Belmonte B, Agostinis C, et al. Prognostic implications of the complement protein C1q in gliomas. Front Immunol. 2019;10:2366. doi:10.3389/fimmu.2019.02366
51. Zhang J, Zhang Y, Wu W, et al. Guanylate-binding protein 2 regulates Drp1-mediated mitochondrial fission to suppress breast cancer cell invasion. Cell Death Dis. 2018;9(11):1127. doi:10.1038/s41419-018-1133-5
52. Dimitrakopoulos C, Vrugt B, Flury R, et al. Identification and validation of a biomarker signature in patients with resectable pancreatic cancer via genome-wide screening for functional genetic variants. JAMA Surg. 2019;154(6):e190484. doi:10.1001/jamasurg.2019.0484
53. Lin Z, Wang H, Zhang Y, et al. Development and validation of a prognostic nomogram to guide decision-making for high-grade digestive neuroendocrine neoplasms. Oncologist. 2019;25(4):e659–e667. doi:10.1634/theoncologist.2019-0566
54. Semenkovich TR, Yan Y, Subramanian M, et al. A clinical nomogram for predicting node-positive disease in esophageal cancer. Ann Surg. 2019;270(3):434–443. doi:10.1097/SLA.0000000000003466
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.