")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Combination of Metformin and Exercise in Management of Metabolic Abnormalities Observed in Type 2 Diabetes Mellitus
Authors Jevtovic F
Received 13 July 2021
Accepted for publication 7 September 2021
Published 16 September 2021 Volume 2021:14 Pages 4043—4057
DOI https://doi.org/10.2147/DMSO.S328694
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Claes-Göran Ostenson
Filip Jevtovic
Department of Kinesiology, College of Health and Human Performance, East Carolina University, Greenville, NC, USA
Correspondence: Filip Jevtovic
East Carolina University; School of Dental Medicine, Ledyard E. Ross Hall; 1851 MacGregor Downs Road, Mail Stop 701, Greenville, NC, 27834, USA
Tel +1 616 844 8323
Fax +1 252 737 7024
Email [email protected]
Abstract: Excess nutrient intake and lack of exercise characterize the problem of obesity and are common factors in insulin resistance (IR). With an increasing number of prediabetic, and type 2 diabetic populations, metformin is still the most prescribed glucose-lowering drug and is often accompanied by recommendations for regular physical exercise. Metformin, by the inhibition of complex 1 of the electron transport chain, and exercise, by increasing energy expenditure, both elicit a low cellular energy state that leads to improvements in glucose control via activation of adenosine 5ʹ monophosphate-activated protein kinase (AMPK). An augmented stimulation of the energy-sensing enzyme AMPK by either of the two modalities leads to an increase in glycogenolysis, glucose uptake, fat oxidation, a decrease in glycogen and protein synthesis, and gluconeogenesis in muscle and the liver, which are remarked as having positive effects on metabolic pathophysiology observed in IR and type 2 diabetes mellitus (T2DM). While both modalities exploit the energy-sensing enzyme AMPK to attain glucose homeostasis, the synergistic effect of these two treatments is not distinctly supported by the literature. Further, an antagonistic dynamic has been observed in cases where metformin and exercise were combined. Reduction of insulin-sensitizing effects of exercise and an overall hindrance of exercise performance and adaptations have been reported and could suggest the possible incongruity of these two modalities. The aim of this review is to elucidate the effect that metformin and exercise have on the management of the metabolic abnormalities observed in T2DM and to provide an insight into the interaction of these two modalities.
Keywords: exercise, metformin, glucose control, type 2 diabetes mellitus, AMPK
Graphical Abstract:

Introduction
Type 2 Diabetes Risk Factors, Prevalence, and Risk of Other Diseases
Energy imbalance in calories consumed and calories expended, an increased intake of energy-dense food, and a sedentary lifestyle play a major role in the development of obesity. In 2016, according to the World Health Organization (WHO),1 obesity (abnormal or excessive fat accumulation that may impair health) defined by body mass index equal to or greater than 30, was seen in 13% of the world’s adult population, while 39% of adults aged 18 years and older were classified as overweight. The early onset and the prevalence of obesity in youth and young adults are likely to translate into a high cumulative incidence of type 2 diabetes mellitus (T2DM) and is supported by the incidence reports from around the world.2 According to the Center for Disease Control and Prevention3 national diabetes report for 2018, about 10.5% or 34.2 million of the US population suffers from diabetes, while about 34.5% of the population categorizes as prediabetic based on their fasting glucose or glycated hemoglobin levels. Further, out of the 34.2 million diagnosed with diabetes, T2DM accounts for approximately 90% to 95% of all diabetes cases in the U.S.3
People suffering from T2DM have a higher prevalence rate of cardiovascular diseases (CVD) when compared to normal, non-diabetic subjects.4 Kivimäki and colleagues5 observed that asymptomatic, and undetected changes of metabolism in obesity, precede T2DM development and increase the risk of cardiometabolic multimorbidity proportionally to the increase in body mass index. Additionally, the risk of CVD is directly proportionate to the rise in plasma glucose levels, even in the prediabetic state, where glucose levels are not sufficient for a diabetes diagnosis.6 Einarson and colleagues7 concluded that approximately 32.2% of all persons suffering from T2DM are affected by CVD, with coronary artery disease and stroke accounting for the majority of incidence. Aside from the morbidity and mortality of CVD, patients suffering from T2DM have an increased incidence of cancer,8 neuropathy,9 nephropathy,10,11 and overall increased risk of all-cause mortality.12,13 Decreasing the incidence of obesity and regulation of blood glucose ought to be the main focus for the management of T2DM, and the prevention of the development of T2DM from the prediabetic state, or obesity alone.
Treatment modalities in T2DM involve lifestyle modification, often in synchrony with oral hypoglycemic, insulin-sensitizing, and other medication that reduces insulin resistance (IR). While exercise and metformin are the most prescribed modalities to manage T2DM due to the similar effects on glucose control; in recent years some studies showed the potential negation of the positive effects that these modalities can have if implemented together.14–27 Because the research in this area is scarce and results are inconsistent, a comprehensive review of the studies is needed to further elucidate the impact of the combination of exercise and metformin on the management of metabolic pathophysiology of T2DM. Additionally, discussion about the effects of exercise and metformin cannot only focus on the management of T2DM but must be extended further to describe the interactions of two modalities.
Type 2 Diabetes Pathophysiology
To better understand T2DM, and the role of IR in the development of diabetes and other cardiometabolic morbidities, it is important to understand the pathophysiology and the chronic changes that T2DM elicits. Understanding the effect of IR on the metabolism of an obese, prediabetic, or diabetic patient, allows for a better understanding and differentiation of the cause(s) of T2DM, along with symptoms that develop as a consequence of such a metabolic abnormality. Fundamentally, T2DM is a metabolic disorder characterized by persistent hyperglycemia that originates from the lower glucose disposal and an increased rate of glucose appearance.12,28,29 Greater rate of glucose appearance stems from the inability of insulin to place a restraint on gluconeogenesis and glycogenolysis in the liver.28 Alternatively, IR and impaired function of insulin in peripheral tissue result in a lower glucose clearance rate.28,29 IR, often defined as a decrease in cell sensitivity to insulin, leads to a compensatory increase in insulin secretion (hyperinsulinemia) to ensure euglycemia.12 With the progression of IR, the failure of the pancreatic islet beta cells to release enough insulin leads to a chronic increase in blood glucose concentration, hyperglycemia, and a diagnosis of T2DM.30 While beta-cell dysfunction and IR affect each other and, in synergy, lead to the development of metabolic abnormalities observed in T2DM, it seems that the development of IR promotes beta-cell exhaustion, leading to beta-cell demise, a decreased beta-cell number, increase in their mass, and cell dedifferentiation.30 This suggests that the progression and treatment of T2DM and IR are dependent on glucose regulation and increased cell insulin sensitivity.
With the progression of the pathophysiology of T2DM, specifically with decreased insulin sensitivity, multiple symptoms such as hormone alterations, mitochondrial dysfunction, and altered fat oxidation arise.29 Decreased insulin-mediated suppression of hormone-sensitive lipase in IR leads to increased hydrolysis of triglycerides, and elevated blood free fatty acids (FFA) levels promoting dyslipidemia, and CVD development.5,29,31,32 This increase in FFA circulation is not only a concern when it comes to the proliferation of CVD disease, but it seems to have a significant effect on insulin sensitivity. An increase in blood FFA was shown to impair glucose uptake/transport33 in hepatic and peripheral tissue,34 as well as skeletal muscle,35 further contributing to hyperglycemia and proliferation of IR. This cycle of decreased insulin suppression of hormone-sensitive lipase, and in return, increased FFA blood concentration, leads to lipid overload. It has been shown that lipid overload of skeletal muscle mitochondria, leads to further mitochondrial dysfunction and a decrease in insulin sensitivity.36 By the inhibition of carnitine palmitoyltransferase-1 (CPT-1), and the decrease in mitochondrial fatty acid oxidation, Wicks et al37 observed an increase in peroxisomal fatty acid oxidation and an increase in mitochondrial biogenesis. This led to improved glucose tolerance, suggesting that a decrease in the concentration of FFA can have a therapeutic effect. With the progression of IR and further damaging of pancreatic beta cells, prediabetic and patients with T2DM suffer from an array of confounding metabolic abnormalities including inflammation, dyslipidemia, hormone alterations, and mitochondrial dysfunction that collectively increase the risk of all-cause mortality. While the metabolic abnormalities observed in T2DM are often highly correlated to a greater risk for the development and the furthering of the other diseases mentioned above, the focus of the treatment of T2DM must stay in the underlying causal factors, specifically the improvement of glycemic control.
With skeletal muscle being responsible for most of the insulin-stimulated glucose uptake, it seems that skeletal muscle IR is a key point for the development of T2DM. Excess nutrient intake and excessive exposure to glucose, FFA, or amino acids proliferate the development of IR.28,36 Further, Saha et al38 showed that an excess of glucose and branched-chain amino acid leucine, while stimulated protein synthesis, induced skeletal muscle IR while simultaneously decreasing the adenosine 5ʹ monophosphate-activated protein kinase (AMPK) activity. Reduced AMPK activity leads to a subsequent decrease in glucose uptake and increases in FFA synthesis fostering the development of IR.24,38 Considering that glucose uptake and increased fatty acid synthesis are observed as contributing factors of IR and subsequently T2DM, regulation of the AMPK activity seems to be a target of interest when developing modalities used to mediate and treat T2DM.
AMPK
AMPK is an energy-sensing enzyme that plays a key role in nutrient regulation and insulin sensitivity.41 As a result of the multifaceted effect that this kinase has on metabolism (Figure 1), it has been actively studied in the context of metabolic disorders including IR. When activated in skeletal muscle, AMPK through downstream targets augments glucose uptake (via increased glucose transporter type (GLUT) 4 translocation), mitochondrial oxidation of long-chain fatty acids (via phosphorylation of acetyl-CoA carboxylase), inhibition of protein synthesis through mammalian target of rapamycin complex 1 (mTORC1), and promotion of mitochondrial biogenesis through activation of peroxisome proliferator-activated receptor γ co-activator-1α (PGC-1α).28,39–44 In addition to skeletal muscle, of the specific interest are the effects of AMPK on adipocytes and hepatocytes considering their role in the pathophysiology of T2DM. Activation of AMPK in the liver leads to an increase in glucose uptake and fatty acid oxidation and inhibits gluconeogenesis, fatty acid, and cholesterol synthesis.28,40 Additionally, activated AMPK increases fatty acid oxidation and attenuates fatty acid synthesis and lipolysis in adipose tissue.28,40 A key factor in the regulation of glucose control stems from the ability of AMPK to increase glucose uptake through insulin-independent pathways.45 Considering the multitude of tissue that AMPK influences and the specific course of action, independent of the faulty insulin pathway observed in IR, AMPK is an important regulator of metabolism and a possible target for the treatment of T2DM.
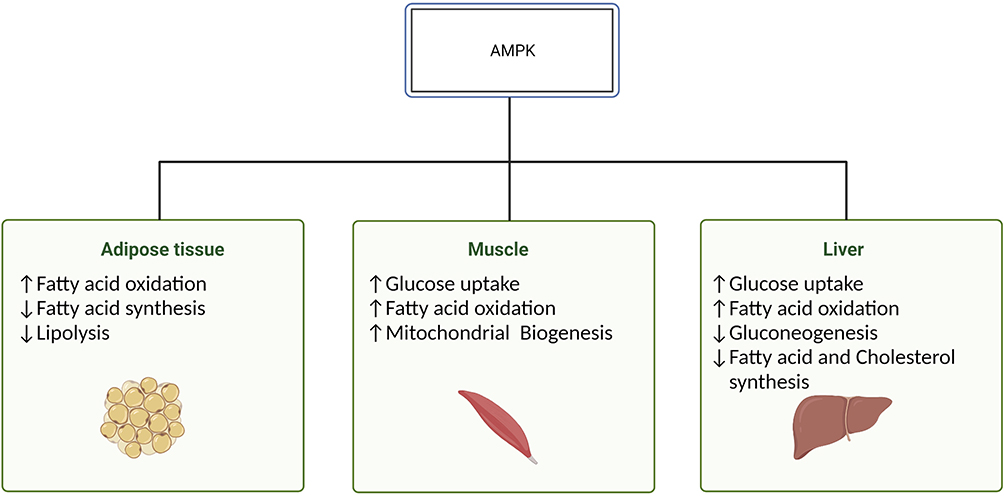 |
Figure 1 Tissue-specific effects of AMPK. Abbreviation: AMPK, adenosine monophosphate-activated protein kinase. Notes: Activation of AMPK promotes energy-producing pathways while terminating the processes that require energy input. In skeletal muscle, AMPK leads to an increase in glucose and fatty acid oxidation, while chronic activation promotes mitochondrial biogenesis. In addition to AMPK-induced increase in glucose uptake and fatty acid oxidation, a decrease in gluconeogenesis and fatty acid and cholesterol synthesis is observed in hepatocytes. Finally, AMPK leads to an increase in fatty acid oxidation and attenuation of fatty acid synthesis and lipolysis. Created with BioRender.com. |
When considering the pathophysiology of T2DM multiple factors need to be recognized to distinguish the state of disease. Additionally, to understand the effects that a modality, which in the case of this review is exercise or metformin or a combination of the two, has on T2DM, metabolic parameters such as glucose control, insulin sensitivity, AMPK activity, as well as changes in body composition and exercise capacity need to be acknowledged.
Exercise
It is widely understood that exercise induces metabolic changes that have a positive effect on T2DM (Figure 2). With exercise, an augmented uptake and oxidation of glucose and fatty acids by active tissue seems to have a favorable effect on IR.41,45–48 Fatty acid and glucose uptake are facilitated predominantly by increased translocation of transmembrane proteins FAT/CD36 and GLUT4 in skeletal muscle.41 It has been observed that during exercise, regulation of fat and glucose uptake via previously mentioned transporters, is through an insulin-independent pathway, following the activation of AMPK.40,41,45,46 Additionally, chronic exercise leads to a long-term upregulation of muscle mitochondrial content and enzymes, as well as the ability for greater fatty acid and glucose uptake.41 While both insulin and exercise increase glucose uptake, according to Hayashi and colleagues,45 the mechanism by which insulin and exercise increase GLUT4 translocation to the surface of the cell differs. Further, Krook and colleagues47 demonstrated an impaired insulin signaling transduction in the skeletal muscle of the T2DM patient at the level of IRS-1, subsequently depressing PI 3-kinase activity and overall glucose transport. While the impairment in insulin-stimulated glucose uptake was observed in T2DM and overweight patients, exercise-induced glucose uptake was normal and unaffected by T2DM,48 which is why exercise is a highly recommended treatment agent in T2DM. The favorable effect of exercise on glucose regulation is attributed mostly to the activation of AMPK via the increase in AMP-to-ATP and ADP-to-ATP ratios.49,50 An increase in AMPK during exercise was demonstrated to lead to a significant increase in glucose and fatty acid uptake during and following exercise.46 In addition to acute changes in glucose and fatty acid uptake, chronic endurance exercise leads to an improved insulin sensitivity associated with the greater basal activity of AMPK.41,51–53 While mechanisms of AMPK activation and effects on metabolism are not completely demonstrated in T2DM, benefits of AMPK activation in T2DM can be associated with an augmented substrate (ie, glucose and fatty acid) uptake and oxidation in active tissue, subsequently promoting glucose homeostasis. Higher activation of AMPK leads to increased insulin-stimulated muscle glucose uptake and insulin sensitivity at rest following the acute bout of exercise.42 Additionally, insulin sensitivity and glucose uptake seem to be improved due to the increase in GLUT4 content. Following exercise, there is an increase in skeletal muscle GLUT4 transcription via PGC-1α-dependent pathway, and this notion has been demonstrated in cultured myotubes and animal models.54,55 This greater ability for glucose transport, due to the upregulation of GLUT4 content and transcription, allows for augmented insulin-stimulated glucose clearance at rest. By enhancing insulin sensitivity and improving glycemic control, exercise positively affects IR and is an indispensable asset for the management of T2DM. While exercise leads to an array of positive changes, improvements in glycemic control can be largely attributed to an augmented AMPK activation.
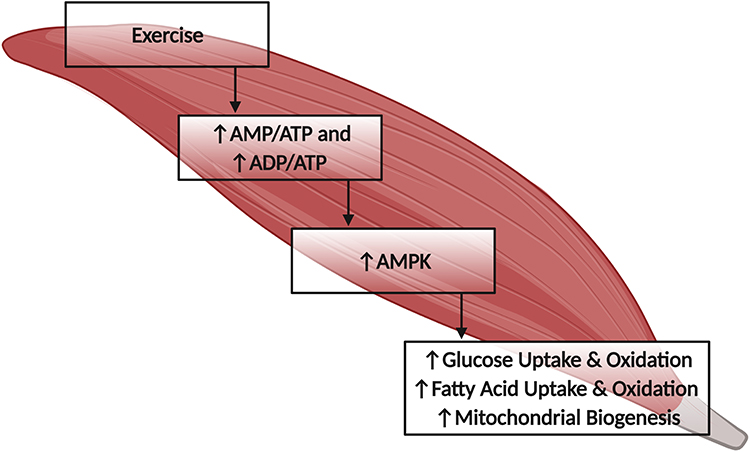 |
Figure 2 Exercise-induced activation of AMPK, and downstream regulation of skeletal muscle metabolism. Abbreviations: AMP, adenosine monophosphate; ADP, adenosine diphosphate; ATP, adenosine triphosphate; AMPK, adenosine monophosphate-activated protein kinase. Notes: Exercise-induced low-energy state leads to an upregulation of AMPK and subsequent increase in glucose and fatty acid uptake and oxidation. Additionally, chronic exercise leads to increase in mitochondrial biogenesis and mitochondrial enzymatic content. Created with BioRender.com. |
In addition to increased glucose uptake, the activation of AMPK leads to increased long-chain fatty acid uptake and oxidation during and following an acute bout of exercise; while increased mitochondrial biogenesis and enzymatic content were observed after following an exercise program.41,43,46 AMPK increases the translocation of FAT/CD36 which as a regulator of fatty acid influx into the cell allows for a greater fatty acid oxidation.40,41,43,46,56 Such changes in mitochondrial content and increased fatty acid oxidation have the potential to attenuate IR resulting from the accumulation of FFA intermediates (lipotoxicity) and improve symptoms of T2DM. Additionally, by increasing the mitochondrial content, chronic training leads to improved exercise capacity that is inversely correlated with IR, making exercise a valuable treatment for IR and T2DM.
Metformin
Metformin belongs to the biguanide family of drugs with insulin-sensitizing properties. While mechanisms of its action are not fully understood, metformin has been and still is a preferred national and international glucose-lowering and insulin-sensitizing pharmacological agent for the treatment of T2DM.57 It has been shown that T2DM patients have increased hepatic glucose output at baseline that is decreased by approximately 75% after taking metformin.58 The primary effects of metformin lie in the decreased hepatic glucose production, while in the tracer studies, it has been observed that there is a minimal impact of this drug on peripheral insulin-mediated glucose uptake.59 An AMPK associated action of metformin is associated with the ability of metformin to decrease the ATP production in mitochondria.60 According to Owen and colleagues,60 and El-Mir et al,61 metformin readily diffuses into the mitochondria to increase its concentration by 1000-fold when compared to the extracellular environment and leads to inhibition of complex 1 of the respiratory chain. Such effects result in suppressed ATP production, and as consequence increased AMP-to-ATP and ADP-to-ATP ratios which are primary signals for the activation of AMPK.49,50 By activation of AMPK as a metabolic regulator there is an increase in nutrient breakdown and decreased hepatic gluconeogenesis, resulting in ameliorated glucose disposal. In addition to increased AMPK activity, a metformin mediated increase in relative levels of AMP was observed to inhibit fructose 1, 6 bisphosphatase and reduce cyclic AMP (cAMP) leading to reduced expression of hepatic gluconeogenic enzymes,62 lowering the endogenous glucose production. While the latter two effects are mediated via an AMPK independent pathway, it has been proposed that the activation of cAMP could be mediated directly by AMPK phosphorylation of cAMP following the metformin administration, as well.63 In addition to a decrease in endogenous glucose production via AMPK dependent and independent pathways, metformin was recently observed to reduce gastrointestinal glucose uptake and increase glucagon-like peptide (GLP-1) secretion.59 Similarly, GLP-1, when secreted promotes satiety, stimulates insulin secretion, inhibits gastric emptying in response to food consumption, and was reported to increase with moderate and high-intensity exercise in both overweight and obese subjects, as well.64 This effect suggests a protective role of both metformin and exercise against GLP-1 resistance associated with T2DM pathophysiology.65
Although seemingly different methods, metformin, and exercise elicit a similar physiological effect via the same energy-sensing enzyme. In both cases, AMPK and changes in energy availability (ie, AMP-to-ATP ratio) can be seen as factors leading to a decrease in endogenous glucose production, an increase in glucose uptake, and overall a decrease in blood glucose concentration. The main difference between metformin and exercise is in the method of increasing the AMP-to-ATP ratio, subsequently influencing the AMPK. In the case of metformin, inhibition of complex 1 of the electron transport chain simulates a low-energy state without an existent increase in energy demand, while in the case of exercise, demand for a greater energy output to accommodate an increasing workload, drives AMPK activation. In addition to AMPK regulated changes, an increase in GLP-1, and decreased net glucose uptake in GI, which is observed in acute exercise and metformin, seem to favor the cooperative action of these two modalities. With such common factors, it is reasonable to assume that these two modalities will have a synergistic effect when taken together. However, although this notion seems plausible, research on this topic is still unclear, with inconclusive and mixed results.14–27
Exercise and Metformin Interaction
Insulin-stimulated glucose uptake can be increased for 3–72h following a single exercise bout.66,67 Mechanisms by which exercise increases insulin-stimulated glucose disposal are often associated with increased muscle blood flow, decreased muscle glycogen, and an increase in enzymes responsible for nonoxidative glucose disposal.68–70 In addition to those, in recent years, an increase in AMPK has been implicated as an important mediator of postexercise insulin sensitivity.52,53,71 Similarly, as previously described, metformin shares AMPK as a common target for glucose regulation, but the combination of these two modalities seems to be unclear and potentially conflicting (Table 1).14–27
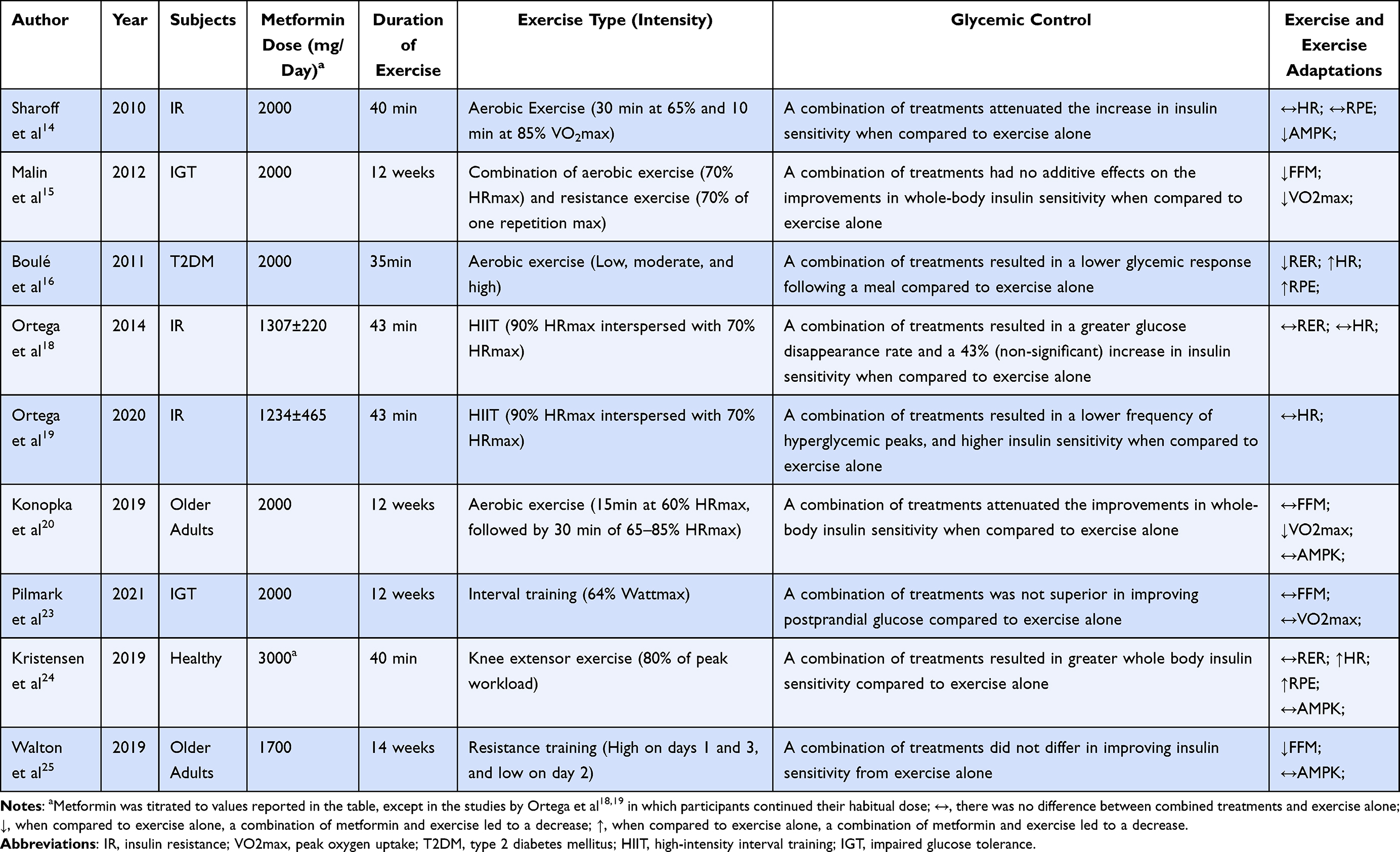 |
Table 1 Characteristics of the Included Studies. Comparison Between the Effects of Combined Treatment and Exercise-Only on Glycemic Control, Exercise, and Exercise Adaptations |
Insulin Sensitivity and Glucose Control
Sharoff et al14 found that insulin-resistant subjects who performed 30 minutes of exercise at 65% peak oxygen uptake (VO2max) followed by 10 minutes of exercise at 85% VO2max had a 52% increase in insulin sensitivity. On the contrary, participants who have been taking 2000 mg/day of metformin for 2 weeks before the exercise bout had no insulin sensitivity change, suggesting that metformin abolished the insulin-sensitizing effects of exercise. In addition to decreased insulin sensitivity, taking metformin resulted in a ~10-fold decrease in muscle AMPK activity when compared to the exercise alone. A decrease in AMPK activity that has been observed by Sharoff and colleagues14 does not match the finding of Kristensen et al24 who observed no significant difference in muscle AMPK activity following exercise with or without metformin. The potential discrepancy between the studies might be due to the difference in duration of metformin treatment and characteristics of participants and exercise. Although Kristensen et al24 reported that acute and short-term metformin “loading” resulted in high plasma and muscle metformin concentrations, prolonged metformin treatment in the study by Sharoff and colleagues14 could have resulted in a superior metformin availability.72 Additionally, intersubject variation in metformin absorption rates could affect metformin availability following an acute metformin administration, resulting in a lower AMPK activation.72 Between the studies, there was a large discrepancy in the participants’ aerobic capacity and metabolic status, as well. Healthy, moderately trained men in the study by Kristensen et al24 had considerably (~50%) higher VO2max when compared to the insulin-resistant subjects (VO2max, 26.8±2.6 mL·kg−1·min−1) in the study by Sharoff et al.14 Further, more dynamic exercise and greater muscle recruitment during cycling at moderate and high intensities could have elicited a higher energetic stress and molecular signaling response when compared to a single-leg knee extensor exercise. This exercise discrepancy is further accentuated when the inconsistencies in aerobic capacity and metabolic state are taken into the account. With higher fitness levels and presumably lower exercise demand, participants in the ladder study potentially had a lesser relative stimulus that would result in the lower AMPK activation. Finally, while not significant, there was a discrepancy in baseline AMPK activity between groups in the study by Sharoff et al14 with higher baseline AMPK observed in the metformin compared to the placebo group. The discrepancy between the baseline AMPK activity could inflate the difference between the AMPK activity within the exercise-only group influencing the magnitude of the effect that the author observed when two groups were compared.
With chronic metformin therapy, Walton et al25 reported a non-significant 21.3% increase in basal phosphorylated AMPK to total AMPK ratio. On the contrary, Konopka et al20 showed a trend but not significant decrease in basal AMPK following 12 weeks of aerobic exercise training, in metformin naïve older adults at risk of T2DM. Both authors failed to observe the expected increase in AMPK following the hindrance of amplified mitochondrial respiration during exercise, suggesting that chronic metformin treatment does not significantly alter the effects of exercise on AMPK levels, regardless of the exercise mode. Although there was no difference in baseline AMPK, Konopka and colleagues20 reported that the addition of metformin attenuated the improvements in skeletal muscle mitochondrial respiration. Considering the importance of mitochondrial capacity in IR, this effect of metformin should be taken into consideration when prescribing the combined treatment. Finally, Konopka et al20 reported an attenuated improvement in whole-body insulin sensitivity when metformin was added to aerobic exercise training. On the contrary, Walton et al25 reported that 12 weeks of resistance exercise combined with metformin did not differ from the exercise alone when it comes to the improvements in insulin sensitivity. A discrepancy in the findings, as suggested by Walton et al25 might be due to different mechanisms that resistance and aerobic exercise use to improve insulin sensitivity. Further, considering these findings, specific effects of metformin may have a greater negative impact on aerobic exercise, when compared to resistance or combination training. In the study by Malin and colleagues,15 improvement in insulin sensitivity following 12 weeks of combined aerobic and resistance exercise, was not significantly different regardless of metformin treatment. Metformin naïve subjects with impaired glucose tolerance were assigned to placebo, metformin, exercise and metformin, and exercise with placebo. Following the 12 weeks of 60–70min of combined moderate-intensity aerobic and resistance program three times a week and/or 2000 mg of metformin daily, Malin and colleagues15 observed no additional benefits of metformin added to exercise. Finally, the combination of metformin and exercise attenuated the improvements in peak aerobic capacity that was positively correlated with the improvements in insulin sensitivity, extending the unfavorable effect of metformin to other metabolic processes. A similar effect of metformin on peak aerobic capacity was reported in the study by Boule et al26 following 22 weeks of combined aerobic and resistance training. Regardless of metformin, a combination of aerobic and resistance training was effective in reducing glycated hemoglobin. Further, a combination of metformin and combined training led to improvements in fasting blood glucose, but this effect was not the case when metformin was withheld. It is important to note that in the studies that incorporated the resistance training, the addition of metformin did not attenuate the improvements in glycemic control as opposed to the aerobic training only in the study by Konopka et al.20 Further, participants in the study by Konopka et al20 and Malin et al15 had a decrease in peak aerobic capacity following 12 weeks of combined treatment; however, only participants who performed aerobic training in combination with metformin had an attenuated improvement in insulin sensitivity when compared to exercise when metformin was withheld. Walton et al25 observed that metformin inhibited the decrease in type 1 fiber frequency following a resistance program. Such an effect could preserve the improvements in insulin sensitivity considering the importance of type 1 fibers in glucose control.73 By preserving type 1 fiber content, it is possible that detriments in peripheral insulin sensitivity correlated to a lower mitochondrial capacity are mitigated. Collectively, the aforementioned studies suggest that detrimental effects of combined treatment might not be restricted to the lack of improvements in AMPK, and might elicit such effects via other metabolic adaptations.
In the study by Pilmark et al23 glucose intolerant, metformin naïve subjects were given either metformin or placebo for 3 weeks after which they engaged in the 12 weeks of aerobic interval training in combination with metformin or placebo. A mixed meal tolerance test was performed at baseline, after medication or placebo only, and following the combination of treatments. Pilmark and colleagues23 reported no additional benefits when metformin was combined with exercise for 12 weeks. Further, the time and treatment-specific effects of metformin and exercise on the postprandial glucose were observed. The author found that in the metformin-treated group, the entire improvement in postprandial glucose occurred as a result of medication, with no additional improvement following exercise training. Inversely, in the exercise-only group, a significant reduction was observed only after the training program. Considering that both groups had a similar improvement from the baseline to the post-exercise regimen, it can be rationalized that improvements in glycemic control can be achieved with exercise alone, regardless of metformin therapy.
Considering both acute and long-term studies, it seems that the combination of metformin and exercise might not be superior to the exercise alone for the metformin naïve subjects. On contrary to the findings in metformin naïve subjects, T2DM patients treated with metformin who participated in the exercise program had a significant improvement in insulin sensitivity, measured by fasting blood glucose, homeostasis model assessment of IR, and glycated hemoglobin.21 According to Abdelbasset,21 patients following 12 weeks of moderate-intensity resistance training had significantly greater improvements than patients in moderate aerobic training or metformin group. Although the resistance program was advantageous in improving insulin sensitivity, both types of exercise were effective in controlling T2DM.21 Considering the lack of a placebo group in this study, it is not possible to compare the effects of combined treatment to exercise alone, but it is worth noting that the addition of exercise to metformin-habituated patients resulted in improvements in insulin sensitivity and aerobic capacity, following a resistance program.
Metformin naïve, type 2 diabetic patients, who completed a single exercise bout after 4 weeks of 2000 mg of metformin daily, had a greater glycemic response to a mixed meal when compared to the same trial without metformin.16 Comparable effect was observed by Myette-Côté et al17 who failed to observe the improvements in glucose-lowering effects of exercise when an acute bout of aerobic training was added to metformin-treated T2DM patients. In this study, Myette-Côté and associates showed that a combination of exercise and metformin resulted in a significantly higher 2-hour postprandial glucose area under the curve, across multiple meals. This effect was observed even if participants skipped their habitual morning or evening dose.17 Although independently metformin and exercise have similar effects on glucose tolerance, the combination of the two is not always favorable, regardless of the duration of metformin treatment. Takao et al74 demonstrated the direct relationship between increased postprandial hyperglycemia and risk for the development of CVD. The aforementioned negative effect that metformin can have on postprandial glucose regulation leads to the mitigation of the positive effects that exercise has on lowering the risk for CVD. This suggests a potentially contradictory rather than synergistic effect when the two treatments are combined, which is why future studies should focus on elucidating the mechanism of interaction of the two treatments.
On contrary to Boulé et al16 and Myette-Côté et al,17 Ortega et al18,19 reported significant difference and improvement in glucose area under the curve and percentage of hyperglycemic peaks when metformin was combined with exercise. Further, Ortega et al18,19 reported greater insulin sensitivity as measured by homeostasis model assessment of IR and intravenous glucose tolerance test, as well as greater glucose disappearance rate when exercise was paired with metformin. The possible disparity in the effects could be attributed to the exercise intensity difference, where Ortega et al18,19 used 4 bouts of 4 minutes at 90% of maximal heart rate (HRmax) interspersed by 3 minutes of cycling at 70% HRmax, while Myette-Côté et al17 implemented a moderate-intensity walking at 85% ventilatory threshold for 50 minutes. The positive effect of metformin in combination with exercise in the two studies by Ortega and colleagues18,19 suggests that exercise intensity and volume could play a role in the interplay of the two treatments, which was further supported by Winding et al.22 Winding and colleagues observed a greater improvement in glycemic control in a high-intensity interval training group despite a ~45% lower training volume when compared to an endurance training group, in metformin-treated subjects. Additionally, metformin-treated type 2 diabetic patients had a greater reduction in blood glucose during exercise and 50 minutes of the recovery period following high-intensity interval training but not moderate-intensity continuous training, in the study by Mendes et al.27 Further, Ortega et al18 showed an energy expenditure of ~300 kcal, which was like the endurance but significantly higher than the interval training group in the study by Winding et al.22 Considering the exercise intensity, and subsequently, the energy expenditure discrepancy, future studies should aim to elucidate the potential energy expenditure and exercise intensity threshold which could influence the interaction of metformin and exercise and mitigate improvements in glucose control. By performing exercise at a higher intensity, Ortega et al18 observed a higher carbohydrate utilization which could result in greater glucose clearance which would appear as a lack of significant change in glucose levels between metformin combined with exercise and in the exercise-only group. A similar pattern was observed by Winding et al22 who reported a greater glucose clearance during high-intensity interval training when compared to endurance training.
Considering the lack of consistency between exercise characteristics, authors exploring the interaction of two treatments should adjust exercise profiles to allow for comparison between studies. Potential interference between exercise modalities could be at the level of the mitochondrial electron transport chain, where exercise-induced energy demand is not met due to the complex 1 inhibition by metformin, resulting in a lower ATP output. Additionally, inferior energy production could lead to an earlier onset of fatigue, due to the accumulation of inorganic phosphate (Pi) and free hydrogen ions (H+), as well as an overall decrease in pH.41 While metformin utilizes disruption of energy production to stimulate the energy-sensing enzyme AMPK, and this mechanism is depicted as valuable in maintaining glucose homeostasis at rest, it seems that such changes in energy availability can be disadvantageous to exercise-induced benefits to glucose regulation and exercise capacity. Additionally, different modes of exercise and the specific mechanism by which they elicit changes in glucose control should be described to address the inconsistency between the effects of resistance and aerobic exercise. Finally, it is important to note and further explore the habituation response to metformin, considering the diverse outcomes that combined treatment elicits in metformin habituated and metformin naïve subjects.
Effects of Metformin on Exercise, and Exercise Adaptations
While it is important to address the effects of metformin on glycemic control and insulin sensitivity, it is critical not to overlook the effects of metformin on exercise capacity and adaptations.14,15,20,23–26,75,76 In particular, the addition of metformin to exercise seems to affect the rate of perceived exertion (RPE), respiratory exchange ratio (RER), heart rate (HR), and VO2max during an exercise session as well as an effect on body composition and peak aerobic capacity following an exercise program.14,15,20,23–26,75,76 In the study by Paul et al75 after 6 weeks of 1000mg of metformin daily, subjects newly diagnosed with metabolic syndrome had a significant, 22% reduction in relative VO2max during a cardiopulmonary exercise test. A similar effect was observed by Malin et al15 and Konopka et al20 in patients with impaired glucose control following 12 weeks of combination or aerobic training. On contrary, lower improvement in aerobic capacity when the treatments are combined was not supported by Pilmark et al.23 Additionally, while a significant decrease in aerobic capacity was not observed in the meta-analysis by Das and colleagues,76 a significant increase in HR was observed in patients (metabolic syndrome, IR, and T2DM) treated with metformin. Further, the author in this meta-analysis reported a decrease in RER and an increase in RPE in the overall population but failed to find any significant effects of metformin in T2DM patients and those with IR. Considering the discrepancy in duration of metformin treatment, the difference in RER and RPE between healthy and subjects with T2DM could be due to habituation of patients to the effects of metformin, which is why this effect is diminished in patients. Further, greater RPE and lower RER in the overall population that was reported by Das et al76 could mean a lowering of exercise intensity for new patients starting their metformin therapy and shift towards low to moderate, rather than high-intensity exercise. According to De Nardi et al77 high-intensity exercise provided greater benefits to the functional capacity in patients with T2DM when compared to moderate-intensity exercise. Additionally, lowering the exercise intensity could result in attenuated AMPK activity, glucose clearance, and improvements in insulin sensitivity during acute exercise. Sharoff et al14 reported lower AMPK activity and content following an acute bout of exercise in metformin habituated insulin-resistant subjects, even without a significant increase in RPE and HR. This would suggest that a significant increase in RPE and HR, as reported in the meta-analysis by Das et al76 can reduce the exercise intensity, and lower the benefits of high-intensity exercise training. Additionally, chronic attenuation of AMPK activity could lead to lower mitochondrial capacity,78 and VO2max,77 and lower skeletal muscle fatty acid metabolism which is often associated with lipotoxicity leading to IR.79 Such effect of metformin was demonstrated in the study by Boule et al26 where metformin non-metformin users had twice as large improvements in VO2max when compared to the metformin-treated patients with type 2 diabetes. Further, the effect of metformin on AMPK could reduce the adaptations in mitochondrial capacity and VO2max, and was observed by Konopka and colleagues.20 Interestingly, in the study by Konopka et al20 an addition of metformin to aerobic exercise training attenuated the increase in VO2max by approximately 50% following 12 weeks of aerobic exercise training, even without a significant reduction of basal AMPK.20 Potential explanation for the lack of the significant difference in muscle AMPK levels between metformin and placebo group could be due to the transient nature of AMPK, and timing (~48 hours after last exercise bout) of the skeletal muscle biopsies in the study. If taken together, Konopka et al20 and Sharoff et al14 suggest that although there is no difference in the basal AMPK, the decrease in AMPK activity during, and immediately following the exercise, as observed by Sharoff and colleagues,14 might play a role when it comes to long term aerobic capacity adaptations. Considering that VO2max has an inverse relationship with all-cause mortality, the negative effect that metformin can have on exercise adaptations can result in the progression of the existing metabolic disease and potentially increase the risk for the development of other comorbidities.
In addition to the reduced improvements of aerobic capacity, studies by Walton et al25 and Malin et al15 show a lack of improvement in total lean mass when metformin is combined with either progressive resistance training or a combination of resistance and aerobic training. Taking into account the nature of T2DM and the central role of skeletal muscle in mediating the adverse effects of IR, the addition of metformin to the resistance training needs to be reconsidered and further explored.
Additionally, Walton et al25 reported inhibition of muscle mass and strength gains following the progressive resistance training program. This lower response to resistance training was consistent with an increase in AMPK, a mTORC1 inhibitory kinase activation.25 Considering the importance of skeletal muscle in glucose homeostasis, future studies should focus on the effects of different types of exercise and provide an insight into the relationship between AMPK activation and potential attenuation of muscle hypertrophy as a result of mTORC1 inhibition.
Considering the above-described effects of combined treatment in comparison with exercise training only, future studies should focus on the timing of dose administration and the combination of specific, potentially lower, dose metformin treatment and exercise. Considering that high-intensity exercise effects were not decreased by a high metformin dose, there is a potential that lowering the dose of metformin could lower the negative effects metformin has on lower and moderate-intensity exercise. Potentially, understanding of metformin dose and exercise intensity, as well as the energy expenditure threshold, could contribute to the development of a dose/response model, allowing for better adjustment of the prescription. Albeit different exercise prescriptions in the described studies are valuable for understanding the effects of metformin across multiple training modes, synchronization of methodologies is encouraged to allow for comparison between the studies. Finally, based on the differential effects of metformin on resistance and aerobic training, understanding the difference in mechanisms by which these modalities affect glucose control is crucial.
Conclusion
Lifestyle, lack of exercise, poor diet were shown to lead to the dysregulation of glucose homeostasis and the development of IR. State of hyperinsulinemia, hyperglycemia and elevated FFA blood levels seem to be the symptoms commonly observed in patients suffering from T2DM. To alleviate some of the detrimental effects that the pathophysiology of T2DM has on patients, and to undermine the increased risk for the development of other cardiometabolic diseases, metformin and exercise are most commonly prescribed. While these treatments have similar targets through which they act, in recent years, research has shown the lack of synergistic effect of the two. Albeit, independently, these modalities deem beneficial in treating T2DM, the combination of metformin and exercise seems not to be advantageous, and potentially unfavorable when compared to the exercise alone.14–27 Based on the varied and inconsistent outcomes of the studies14–27 discussed in this review, the combined effect of these two modalities requires further investigation. On the contrary, authors have observed the negative effect of the combination of two modalities resulting in lower RER, HR, VO2max, and increase RPE during exercise. These effects result in a prescription of lower intensity exercise for patients suffering from T2DM, depriving them of well-documented benefits of high-intensity exercise. Considering the differing results, future research should focus on elucidating the effects of timing and amount of metformin and withholding of the dose on exercise days. Understanding the relationship between the timing of metformin and exercise will help guide future prescriptions and help mitigate the less favorable outcomes that are observed when these two modalities are combined. Considering that studies using moderate-intensity exercise showed a negative effect when metformin was added to exercise, future research should focus on determining if higher exercise intensities lower the negative effect of the combined treatment. Additionally, future research should strive to elucidate if there is a relationship between the dosage of metformin and exercise intensity and/or type and determine if increases in metformin dosage need to be congruent with increases of exercise intensity. Finally, considering the previously reported influence of lifestyle-modification in the prevention of T2DM,80 and incongruency regarding the effects of combined treatment reported in this review, prescription of metformin should be reevaluated if implemented together with exercise.
Funding
No source of funding.
Disclosure
The author declares that they have no conflicts of interest associated with this work.
References
1. World Health Organization. Obesity and overweight. Available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight.
2. Gregg EW, Shaw JE. Global health effects of overweight and obesity. N Engl J Med. 2017;377(1):80–81. doi:10.1056/NEJMe1706095
3. Centers for Disease Control and Prevention. National diabetes statistics report; 2020. Available from: https://www.cdc.gov/diabetes/data/statistics-report/index.html.
4. Emerging Risk Factors Collaboration. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375(9733):2215–2222. doi:10.1016/S0140-6736(10)60484-9
5. Kivimäki M, Kuosma E, Ferrie JE, et al. Overweight, obesity, and risk of cardiometabolic multimorbidity: pooled analysis of individual-level data for 120 813 adults from 16 cohort studies from the USA and Europe. Lancet Public Health. 2017;2(6):e277–e285. doi:10.1016/S2468-2667(17)30074-9
6. Singh GM, Danaei G, Farzadfar F, et al. The age-specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: a pooled analysis. PLoS One. 2013;8(7):e65174. doi:10.1371/journal.pone.0065174
7. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc Diabetol. 2018;17(1):83. doi:10.1186/s12933-018-0728-6
8. Wang M, Yang Y, Liao Z. Diabetes and cancer: epidemiological and biological links. World J Diabetes. 2020;11(6):227–238. doi:10.4239/wjd.v11.i6.227
9. Sun J, Wang Y, Zhang X, Zhu S, He H. Prevalence of peripheral neuropathy in patients with diabetes: a systematic review and meta-analysis. Prim Care Diabetes. 2020;14(5):435–444. doi:10.1016/j.pcd.2019.12.005
10. Rabieenia E, Jalali R, Mohammadi M. Prevalence of nephropathy in patients with type 2 diabetes in Iran: a systematic review and meta-analysis based on geographic information system (GIS). Diabetes Metab Syndr. 2020;14(5):1543–1550. doi:10.1016/j.dsx.2020.08.007
11. John S. Complication in diabetic nephropathy. Diabetes Metab Syndr. 2016;10(4):247–249. doi:10.1016/j.dsx.2016.06.005
12. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. 2014;383(9922):1068–1083. doi:10.1016/S0140-6736(13)62154-6
13. Tancredi M, Rosengren A, Svensson AM, et al. Excess mortality among persons with type 2 diabetes. N Engl J Med. 2015;373(18):1720–1732. doi:10.1056/NEJMoa1504347
14. Sharoff CG, Hagobian TA, Malin SK, et al. Combining short-term metformin treatment and one bout of exercise does not increase insulin action in insulin-resistant individuals. Am J Physiol Endocrinol Metab. 2010;298(4):E815–E823. doi:10.1152/ajpendo.00517.2009
15. Malin SK, Gerber R, Chipkin SR, Braun B. Independent and combined effects of exercise training and metformin on insulin sensitivity in individuals with prediabetes. Diabetes Care. 2012;35(1):131–136. doi:10.2337/dc11-0925
16. Boulé NG, Robert C, Bell GJ, et al. Metformin and exercise in type 2 diabetes: examining treatment modality interactions. Diabetes Care. 2011;34(7):1469–1474. doi:10.2337/dc10-2207
17. Myette Cote É, Terada T, Boulé NG. The effect of exercise with or without metformin on glucose profiles in type 2 diabetes: a pilot study. Can J Diabetes. 2016;40(2):173–177. doi:10.1016/j.jcjd.2015.08.015
18. Ortega JF, Hamouti N, Fernández-Elías VE, de Prada MV, Martínez-Vizcaíno V, Mora-Rodríguez R. Metformin does not attenuate the acute insulin-sensitizing effect of a single bout of exercise in individuals with insulin resistance. Acta Diabetol. 2014;51(5):749–755. doi:10.1007/s00592-014-0580-4
19. Ortega JF, Morales-Palomo F, Ramirez-Jimenez M, Moreno-Cabañas A, Mora-Rodríguez R. Exercise improves metformin 72-h glucose control by reducing the frequency of hyperglycemic peaks. Acta Diabetol. 2020;57(6):715–723. doi:10.1007/s00592-020-01488-7
20. Konopka AR, Laurin JL, Schoenberg HM, et al. Metformin inhibits mitochondrial adaptations to aerobic exercise training in older adults. Aging Cell. 2019;18(1):e12880. doi:10.1111/acel.12880
21. Abdelbasset WK. Resistance exercise versus aerobic exercise combined with metformin therapy in the treatment of type 2 diabetes: a 12-week comparative clinical study. Endocr Metab Immune Disord Drug Targets. 2020;20. doi:10.2174/1871530320999200918143227
22. Winding KM, Munch GW, Iepsen UW, Van Hall G, Pedersen BK, Mortensen SP. The effect on glycaemic control of low-volume high-intensity interval training versus endurance training in individuals with type 2 diabetes. Diabetes Obes Metab. 2018;20(5):1131–1139. doi:10.1111/dom.13198
23. Pilmark NS, Lyngbæk M, Oberholzer L, et al. The interaction between metformin and physical activity on postprandial glucose and glucose kinetics: a randomised, clinical trial. Diabetologia. 2021;64(2):397–409. doi:10.1007/s00125-020-05282-6
24. Kristensen JM, Lillelund C, Kjøbsted R, et al. Metformin does not compromise energy status in human skeletal muscle at rest or during acute exercise: a randomised, crossover trial. Physiol Rep. 2019;7(23):e14307. doi:10.14814/phy2.14307
25. Walton RG, Dungan CM, Long DE, et al. Metformin blunts muscle hypertrophy in response to progressive resistance exercise training in older adults: a randomized, double-blind, placebo-controlled, multicenter trial: the MASTERS trial. Aging Cell. 2019;18(6):e13039. doi:10.1111/acel.13039
26. Boulé NG, Kenny GP, Larose J, Khandwala F, Kuzik N, Sigal RJ. Does metformin modify the effect on glycaemic control of aerobic exercise, resistance exercise or both? Diabetologia. 2013;56(11):2378–2382. doi:10.1007/s00125-013-3026-6
27. Mendes R, Sousa N, Themudo-Barata JL, Reis VM. High-intensity interval training versus moderate-intensity continuous training in middle-aged and older patients with type 2 diabetes: a randomized controlled crossover trial of the acute effects of treadmill walking on glycemic control. Int J Environ Res Public Health. 2019;16(21):4163. doi:10.3390/ijerph16214163
28. Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. AMPK activation: a therapeutic target for type 2 diabetes? Diabetes Metab Syndr Obes. 2014;7:241–253.
29. Castro AV, Kolka CM, Kim SP, Bergman RN. Obesity, insulin resistance and comorbidities? Mechanisms of association. Arq Bras Endocrinol Metabol. 2014;58(6):600–609. doi:10.1590/0004-2730000003223
30. Mezza T, Cinti F, Cefalo CMA, Pontecorvi A, Kulkarni RN, Giaccari A. β-cell fate in human insulin resistance and type 2 diabetes: a perspective on islet plasticity. Diabetes. 2019;68(6):1121–1129. doi:10.2337/db18-0856
31. Lan YL, Lou JC, Lyu W, Zhang B. Update on the synergistic effect of HSL and insulin in the treatment of metabolic disorders. Ther Adv Endocrinol Metab. 2019;10:2042018819877300. doi:10.1177/2042018819877300
32. Matsuzaka T, Shimano H. New perspective on type 2 diabetes, dyslipidemia and non-alcoholic fatty liver disease. J Diabetes Investig. 2020;11(3):532–534. doi:10.1111/jdi.13258
33. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133–2223. doi:10.1152/physrev.00063.2017
34. Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK. Role of free fatty acids in endothelial dysfunction. J Biomed Sci. 2017;24(1):50. doi:10.1186/s12929-017-0357-5
35. Rachek LI. Free fatty acids and skeletal muscle insulin resistance. Prog Mol Biol Transl Sci. 2014;121:267–292.
36. Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. doi:10.1016/j.cmet.2007.10.013
37. Wicks SE, Vandanmagsar B, Haynie KR, et al. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc Natl Acad Sci U S A. 2015;112(25):E3300–E3309. doi:10.1073/pnas.1418560112
38. Saha AK, Xu XJ, Lawson E, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. 2010;59(10):2426–2434. doi:10.2337/db09-1870
39. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–135. doi:10.1038/nrm.2017.95
40. Wang Q, Liu S, Zhai A, Zhang B, Tian G. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol Pharm Bull. 2018;41(7):985–993. doi:10.1248/bpb.b17-00724
41. Mougios V. Exercise Biochemistry. Champaign, IL: Human Kinetics; 2020.
42. Kjøbsted R, Munk-Hansen N, Birk JB, et al. Enhanced muscle insulin sensitivity after contraction/exercise is mediated by AMPK. Diabetes. 2017;66(3):598–612. doi:10.2337/db16-0530
43. O’Neill HM, Holloway GP, Steinberg GR. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol Cell Endocrinol. 2013;366(2):135–151.
44. Sadria M, Layton AT. Interactions among mTORC, AMPK and SIRT: a computational model for cell energy balance and metabolism. Cell Commun Signal. 2021;19(1):57. doi:10.1186/s12964-021-00706-1
45. Hayashi T, Wojtaszewski JF, Goodyear LJ. Exercise regulation of glucose transport in skeletal muscle. Am J Physiol. 1997;273(6):E1039–E1051.
46. Lundsgaard AM, Fritzen AM, Kiens B. Molecular regulation of fatty acid oxidation in skeletal muscle during aerobic exercise. Trends Endocrinol Metab. 2018;29(1):18–30. doi:10.1016/j.tem.2017.10.011
47. Krook A, Björnholm M, Galuska D, et al. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes. 2000;49(2):284–292. doi:10.2337/diabetes.49.2.284
48. Kennedy JW, Hirshman MF, Gervino EV, et al. Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes. 1999;48(5):1192–1197. doi:10.2337/diabetes.48.5.1192
49. Ke R, Xu Q, Li C, Luo L, Huang D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int. 2018;42(4):384–392. doi:10.1002/cbin.10915
50. Coccimiglio IF, Clarke DC. ADP is the dominant controller of AMP-activated protein kinase activity dynamics in skeletal muscle during exercise. PLoS Comput Biol. 2020;16(7):e1008079. doi:10.1371/journal.pcbi.1008079
51. Musi N, Fujii N, Hirshman MF, et al. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes. 2001;50(5):921–927. doi:10.2337/diabetes.50.5.921
52. Frøsig C, Jørgensen SB, Hardie DG, Richter EA, Wojtaszewski JF. 5ʹ-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am J Physiol Endocrinol Metab. 2004;286(3):E411–E417. doi:10.1152/ajpendo.00317.2003
53. Vind BF, Pehmøller C, Treebak JT, et al. Impaired insulin-induced site-specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise training. Diabetologia. 2011;54(1):157–167. doi:10.1007/s00125-010-1924-4
54. Wende AR, Schaeffer PJ, Parker GJ, et al. A role for the transcriptional coactivator PGC-1alpha in muscle refueling. J Biol Chem. 2007;282(50):36642–36651. doi:10.1074/jbc.M707006200
55. Z Michael LF, Wu Z, Cheatham RB, et al. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A. 2001;98(7):3820–3825. doi:10.1073/pnas.061035098
56. Yoshida Y, Jain SS, McFarlan JT, Snook LA, Chabowski A, Bonen A. Exercise- and training-induced upregulation of skeletal muscle fatty acid oxidation are not solely dependent on mitochondrial machinery and biogenesis. J Physiol. 2013;591(18):4415–4426. doi:10.1113/jphysiol.2012.238451
57. Bailey CJ. Metformin: historical overview. Diabetologia. 2017;60(9):1566–1576. doi:10.1007/s00125-017-4318-z
58. Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333(9):550–554. doi:10.1056/NEJM199508313330903
59. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi:10.1007/s00125-017-4342-z
60. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. doi:10.1042/bj3480607
61. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–228. doi:10.1074/jbc.275.1.223
62. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signaling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. doi:10.1038/nature11808
63. Johanns M, Lai YC, Hsu MF, et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat Commun. 2016;7:10856. doi:10.1038/ncomms10856
64. Hamasaki H. Exercise and glucagon-like peptide-1: does exercise potentiate the effect of treatment? World J Diabetes. 2018;9(8):138–140. doi:10.4239/wjd.v9.i8.138
65. Herzberg-Schäfer S, Heni M, Stefan N, Häring HU, Fritsche A. Impairment of GLP1-induced insulin secretion: role of genetic background, insulin resistance and hyperglycaemia. Diabetes Obes Metab. 2012;14(Suppl 3):85–90. doi:10.1111/j.1463-1326.2012.01648.x
66. Rynders CA, Weltman JY, Jiang B, et al. Effects of exercise intensity on postprandial improvement in glucose disposal and insulin sensitivity in prediabetic adults. J Clin Endocrinol Metab. 2014;99(1):220–228. doi:10.1210/jc.2013-2687
67. Ryan BJ, Schleh MW, Ahn C, et al. Moderate-intensity exercise and high-intensity interval training affect insulin sensitivity similarly in obese adults. J Clin Endocrinol Metab. 2020;105(8):e2941–e2959. doi:10.1210/clinem/dgaa345
68. Hingst JR, Bruhn L, Hansen MB, et al. Exercise-induced molecular mechanisms promoting glycogen supercompensation in human skeletal muscle. Mol Metab. 2018;16:24–34. doi:10.1016/j.molmet.2018.07.001
69. Kido K, Ato S, Yokokawa T, Makanae Y, Sato K, Fujita S. Acute resistance exercise-induced IGF1 expression and subsequent GLUT4 translocation. Physiol Rep. 2016;4(16):e12907. doi:10.14814/phy2.12907
70. Dela F, Ingersen A, Andersen NB, et al. Effects of one-legged high-intensity interval training on insulin-mediated skeletal muscle glucose homeostasis in patients with type 2 diabetes. Acta Physiol. 2019;226(2):e13245. doi:10.1111/apha.13245
71. Malin SK, Braun B. Impact of metformin on exercise-induced metabolic adaptations to lower type 2 diabetes risk. Exerc Sport Sci Rev. 2016;44(1):4–11. doi:10.1249/JES.0000000000000070
72. Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98. doi:10.2165/11534750-000000000-00000
73. Albers PH, Pedersen AJT, Birk JB, et al. Human muscle fiber type–specific insulin signaling: impact of obesity and type 2 diabetes. Diabetes. 2014;64(2):485–497. doi:10.2337/db14-0590
74. Takao T, Suka M, Yanagisawa H, Iwamoto Y. Impact of postprandial hyperglycemia at clinic visits on the incidence of cardiovascular events and all-cause mortality in patients with type 2 diabetes. J Diabetes Investig. 2017;8(4):600–608. doi:10.1111/jdi.12610
75. Paul AA, Dkhar SA, Kamalanathan S, et al. Effect of metformin on exercise capacity in metabolic syndrome. Diabetes Metab Syndr. 2017;11(Suppl 1):S403–S406. doi:10.1016/j.dsx.2017.03.025
76. Das S, Behera SK, Srinivasan A, et al. Effect of metformin on exercise capacity: a meta-analysis. Diabetes Res Clin Pract. 2018;144:270–278. doi:10.1016/j.diabres.2018.08.022
77. De Nardi AT, Tolves T, Lenzi TL, Signori LU, da Silva AM. High-intensity interval training versus continuous training on physiological and metabolic variables in prediabetes and type 2 diabetes: a meta-analysis. Diabetes Res Clin Pract. 2018;137:149–159. doi:10.1016/j.diabres.2017.12.017
78. Daussin FN, Zoll J, Ponsot E, et al. Training at high exercise intensity promotes qualitative adaptations of mitochondrial function in human skeletal muscle. J Appl Physiol. 2008;104(5):1436–1441. doi:10.1152/japplphysiol.01135.2007
79. Marcinko K, Steinberg GR. The role of AMPK in controlling metabolism and mitochondrial biogenesis during exercise. Exp Physiol. 2014;99(12):1581–1585. doi:10.1113/expphysiol.2014.082255
80. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.