")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 6
Combined hepatocellular cholangiocarcinoma (cHCC-CC): an update of genetics, molecular biology, and therapeutic interventions
Authors Stavraka C , Rush H, Ross P
Received 4 September 2018
Accepted for publication 8 November 2018
Published 28 December 2018 Volume 2019:6 Pages 11—21
DOI https://doi.org/10.2147/JHC.S159805
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Chara Stavraka,1,* Hannah Rush,1,* Paul Ross1,2
1Department of Medical Oncology, Guy’s Cancer, Guy’s & St Thomas’ NHS Foundation Trust, London, UK; 2Department of Oncology, King’s College Hospital NHS Foundation Trust, London, UK
*These authors contributed equally to this work
Abstract: Combined hepatocellular cholangiocarcinoma (CC) is a rare and aggressive primary hepatic malignancy with significant histological and biological heterogeneity. It presents with more aggressive behavior and worse survival outcomes than either hepatocellular carcinoma or CC and remains a diagnostic challenge. An accurate diagnosis is crucial for its optimal management. Major hepatectomy with hilar node resection remains the mainstay of treatment in operable cases. Advances in the genetic and molecular characterization of this tumor will contribute to the better understanding of its pathogenesis and shape its future management.
Keywords: cholangiocarcinoma, hepatocellular carcinoma, combined, genetics, molecular biology, treatment, mixed liver tumors, primary liver cancer
Introduction
Combined or mixed hepatocellular cholangiocarcinoma (cHCC-CC) is a distinct type of primary liver cancer sharing unequivocal phenotypical characteristics of both hepatocellular carcinoma (HCC) and cholangiocarcinoma (CC).1,2 It is a rare entity with a variably reported incidence between 0.4% and 14.2% across a number of studies.1,3–6 This is considered to be an underestimation not only due to the diagnostic challenges associated with this malignancy but also due to a previous lack of consensus on the nomenclature.7
The histogenesis and natural history of this rare malignancy remain elusive;8 however, the widely accepted origin of cHCC-CC is a common hepatic stem cell.9,10 There is conflicting evidence in the existing literature with regard to the epidemiological and clinical features of cHCC-CC. Several studies have suggested similarities to HCC,11–13 whereas others related it to CC.1,6 There is also growing evidence suggesting that cHCC-CC has intermediate clinical features between HCC and CC,14 which is also reflected in patient survival outcomes. cHCC-CC displays a rather aggressive behavior and is associated with poorer prognosis compared to HCC and more favorable than CC in patients undergoing liver resection.12,15–18 An accurate perioperative diagnosis is of paramount importance and directs the consequent surgical management of this tumor, with major hepatectomy being the best therapeutic approach.17,18 The management of this tumor is hindered by the lack of international guidelines, while the role and indications of liver transplantation (LT) remain equivocal.18,19
Here we present a review on this challenging primary hepatic tumor with a particular focus on its genetics, molecular biology, and therapeutic interventions.
Classification
The initial description of cHCC-CC dates back to 1903, but was more comprehensively studied later in 1949 when Allen and Lisa classified it in three different histological types (type A, B, and C) depending on the presence of HCC and CC: at different sites in the same liver, at adjacent sites, or in the same tumor, respectively.4 Subsequently, Goodman et al reclassified these tumors into type I (collision) characterized by the coincidental existence of both HCC and CC in the same liver, type II (transitional) characterized by the presence of areas of transition between HCC and CC, and type III (fibrolamellar), which resemble the fibrolamellar variant of HCC with the additional presence of pseudo-glands producing mucin.1 WHO recognized cHCC-CC as a distinct entity and further classified it into two main types: the classical type, which is characterized by intermixed areas of typical HCC and CC and the presence of transition zones with intermediate morphology of both types, and the type with stem cell features, which is less common and further subdivided into typical, intermediate, and cholangiocellular (CLC) subtype (Table 1).2
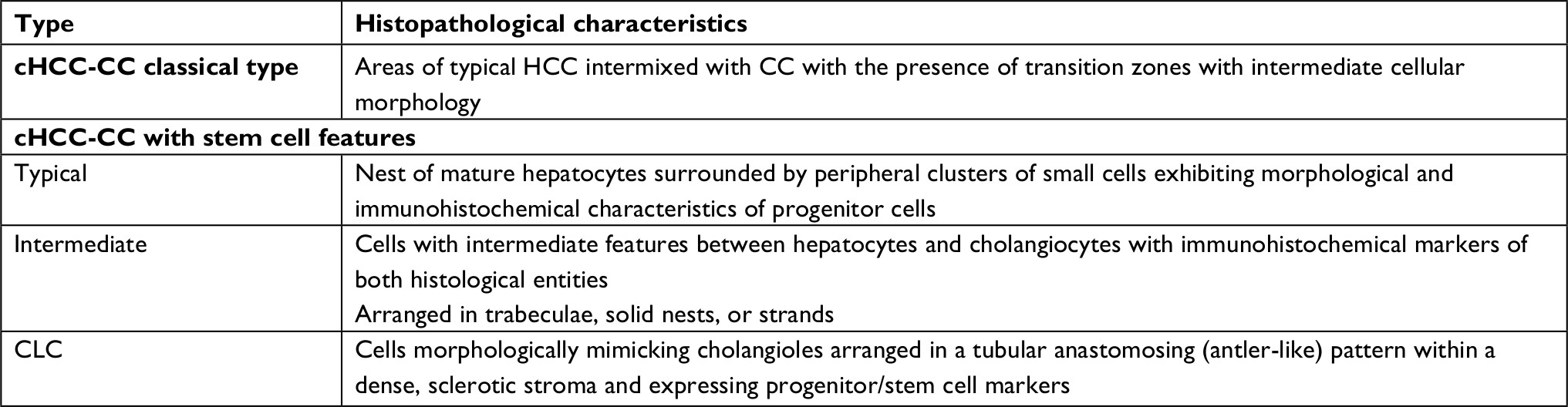 | Table 1 The 2010 WHO classification of cHCC-CC. Adapted with permission from Bosman, FT, Carneiro,F, Hruban, RH, Theise, ND. World Health Organization Classification of Tumours of the Digestive System. IARC, Lyon, 2010.2 Abbreviations: CC, cholangiocarcinoma; CLC, cholangiocellular; cHCC-CC, combined hepatocellular cholangiocarcinomas; HCC, hepatocellular carcinoma. |
Certain histopathological criteria have been established for the definitive diagnosis of cHCC-CC, which require the presence of fully differentiated components of hepatocellular and CC intimately mixed with concurrent evidence of transition zones comprising cells with intermediate morphology.2 This distinguishes it from HCC and CCs found in the same liver lobe, which represent collision tumors.
Epidemiology and clinical profile
The demographic and clinical profile of combined HCC is not yet fully characterized. Due to the rarity of this malignancy, the majority of data are generated from single-center retrospective studies with relatively small cohort sizes and significant variations in the studied populations. Although risk factors for HCC and CC have been established, this is not the case for cHCC-CC, and this yet remains an elusive matter.6,20 The most common risk factors for HCC include liver cirrhosis, infection with hepatotropic viruses such as Hepatitis B Virus (HBV), Hepatitis C virus (HBC) and Hepatitis D virus (HDV), alcoholic liver disease, and nonalcoholic fatty liver disease.21,22 Risk factors associated with CC involve primary sclerotic cholangitis, liver fluke infestation, hepatolithiasis, and exposure to Thorotrast.23 A number of studies conducted in Asian patients have demonstrated similarities between cHCC-CC and HCC, which involve strong male predominance, underlying liver cirrhosis, and serologically confirmed hepatitis.3,24–28 On the contrary, several studies originating from Western countries supported a resemblance between cHCC-CC and CC with no gender predisposition and absence of hepatitis.1,6,29,30 To add to the existing ambiguity, other studies have suggested that cHCC-CC has a distinct clinical profile with intermediate characteristics to those of HCC and CC.3,14 This inconsistency is further compounded by the previous absence of accurate histological diagnostic criteria for cHCC-CC.
Histogenesis
The histogenesis of cHCC-CC remains elusive and has been a subject of debate. The most prominent hypothesis is that it derives from bipotent hepatic progenitor cells, which are intermediate stem cells capable of undergoing bidirectional differentiation into hepatocytes and bile duct epithelial cells.31–33 However, Moeini et al suggested that cHCC-CCs may be derived from more than one cell of origin. They performed gene expression profiling showing a biliary committed precursor for CLC type and a biphenotypic progenitor-like precursor for the classical and other stem cell subtypes.34
The monoclonal origin of cHCC-CC was supported by Fujii et al35 who used microdissection and DNA extraction showing that both hepatocellular and CLC components of these tumors share identical allelic loses.35 On a similar note, another study demonstrated that a primary cell line derived from resected cHCC-CC could differentiate into either HCC or CC by changing the different growth conditions.36
Diagnosis
Histopathology and IHC
A definitive diagnosis of cHCC-CC can only be made after histopathological assessment of a representative biopsy. In clinical practice, this may mean that a single pass biopsy results in an incorrect diagnosis due to sampling a nonrepresentative area. Therefore, when a diagnosis of cHCC-CC is suspected, extensive biopsies should be considered to increase diagnostic accuracy. Despite this, diagnosis may not be established until histopathological examination of the resected specimen.
To meet the diagnostic criteria, the sample must show clear evidence of hepatocellular and biliary differentiation with the two types of tumor cells intermingling and transition zones where the cells demonstrate intermediate morphology (Figure 1).2 Patients may have a collision tumor with both HCC and intrahepatic cholangiocarcinoma (ICC), but if there are separate foci of disease with no integration of tumor cells and no transition zones then this does not constitute a true cHCC-CC. Tumors may have a preponderance of either tumor type, which will in turn influence the features of the disease.
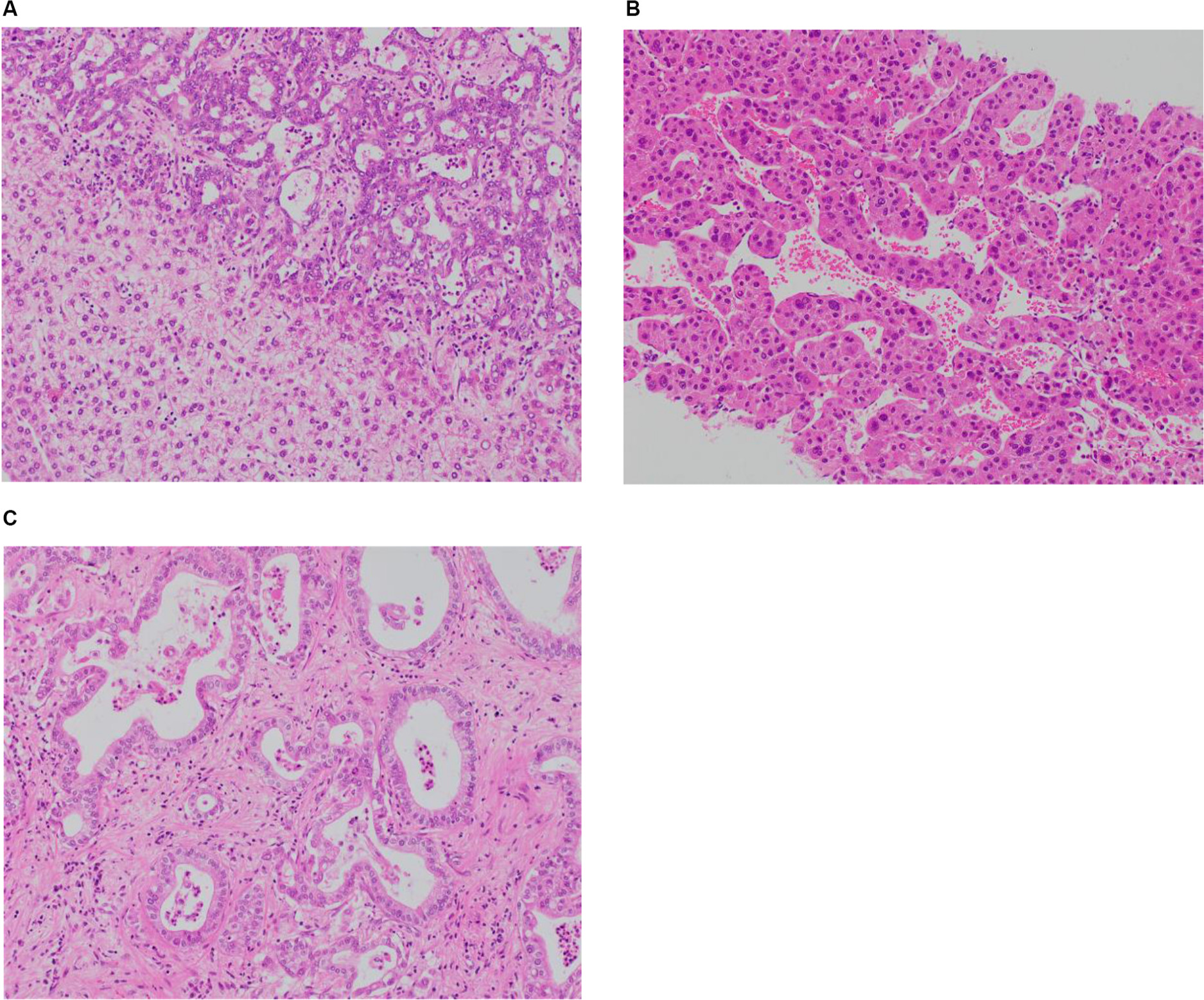 | Figure 1 (A) Combined hepatocellular and cholangiocellular carcinoma. A single tumor nodule shows two different histological components, one with glandular differentiation and biliary immunoprofile consistent with cholangiocarcinoma (upper area of the picture) and one with a well-to-moderately differentiated hepatocellular carcinoma (lower part of the picture); H&E staining. (B) Moderately differentiated HCC, trabecular pattern. Cellular variability with scattered large hyperchromatic nuclei; H&E staining, 100×. (C) Moderately differentiated intrahepatic cholangiocarcinoma. Glandular structures are variable in size and shape, in a sclerosed stroma; H&E staining, 100×. |
HCC differentiation is determined by the presence of bile-producing cells, Mallory-Denk bodies, bile canaliculi, and a pseudoglandular/trabecular growth pattern. CC differentiation is characterized by mucin-producing biliary epithelium forming true glandular structures and surrounding desmoplastic stroma.37 cHCC-CC demonstrates independent biphenotypic differentiation; each component can be well to poorly differentiated.
cHCC-CCs also demonstrate an immunohistochemistry (IHC) profile consistent with both hepatic and biliary phenotypes. For the HCC component, this would include positive staining for carcinoembryonic antigen (CEA), HepPar1, CD-10, CAM5.2, and glypican-3. For the CC component, this includes positive staining for mucin, which is essential to demonstrate the biliary component, CK7, CK19, and AE1.38 Transition zones typically stain for CC cytokeratins CK7, CK19, and the hepatocellular marker HepPar-1. The presence of albumin mRNA detected by in situ hybridization is an additional hepatocellular marker seen in the transition zone that may differentiate the tumor from ICC.30 The cHCC-CC with stem cell features subtype will also characteristically stain positive for CK7, CK10, CD56, c-KIT, NCAM, DLK-1, and EpCAM.39 However, IHC does not differentiate between HCC with inflammation of the biliary tree vs cHCC-CC. Examination of the biopsy should assess for the presence of desmoplastic stroma rather than the inflammatory cells of ductal reactivity, in order to confirm that the diagnosis is cHCC-CC.31
Raised alpha-fetoprotein (AFP) is associated with HCC and raised Ca19–9 associated with CC. In patients with a radiological diagnosis of either HCC or CC, a discordant pattern of tumor markers or a pattern of synchronously raised tumor markers should be a warning to consider the diagnosis of cHCC-CC.2,38 Patients with cHCC-CC tend to have a more modestly raised AFP than patients with HCC.
Imaging
A radiological diagnosis of cHCC-CC can be difficult to make as this tumor presents with heterogeneous imaging characteristics. The presence of overlapping radiological features of HCC and CC mandates a biopsy for definitive diagnosis. The predominant histologic component within the tumor will determine the predominant radiological features.
On ultrasound, the appearances of cHCC-CC are nonspecific. Lesions may be visualized as a round hypoechoic mass with a central hyperechoic focus or a heterogeneous hypoechoic mass.40 While ultrasound is unlikely to be sufficient to diagnose cHCC-CC, it can be of use to guide biopsy toward appropriate lesions and thus play a role in the evaluation of such patients.
The computed tomography (CT) appearances vary according to the proportion and distribution of HCC and CC components and also according to the subtype of CC. Features suggestive of an HCC on CT imaging include arterial phase diffuse enhancement, portal venous washout, and enhanced pseudocapsule on delayed imaging. On the other hand, features suggestive of a CC on CT imaging are arterial peripheral ring enhancement, progressive fibrous stroma central enhancement, dilation of the biliary tree, and retraction of the capsule.41 cHCC-CC may show any of these features to a mixed degree.
Multiple studies have examined MRI appearances of cHCC-CC with mixed results. Some report that the tumor more closely resembles HCC,42 while others suggest that MRI appearances are more similar to ICC or metastases.43,44 This likely reflects the heterogeneous nature of the disease and different cohorts studied. Important MRI features that may indicate a diagnosis of cHCC-CC include the presence of washout, washout and progression in the same lesion, intralesional fat, and hemorrhage.42 On T1w imaging, cHCC-CC appears hypointense, while on T2w imaging, lesions demonstrate an intermediate to high signal intensity with or without a central hypointense focus.42
The MRI contrast agent gadoxetic acid has both perfusion-selective and hepatocyte-selective features to help distinguish between cHCC-CC and ICC or metastases. The features of cHCC-CC with contrast-enhanced MRI are still diverse due to the heterogeneous histological features; therefore, the use of contrast cannot always differentiate between HCC and cHCC-CC.45
Table 2 summarizes two proposed classification systems of cHCC-CC based on CT patterns of enhancement. Aoki’s type A and Sanada’s type 3 pattern are the most suggestive of cHCC-CC on imaging and tumors displaying that these characteristics should undergo further evaluation.27,46
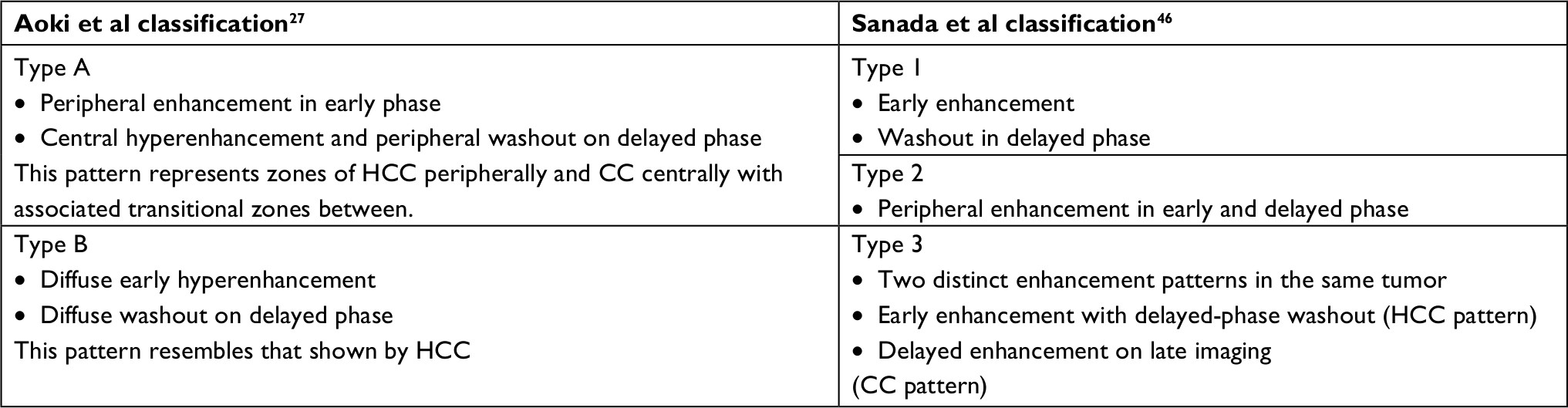 | Table 2 Proposed radiological classifications based on enhancement patterns Abbreviations: CC, cholangiocarcinoma; HCC, hepatocellular carcinoma. |
More widely adopted is the Liver Imaging-Reporting and Data System (Li-RADS) providing a standardized approach to reporting all liver lesions on the basis of MRI and CT imaging.47 This system was developed in an attempt to standardize radiological criteria for diagnosing HCC. Lesions are labeled with increasing numbers representing increasing likelihood that a lesion is HCC – from LR-1 at 0% probability to LR-5 at 100% probability. Intrahepatic malignancy that is not HCC is labeled as LR-M. Subsequent analysis supports that this system differentiates between HCC and cHCC-CC. The majority of cHCC-CC cases reviewed against the Li-RADS criteria demonstrated ancillary features favoring non-HCC malignancy, sufficient for them to be recorded as LR-M.47 In their study, Potretzke et al reviewed 61 cases of biphenotypic primary liver carcinomas using Li-RADS, version 2014, demonstrating that the addition of ancillary features to major features had superior diagnostic accuracy over the sole use of major features.48
Studies suggest that radiological features can be used to provide prognostic information about cHCC-CC. Tumors, which are radiologically HCC predominant, have a better prognosis from ICC-predominant subtypes.49
Genetic characterization of cHCC-CC demonstrates a diverse range of mutations, which overlap with mutations seen in HCC and CC. Understanding the genetic signature specific to cHCC-CC could help differentiate between these diagnoses. Genetic profiling could help define subtypes with cHCC-CC and identify therapeutic targets. It could also provide information on etiology, histogenesis, and prognosis. Genetic and epigenetic evaluation of tumors has been carried out in HCC and CC, but only small studies have currently been undertaken in cHCC-CC.
In cHCC-CC, the genetic landscape is similar across both HCC and CC components, proving that this is a distinct tumor type rather than a collision of two separate cancers. Whole-genome analysis of copy number variations comparing the HCC and CC components of the same tumor found a concordant trend between copy number gain or loss in both components.50
In order to identify potential therapeutic targets, mutations within particular signaling pathways involved in carcinogenesis have been evaluated. This demonstrates cHCC-CC has features in common with both HCC and ICC. Pathways identified in patients with cHCC-CC that may provide therapeutic targets include MAPK, P53, pI3K–AKT–mTOR, and the Notch–Hedgehog pathway.51
While some mutations are common to all primary liver cancers, other mutations are more specific and, if identified, may aid diagnosis. For example, mutations in tumor-suppressor gene TP53 are seen in all tumor types, although it is more prevalent in cHCC-CC (27%–100%) and HCC (26%) in comparison with CC (0%–11%).50,51 These are summarized in Table 3.
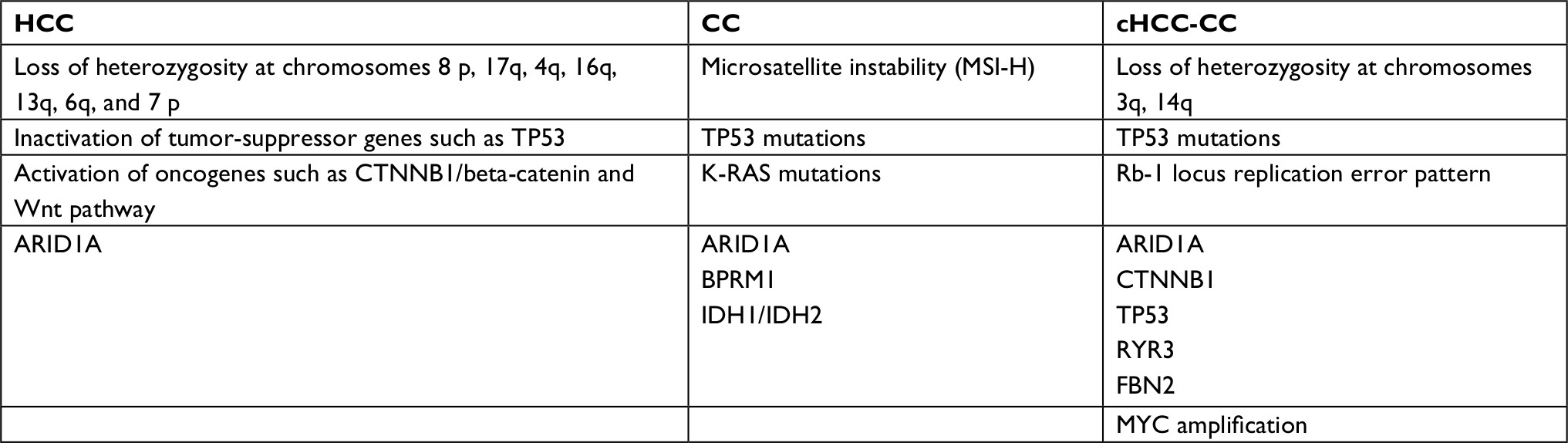 | Table 3 Common genetic alterations seen in HCC, CC, and cHCC-CC41,50,85 Abbreviations: CC, cholangiocarcinoma; cHCC-CC, combined hepatocellular cholangiocarcinoma; HCC, hepatocellular carcinoma; MSI-H, microsatellite instability high. |
Gene profiling by microarray confirms that cHCC-CC displays a combination of the upregulated and downregulated genes seen in HCC and ICC. There are a significant number of differently expressed genes between ICC and HCC, and there is a less marked yet still significant difference between ICC and cHCC-CC supporting that these tumors are distinct entities.52 The pattern of differentially expressed genes as assessed by microarray could allow for more accurate diagnosis in cases of diagnostic uncertainty.
A targeted gene panel revealed that mutations in genes KRAS, ARID1A, TERT promoter, and TP53 are associated with different clinical phenotypes of cHCC-CC (Table 4).53 For example, patients with ARID1A gene mutations were more likely to report a history of alcohol excess. Similar correlations with mutations in ARID1A are seen in HCC and ICC. Studies suggest that ARID1A mutations occur in response to oxidative stress, which may be induced by alcoholic liver disease. Recently, mutations to IDH1/H2 have been identified in HCC tumors.54 These tumors had a histopathological picture of HCC but clinical and genetic features of CC and HCC. This generated the hypothesis that IDH1/H2 mutations could shift primary liver tumors toward a biliary phenotype.
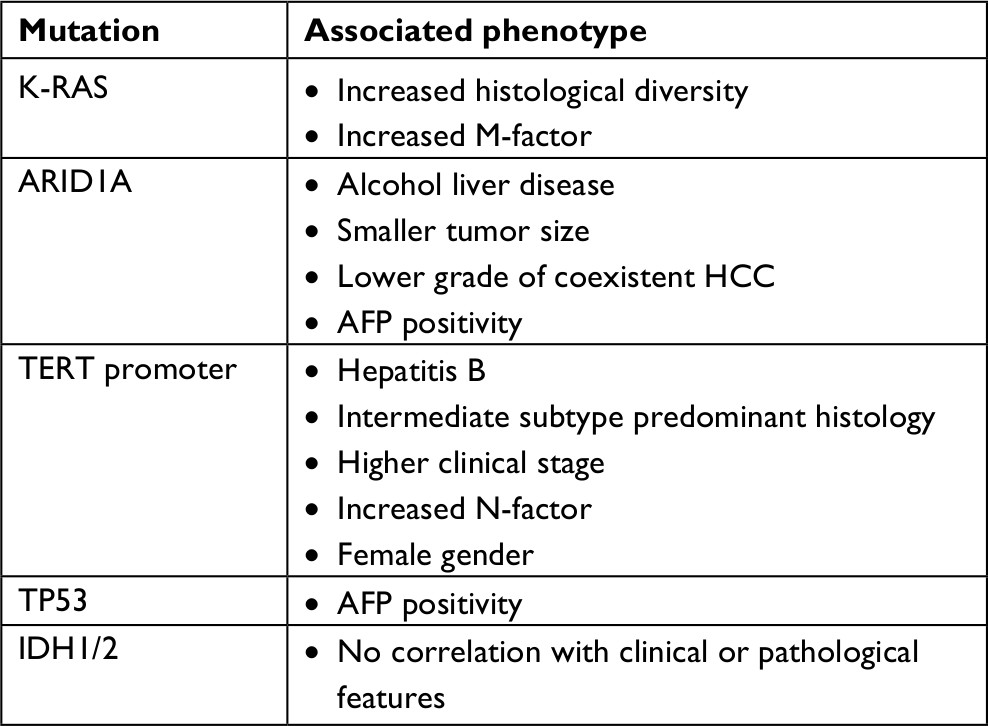 | Table 4 Mutational landscape of cHCC-CC with clinical–pathological phenotype correlates53 Abbreviation: AFP, alpha fetoprotein; cHCC-CC, combined hepatocellular cholangiocarcinoma; HCC, hepatocellular carcinoma; IDH, isocitrate dehydrogenase; TERT, telomerase reverse transcriptase. |
Patients with mutations in the TERT (telomerase reverse transcriptase) promoter gene were more likely to have a history of underlying hepatitis B. TERT promoter mutations are also commonly seen as an early mutation in patients with HCC on a background of chronic hepatitis.53 Specific mutations in TP53 are associated with a shorter prognosis in patients with HCC. The same mutations have been identified in patients with cHCC-CC. The data do not currently confirm whether these mutations are prognostic markers in cHCC-CC also.51
Recognition of these mutations may give clues on the etiology of the tumor. However, confounding factors and causal links have not been established from current cHCC-CC studies. Although our knowledge of the genetics of cHCC-CC is increasing, much of the data are from small case series, bringing into question their reliability and how easily they can be generalized to different populations. Conflicting results on which mutations are diagnostic of cHCC-CC need to be clarified. Further studies to evaluate the significance of genetic and signaling pathway mutations may lead to a better understanding of pathogenesis and potential therapeutic targets.
Molecular biology
The molecular profile of cHCC-CCs remains incompletely characterized. Woo et al performed gene expression profiling on HCC, CC, and cHCC-CC human samples identifying a subtype of HCC with CC traits, which was associated with worse survival outcomes and suggesting a phenotypical overlap between HCC, CC, and cHCC-CC.55 Coulouarn et al profiled 20 tissue samples of histologically confirmed cHCC-CC showing that it has stem cell progenitor features and is characterized by a downregulation of an HNF4-driven hepatocyte differentiation program and a commitment to the biliary lineage. Activation of TGFβ and Wnt/β-catenin signaling pathways was also observed.56 TGFβ pathway is known to be involved in biliary differentiation and in epithelial-mesenchymal transition, whereas Wnt/β-catenin pathway plays a key role in regulating the fate of hepatoblasts toward a biliary ductal morphogenesis.57,58 Furthermore, integrative genomic analysis in the same study demonstrated that cHCC-CC shares common characteristics with a subset of HCC with stem cell traits.56
However, genome-wide allelotyping of classical cHCC-CC, by Cazals-Hatem et al, showed that these tumors exhibit high level of chromosomal instability and are genetically closer to CC than to HCC.14 A more recent comprehensive molecular characterization of cHCC-CCs was performed on 18 human tissue samples suggesting that they represent a heterogeneous group of tumors, which can be categorized into two main subclasses: 1) the classical subclass, which shares components of both HCC and CC arising from a common clonal origin, and 2) the stem cell subclass, which is associated with a progenitor-like phenotype and a more aggressive behavior. The latter is characterized by upregulation of proliferative signaling pathways (MYC, mTORC, and NOTCH). Interestingly, in this study, the CLC carcinoma was found to be a distinct entity with a biliary molecular profile and no HCC traits. It is characterized by chromosomal stability and active TGFβ signaling.34
Therapeutic interventions
Surgery
Surgical resection is the only curative option for patients with cHCC-CC. The feasibility of surgery is dictated by several factors including patient’s overall physical condition, the extent of the tumor, and anatomical characteristics. The aim is to completely excise the lesion with clear margins and the least possible impact on the liver function, as significant liver impairment is associated with poor survival outcomes.25
cHCC-CCs demonstrate characteristics of both tumors, showing hepatic and portal venous infiltration similar to HCC and also metastasizing to the lymph nodes on a similar pattern to CC. Therefore, major hepatic resection with hilar node dissection remains the recommended treatment in noncirrhotic patients with no distant metastases.59 In cases of liver cirrhosis, limited resection may be considered to maintain adequate residual liver function.25 The additional benefit of lymphadenectomy in the overall survival (OS) remains a controversial issue.60–62 Despite the risks and the complexity of major liver resection, fit patients with noncirrhotic liver have been found to tolerate it well.63 A recent study suggested that in patients with similar tumor characteristics, age should not be considered a limiting factor for aggressive liver surgery as both investigated age groups had similar survival outcomes.64 However, it should be taken into account that an age cut-off of 60 years was used for patient stratification in this study with the elderly patients having a mean age of 67.5 years and the younger 47 years. In patients with multiple comorbidities or liver cirrhosis, aggressive liver surgery could be detrimental; therefore, careful selection of the optimal surgical candidates is of paramount importance. The Child-Turgotte-Pugh and the Model for End-Stage Liver Disease scores have been used as predictors of postoperative mortality post liver resection. The latter is calculated from serum bilirubin, creatinine, and the international normalized ratio and has been preferable due to its more objective variables.65,66
Despite the curative aim of liver resection, cHCC-CC shows an aggressive behavior with high recurrence rates and an average 5-year survival rate of 30%.16,17,30 The majority of studies so far have demonstrated worse postsurgical survival outcomes for patients with cHCC-CC compared to those with HCC but better than CC;3,11,16,17 however, in some cases, surgical outcomes were even worse than those for CC.12,15 This behavior could be underpinned by the distinct biological features of cHCC-CC, which need to be further elucidated.
Liver transplantation
The role of LT as a potential treatment option for cHCC-CC has been investigated in a number of studies limited by their retrospective nature, their small patient numbers, and also by the fact that, in the majority of cases, final diagnosis of cHCC-CC was only established postoperatively. Groeschl et al performed a retrospective analysis of the Surveillance Epidemiology and End Results (SEER) database identifying 1,147 patients with HCC and 19 with cHCC-CC and compared survival outcomes after LT. They observed that patients with cHCC-CC had inferior 5-year OS rates compared to those with HCC (48% vs 78%, P=0.01).67 Similarly, Garancini et al interrogated the SEER database identifying 465 patients with cHCC-CC who had undergone minor or major hepatectomy, or LT. Survival outcomes post LT were again less favorable than for patients with HCC (41.1% vs 67%, P<0.001). LT did not offer a survival benefit compared to major hepatectomy (MJH) and minor hepatectomy (MNH) in the multivariate analysis (HR: 0.28, 0.25, 0.29, respectively); therefore, they questioned the efficacy of LT and supported MJH as the optimal treatment approach for cHCC-CC.18 The inferiority of LT in cHCC-CC patients was further supported by Vilchez et al, in their analysis of the united network for organ sharing (UNOS) database. They found a 5-year OS of 40% in cHCC-CC patients compared to 62% in HCC patients (P=0.002).68 Park et al, in their retrospective analysis of 15 patients who underwent LT with a pretransplant diagnosis of HCC, reported that cHCC-CC was associated with high relapse rates within the first year.69
Several smaller case studies have also been published with none conferring a clear benefit of LT to hepatic resection in this patient population.19,70–73 Magistri et al performed a systematic review of the literature to address the same question, concluding that LT should not be considered for the management of cHCC-CC.74 Considering all published evidence to date, major hepatic resection remains the best treatment option for resectable cHCC-CC. The role of LT in this tumor type is yet to be defined with more studies and better patient selection.
Nonsurgical treatment options
In cases of inoperable or recurrent cHCC-CCs, there are nonsurgical treatment options, which include transarterial chemoembolization (TACE), radioembolization, hepatic arterial infusional chemotherapy, ablative therapies, and systemic chemotherapy.25
Limited data exist on the efficacy and benefits of liver-directed therapies. A small retrospective study evaluated survival outcomes in 18 patients with inoperable cHCC-CC who received liver-directed therapy (TACE, radioembolization, or hepatic arterial infusional chemotherapy) from a total cohort of 79 patients.75 Those receiving liver-directed treatment had larger tumors than those undergoing surgical treatment (mean tumor size 8.9 vs 5.8 cm), more frequent satellite lesions (83% vs 32%), and higher incidence of lymph node metastases (33% vs 8%). Liver-directed therapy resulted in an overall partial response rate of 47% (50% with radioembolization, 20% with TACE, and 66% with hepatic arterial infusional chemotherapy) with median progression-free survival and OS of 8.3 and 16.0 months, respectively. Despite being limited by its retrospective nature and the small number of patients, this study demonstrates that liver-directed treatments could offer a survival benefit and potentially downstage patients for surgical treatment.75 However, more research is needed to further support the validity of these outcomes.
TACE has been the mainstay for palliative management of HCC, and its efficacy is highly dependent on tumor vascularity due to the intravascular delivery of the embolic and chemotherapeutic agents. However, due to the histological heterogeneity of cHCC-CCs76 and the fact that they are usually more fibrotic and less vascular than HCCs, the benefit of TACE is debatable. The efficacy of TACE on primary nonresectable and recurrent cHCC-CCs was investigated in two small retrospective studies, concluding that response and consequently prognosis are highly related to tumor vascularity. Survival outcomes were poorer than those seen for patients with HCC.26,77
Systemic chemotherapy
A standard treatment has not yet been established for advanced nonresectable cHCC-CCs, and the role of systemic chemotherapy remains unclear and associated with unfavorable outcomes.75,78,79 Due to the rarity of this tumor, current evidence is limited and relies on case reports and small retrospective studies, while treatment decisions are extrapolated from HCC and CC.80,81 A recent multicenter retrospective analysis involving 36 patients evaluated several first-line treatments including gemcitabine/cisplatin, fluorouracil/cisplatin, and sorafenib and showed poorer OS upon treatment with sorafenib monotherapy than for treatment with platinum-containing regimens (HR: 15.83 [95% CI 2.25–111.43], P=0.006). In this study, patients had an OS of 8.9 months across all treatments.78 In their retrospective analysis of seven patients, Rogers et al showed similar survival outcomes (median OS: 8.3 months) and supported the lack of efficacy of sorafenib in this tumor type. Disease control was achieved in three patients who received gemcitabine plus platinum with or without bevacizumab as first or second line of treatment.79
It is of interest that cHCC-CCs do not seem to respond to systemic treatments that are effective in the two separate malignancies, which underlines the discrete entity of these tumors. Platinum-containing regimens such as gemcitabine and cisplatin seem to be more promising among other available treatments; however, prospective trials are needed to identify a standard of care for unresectable cHCC-CCs.
Future perspectives
The failure of standard surgical and systemic treatments to tackle these phenotypically heterogeneous tumors requires new approaches for their optimal management. With the advent of omics and their integration in translational research, dissecting the mutational landscape of cHCC-CCs in view of developing molecularly targeted treatments should be a tangible goal. A great step toward this goal has been the successful development of primary liver cancer-derived organoids, which retain the histological characteristics and the genomic landscape of the original tumor.82 These can be utilized not only for the identification of novel biomarkers but also for the screening of potential molecular targeted treatments. Of note is the identification of ERK inhibition as a potential target in a subset of HCC and CC patients. The use of organoids could revolutionize the biomarker discovery and drug testing for cHCC-CCs leading to novel treatments in the near future.
Furthermore, promising data stemming from ongoing clinical trials on HCC investigating several combinations of immunotherapy agents and the combination of immunotherapy with liver ablation indicate that this could be a potential therapeutic avenue to be explored also for cHCC-CCs.83,84
Conclusion
cHCC-CC is a rare, aggressive primary liver malignancy with poorer prognosis than HCC and CC. An accurate preoperative diagnosis is of key importance for its optimal management. However, this aspect remains a significant challenge due to the heterogeneity of its clinical and demographic features, indistinct imaging characteristics as well as the inconsistent application of histopathological criteria. The combination of imaging along with serum levels of tumor markers raise the suspicion of cHCC-CC, but tumor biopsies are required for a definite diagnosis. The final diagnosis of cHCC-CC is based on immunohistopathology and can be facilitated by genetic analyses. There is ongoing progress in the identification of the mutational landscape of cHCC-CC, which can further characterize these tumors and result in the development of new targeted treatment. Continued research is required to define the roles of both advanced imaging and molecular analysis in the diagnostic and therapeutic algorithms of cHCC-CC. Major hepatectomy and hilar lymph node resection remain the standard of care in operable disease. LT has not been found to be of benefit in cHCC-CC; however, more clinical data are required to obtain a conclusive outcome. Similarly, the role of liver-directed therapies and systemic treatment remains limited and needs to be further investigated in the context of prospective clinical trials.
Acknowledgment
Pictures kindly provided by Dr Rosa Miquel, consultant histopathologist, Liver Histopathology Laboratory, King’s College Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
Goodman ZD, Ishak KG, Langloss JM, Sesterhenn IA, Rabin L. Combined hepatocellular-cholangiocarcinoma. A histologic and immunohistochemical study. Cancer. 1985;55(1):124–135. | ||
Bosman FT, Carneiro F, Hruban R, Theise ND. WHO Classification of Tumours of the Digestive System. IARC Press: Lyon; 2010. | ||
Koh KC, Lee H, Choi MS, et al. Clinicopathologic features and prognosis of combined hepatocellular cholangiocarcinoma. Am J Surg. 2005;189(1):120–125. | ||
Allen RA, Lisa JR. Combined liver cell and bile duct carcinoma. Am J Pathol. 1949;25(4):647–655. | ||
Wachtel MS, Zhang Y, Xu T, Chiriva-Internati M, Frezza EE. Combined hepatocellular cholangiocarcinomas; analysis of a large database. Clin Med Pathol. 2008;1:S500–S547. | ||
Jarnagin WR, Weber S, Tickoo SK, et al. Combined hepatocellular and cholangiocarcinoma: demographic, clinical, and prognostic factors. Cancer. 2002;94(7):2040–2046. | ||
Brunt E, Aishima S, Clavien PA, et al. cHCC-CCA: consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentiation. Hepatology. 2018;68(1):113–126. | ||
Wang AQ, Zheng YC, Du J, et al. Combined hepatocellular cholangiocarcinoma: controversies to be addressed. World J Gastroenterol. 2016;22(18):4459–4465. | ||
Kim H, Park C, Han KH, et al. Primary liver carcinoma of intermediate (hepatocyte–cholangiocyte) phenotype. J Hepatol. 2004;40(2):298–304. | ||
Zhang F, Chen XP, Zhang W, et al. Combined hepatocellular cholangiocarcinoma originating from hepatic progenitor cells: immunohistochemical and double-fluorescence immunostaining evidence. Histopathology. 2008;52(2):224–232. | ||
Lee WS, Lee KW, Heo JS, et al. Comparison of combined hepatocellular and cholangiocarcinoma with hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Surg Today. 2006;36(10):892–897. | ||
Zuo HQ, Yan LN, Zeng Y, et al. Clinicopathological characteristics of 15 patients with combined hepatocellular carcinoma and cholangiocarcinoma. Hepatobiliary Pancreat Dis Int. 2007;6(2):161–165. | ||
Lee CH, Hsieh SY, Chang CJ, Lin YJ. Comparison of clinical characteristics of combined hepatocellular-cholangiocarcinoma and other primary liver cancers. J Gastroenterol Hepatol. 2013;28(1):122–127. | ||
Cazals-Hatem D, Rebouissou S, Bioulac-Sage P, et al. Clinical and molecular analysis of combined hepatocellular-cholangiocarcinomas. J Hepatol. 2004;41(2):292–298. | ||
Lee JH, Chung GE, Yu SJ, et al. Long-term prognosis of combined hepatocellular and cholangiocarcinoma after curative resection comparison with hepatocellular carcinoma and cholangiocarcinoma. J Clin Gastroenterol. 2011;45(1):69–75. | ||
Yoon YI, Hwang S, Lee YJ, et al. Postresection outcomes of combined hepatocellular carcinoma-cholangiocarcinoma, hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J Gastrointest Surg. 2016;20(2):411–420. | ||
Yin X, Zhang BH, Qiu SJ, et al. Combined hepatocellular carcinoma and cholangiocarcinoma: clinical features, treatment modalities, and prognosis. Ann Surg Oncol. 2012;19(9):2869–2876. | ||
Garancini M, Goffredo P, Pagni F, et al. Combined hepatocellular-cholangiocarcinoma: a population-level analysis of an uncommon primary liver tumor. Liver Transpl. 2014;20(8):952–959. | ||
Jung DH, Hwang S, Song GW, et al. Longterm prognosis of combined hepatocellular carcinoma-cholangiocarcinoma following liver transplantation and resection. Liver Transpl. 2017;23(3):330–341. | ||
Ng IO, Shek TW, Nicholls J, Ma LT. Combined hepatocellular-cholangiocarcinoma: a clinicopathological study. J Gastroenterol Hepatol. 1998;13(1):34–40. | ||
Ghouri YA, Mian I, Rowe JH. Review of hepatocellular carcinoma: epidemiology, etiology, and carcinogenesis. J Carcinog. 2017;16:1–1. | ||
Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist. 2010;15(Suppl 4):14–22. | ||
Khan SA, Toledano MB, Taylor-Robinson SD. Epidemiology, risk factors, and pathogenesis of cholangiocarcinoma. HPB. 2008;10(2):77–82. | ||
Taguchi J, Nakashima O, Tanaka M, Hisaka T, Takazawa T, Kojiro M. A clinicopathological study on combined hepatocellular and cholangiocarcinoma. J Gastroenterol Hepatol. 1996;11(8):758–764. | ||
Kassahun WT, Hauss J. Management of combined hepatocellular and cholangiocarcinoma. Int J Clin Pract. 2008;62(8):1271–1278. | ||
Kim JH, Yoon HK, Ko GY, et al. Nonresectable combined hepatocellular carcinoma and cholangiocarcinoma: analysis of the response and prognostic factors after transcatheter arterial chemoembolization. Radiology. 2010;255(1):270–277. | ||
Aoki K, Takayasu K, Kawano T, et al. Combined hepatocellular carcinoma and cholangiocarcinoma: clinical features and computed tomographic findings. Hepatology. 1993;18(5):1090–1095. | ||
Yano Y, Yamamoto J, Kosuge T, et al. Combined hepatocellular and cholangiocarcinoma: a clinicopathologic study of 26 resected cases. Jpn J Clin Oncol. 2003;33(6):283–287. | ||
Bhagat V, Javle M, Yu J, et al. Combined hepatocholangiocarcinoma: case-series and review of literature. Int J Gastrointest Cancer. 2006;37(1):27–34. | ||
Tickoo SK, Zee SY, Obiekwe S, et al. Combined hepatocellular-cholangiocarcinoma: a histopathologic, immunohistochemical, and in situ hybridization study. Am J Surg Pathol. 2002;26(8):989–997. | ||
Yeh MM. Pathology of combined hepatocellular-cholangiocarcinoma. J Gastroenterol Hepatol. 2010;25(9):1485–1492. | ||
Theise ND, Yao JL, Harada K, et al. Hepatic “stem cell” malignancies in adults: four cases. Histopathology. 2003;43(3):263–271. | ||
Wu PC, Fang JW, Lau VK, Lai CL, Lo CK, Lau JY. Classification of hepatocellular carcinoma according to hepatocellular and biliary differentiation markers. Clinical and biological implications. Am J Pathol. 1996;149(4):1167–1175. | ||
Moeini A, Sia D, Zhang Z, et al. Mixed hepatocellular cholangiocarcinoma tumors: cholangiolocellular carcinoma is a distinct molecular entity. J Hepatol. 2017;66(5):952–961. | ||
Fujii H, Zhu XG, Matsumoto T, et al. Genetic classification of combined hepatocellular-cholangiocarcinoma. Hum Pathol. 2000;31(9):1011–1017. | ||
Yano H, Iemura A, Haramaki M, et al. A human combined hepatocellular and cholangiocarcinoma cell line (KMCH-2) that shows the features of hepatocellular carcinoma or cholangiocarcinoma under different growth conditions. J Hepatol. 1996;24(4):413–422. | ||
Maeda T, Adachi E, Kajiyama K, Sugimachi K, Tsuneyoshi M. Combined hepatocellular and cholangiocarcinoma: proposed criteria according to cytokeratin expression and analysis of clinicopathologic features. Hum Pathol. 1995;26(9):956–964. | ||
O’Connor K, Walsh JC, Schaeffer DF. Combined hepatocellular-cholangiocarcinoma (cHCC-CC): a distinct entity. Ann Hepatol. 2014;13(3):317–322. | ||
Gera S, Ettel M, Acosta-Gonzalez G, Xu R. Clinical features, histology, and histogenesis of combined hepatocellular-cholangiocarcinoma. World J Hepatol. 2017;9(6):300–309. | ||
Choi BI, Han JK, Kim YI, et al. Combined hepatocellular and cholangiocarcinoma of the liver: sonography, CT, angiography, and iodized-oil CT with pathologic correlation. Abdom Imaging. 1994;19(1):43–46. | ||
Maximin S, Ganeshan DM, Shanbhogue AK, et al. Current update on combined hepatocellular-cholangiocarcinoma. Eur J Radiol Open. 2014;1:40–48. | ||
Sammon J, Fischer S, Menezes R, et al. MRI features of combined hepatocellular-cholangiocarcinoma versus mass forming intrahepatic cholangiocarcinoma. Cancer Imaging. 2018;18(1):8. | ||
Fowler KJ, Sheybani A, Parker RA 3rd, et al. Combined hepatocellular and cholangiocarcinoma (biphenotypic) tumors: imaging features and diagnostic accuracy of contrast-enhanced CT and MRI. Am J Roentgenol. 2013;201(2):332–339. | ||
Wells ML, Venkatesh SK, Chandan VS, et al. Biphenotypic hepatic tumors: imaging findings and review of literature. Abdom Imaging. 2015;40(7):2293–2305. | ||
Park SH, Lee SS, Yu E, et al. Combined hepatocellular-cholangiocarcinoma: gadoxetic acid-enhanced MRI findings correlated with pathologic features and prognosis. J Magn Reson Imaging. 2017;46(1):267–280. | ||
Sanada Y, Shiozaki S, Aoki H, Takakura N, Yoshida K, Yamaguchi Y. A clinical study of 11 cases of combined hepatocellular–cholangiocarcinoma: assessment of enhancement patterns on dynamics computed tomography before resection. Hepatol Res. 2005;32(3):185–195. | ||
Jeon SK, Joo I, Lee DH, et al. Combined hepatocellular cholangiocarcinoma: LI-RADS v2017 categorisation for differential diagnosis and prognostication on gadoxetic acid-enhanced MR imaging. Eur Radiol. Epub 2018 Jun 28. | ||
Potretzke TA, Tan BR, Doyle MB, Brunt EM, Heiken JP, Fowler KJ. Imaging features of biphenotypic primary liver carcinoma (hepatocholangiocarcinoma) and the potential to mimic hepatocellular carcinoma: LI-RADS analysis of CT and MRI features in 61 cases. Am J Roentgenol. 2016;207(1):25–31. | ||
He C, Mao Y, Wang J, et al. The Predictive value of staging systems and inflammation scores for patients with combined hepatocellular cholangiocarcinoma after surgical resection: a retrospective study. J Gastrointest Surg. 2018;22(7):1239–1250. | ||
You HL, Weng SW, Li SH, et al. Copy number aberrations in combined hepatocellular carcinoma and cholangiocarcinoma. Exp Mol Pathol. 2012;92(3):281–286. | ||
Liu ZH, Lian BF, Dong QZ, et al. Whole-exome mutational and transcriptional landscapes of combined hepatocellular cholangiocarcinoma and intrahepatic cholangiocarcinoma reveal molecular diversity. Biochim Biophys Acta. 1864;2018((6 Pt B)):2360–2368. | ||
Xue TC, Zhang BH, Ye SL, Ren ZG. Differentially expressed gene profiles of intrahepatic cholangiocarcinoma, hepatocellular carcinoma, and combined hepatocellular-cholangiocarcinoma by integrated microarray analysis. Tumor Biol. 2015;36(8):5891–5899. | ||
Sasaki M, Sato Y, Nakanuma Y. Mutational landscape of combined hepatocellular carcinoma and cholangiocarcinoma, and its clinicopathological significance. Histopathology. 2017;70(3):423–434. | ||
The Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327–1341.e23. | ||
Woo HG, Lee JH, Yoon JH, et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res. 2010;70(8):3034–3041. | ||
Coulouarn C, Cavard C, Rubbia-Brandt L, et al. Combined hepatocellular-cholangiocarcinomas exhibit progenitor features and activation of Wnt and TGFbeta signaling pathways. Carcinogenesis. 2012;33(9):1791–1796. | ||
Decaens T, Godard C, de Reyniès A, et al. Stabilization of beta-catenin affects mouse embryonic liver growth and hepatoblast fate. Hepatology. 2008;47(1):247–258. | ||
Tan X, Yuan Y, Zeng G, et al. Beta-catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47(5):1667–1679. | ||
Kim KH, Lee SG, Park EH, et al. Surgical treatments and prognoses of patients with combined hepatocellular carcinoma and cholangiocarcinoma. Ann Surg Oncol. 2009;16(3):623–629. | ||
Nakamura S, Suzuki S, Sakaguchi T, et al. Surgical treatment of patients with mixed hepatocellular carcinoma and cholangiocarcinoma. Cancer. 1996;78(8):1671–1676. | ||
Sasaki A, Kawano K, Aramaki M, et al. Clinicopathologic study of mixed hepatocellular and cholangiocellular carcinoma: modes of spreading and choice of surgical treatment by reference to macroscopic type. J Surg Oncol. 2001;76(1):37–46. | ||
Ercolani G, Grazi GL, Ravaioli M, et al. The role of lymphadenectomy for liver tumors: further considerations on the appropriateness of treatment strategy. Ann Surg. 2004;239(2):202–209. | ||
Bismuth H, Majno PE. Hepatobiliary surgery. J Hepatol. 2000;32(1 Suppl):208–224. | ||
Tao CY, Liu WR, Jin L, et al. Surgical treatment of combined hepatocellular-cholangiocarcinoma is as effective in elderly patients as it is in younger patients: a propensity score matching analysis. J Cancer. 2018;9(6):1106–1112. | ||
Northup PG, Wanamaker RC, Lee VD, Adams RB, Berg CL. Model for End-Stage Liver Disease (MELD) predicts nontransplant surgical mortality in patients with cirrhosis. Ann Surg. 2005;242(2):244–251. | ||
Malinchoc M, Kamath PS, Gordon FD, Peine CJ, Rank J, Ter Borg PC. A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology. 2000;31(4):864–871. | ||
Groeschl RT, Turaga KK, Gamblin TC. Transplantation versus resection for patients with combined hepatocellular carcinoma-cholangiocarcinoma. J Surg Oncol. 2013;107(6):608–612. | ||
Vilchez V, Shah MB, Daily MF, et al. Long-term outcome of patients undergoing liver transplantation for mixed hepatocellular carcinoma and cholangiocarcinoma: an analysis of the UNOS database. HPB. 2016;18(1):29–34. | ||
Park YH, Hwang S, Ahn CS, et al. Long-term outcome of liver transplantation for combined hepatocellular carcinoma and cholangiocarcinoma. Transpl Proc. 2013;45(8):3038–3040. | ||
Song S, Moon HH, Lee S, et al. Comparison between resection and transplantation in combined hepatocellular and cholangiocarcinoma. Transpl Proc. 2013;45(8):3041–3046. | ||
Bergquist JR, Groeschl RT, Ivanics T, et al. Mixed hepatocellular and cholangiocarcinoma: a rare tumor with a mix of parent phenotypic characteristics. HPB. 2016;18(11):886–892. | ||
Maganty K, Levi D, Moon J, et al. Combined hepatocellular carcinoma and intrahepatic cholangiocarcinoma: outcome after liver transplantation. Dig Dis Sci. 2010;55(12):3597–3601. | ||
Wu CH, Yong CC, Liew EH, et al. Combined hepatocellular carcinoma and cholangiocarcinoma: diagnosis and prognosis after resection or transplantation. Transpl Proc. 2016;48(4):1100–1104. | ||
Magistri P, Tarantino G, Serra V, Guidetti C, Ballarin R, di Benedetto F. Liver transplantation and combined hepatocellular-cholangiocarcinoma: feasibility and outcomes. Dig Liver Dis. 2017;49(5):467–470. | ||
Fowler K, Saad NE, Brunt E, et al. Biphenotypic primary liver carcinomas: assessing outcomes of hepatic directed therapy. Ann Surg Oncol. 2015;22(13):4130–4137. | ||
de Vito C, Sarker D, Ross P, Heaton N, Quaglia A. Histological heterogeneity in primary and metastatic classic combined hepatocellular-cholangiocarcinoma: a case series. Virchows Archiv. 2017;471(5):619–629. | ||
Na SK, Choi GH, Lee HC, et al. The effectiveness of transarterial chemoembolization in recurrent hepatocellular-cholangiocarcinoma after resection. PLoS One. 2018;13(6):e0198138. | ||
Kobayashi S, Terashima T, Shiba S, et al. Multicenter retrospective analysis of systemic chemotherapy for unresectable combined hepatocellular and cholangiocarcinoma. Cancer Sci. 2018;109(8):2549–2557. | ||
Rogers JE, Bolonesi RM, Rashid A, et al. Systemic therapy for unresectable, mixed hepatocellular-cholangiocarcinoma: treatment of a rare malignancy. J Gastrointest Oncol. 2017;8(2):347–351. | ||
Chi M, Mikhitarian K, Shi C, Goff LW. Management of combined hepatocellular-cholangiocarcinoma: a case report and literature review. Gastrointest Cancer Res. 2012;5(6):199–202. | ||
Kim GM, Jeung H-C, Kim D, et al. A case of combined hepatocellular-cholangiocarcinoma with favorable response to systemic chemotherapy. Cancer Res Treat. 2010;42(4):235–238. | ||
Broutier L, Mastrogiovanni G, Verstegen MM, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23(12):1424–1435. | ||
Sprinzl MF, Galle PR. Current progress in immunotherapy of hepatocellular carcinoma. J Hepatol. 2017;66(3):482–484. | ||
Duffy AG, Ulahannan SV, Makorova-Rusher O, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol. 2017;66(3):545–551. | ||
Liu CL, Fan ST, Lo CM, et al. Hepatic resection for combined hepatocellular and cholangiocarcinoma. Arch Surg. 2003;138(1):86–90. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.