")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Combination of erlotinib and a PARP inhibitor inhibits growth of A2780 tumor xenografts due to increased autophagy
Authors Sui H, Shi C, Yan Z, Li H
Received 2 February 2015
Accepted for publication 9 March 2015
Published 22 June 2015 Volume 2015:9 Pages 3183—3190
DOI https://doi.org/10.2147/DDDT.S82035
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Hongying Sui, Caixia Shi, Zhipeng Yan, Hucheng Li
Department of Gynecological Oncology, Hunan Cancer Hospital, The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha City, People’s Republic of China
Background: Ovarian cancer is the leading cause of death in women with gynecological malignancy worldwide. Despite multiple new approaches to treatment, relapse remains almost inevitable in patients with advanced disease. The poor outcome of advanced ovarian cancer treated with conventional therapy stimulated the search for new strategies to improve therapeutic efficacy. Although epidermal growth factor receptor (EGFR) and poly(ADP-ribose) polymerase (PARP) inhibitors have known activity in advanced ovarian cancer, the effect of combined therapy against EGFR and PARP in this population has not been reported. In the current study, we investigated the mechanisms of erlotinib used alone or in combination with olaparib (AZD2281), a potent inhibitor of PARP, in an EGFR-overexpressing ovarian tumor xenograft model.
Methods: A2780 (EGFR-overexpressing, BRCA1/2 wild-type) cells were subcutaneously injected into nude mice, which were then randomly assigned to treatment with vehicle, erlotinib, AZD2281, or erlotinib + AZD2281, for up to 3 weeks. All mice were then sacrificed and tumor tissues were subjected to Western blot analysis and monodansylcadervarine staining (for analysis of autophagy).
Results: Erlotinib could slightly inhibit growth of A2780 tumor xenografts, and AZD2281 alone had similar effects on tumor growth. However, the combination treatment had a markedly enhanced antitumor effect. Western blot analysis revealed that treatment with erlotinib could significantly reduce the phosphorylation level of ERK1/2 and AKT in A2780 tumor tissue. Of interest, monodansylcadervarine staining showed that the autophagic effects were substantially enhanced when the agents were combined, which may be due to downregulation of apoptosis.
Conclusion: These results suggest that combination of a selective EGFR inhibitor and a PARP inhibitor is effective in ovarian cancer A2780 xenografts, and depends on enhanced autophagy.
Keywords: erlotinib, poly(ADP-ribose) polymerase, PARP, olaparib, tyrosine kinase inhibitor, ovarian cancer, autophagy, apoptosis
Introduction
Ovarian cancer is the leading cause of death in women with gynecological cancer. Currently, the standard therapy involves combining maximal cytoreductive surgery with chemotherapy that consists of a platinum agent and a taxane compound.1 However, more than 70% of patients with ovarian cancer are diagnosed late and suffer high morbidity and mortality. The majority of cases are diagnosed in the advanced stages, with a 5-year survival rate of only 26.9%.2 The poor long-term prognosis is due to recurrence and lack of effective second-line chemotherapeutic regimens. Thus, targeted therapies and novel immunotherapeutic approaches might improve the clinical outcome.
The epidermal growth factor receptor (EGFR) has been suggested as a promising target since up to 70% of ovarian cancers are EGFR-positive, and overexpression of EGFR develops during cancer progression and correlates with a poor prognosis.3,4 Preclinical studies suggest that inhibition of this target might have antitumor activity and reverse chemoresistance.5,6 However, single-agent studies of EGFR tyrosine kinase inhibitors (erlotinib and gefitinib), as well as monoclonal antibodies against EGFR (cetuximab, panitumumab, and matuzumab) have shown only modest efficacy.7,8 These studies indicate that EGFR targeting in ovarian cancer does not have adequate clinical benefit.
The poly(ADP)-ribose polymerases (PARPs) are a large family of multifunctional enzymes. PARP-1 is the most abundant isoform and plays a key role in repairing single-strand breaks in DNA through the base excision repair pathway.9 Clinical studies have confirmed the activity of PARP inhibitors in patients with ovarian cancer and germline BRCA1/2 mutations.10 However, recent clinical data indicate that a subset of patients who develop sporadic ovarian cancer (with wild-type BRCA1/2) may also respond to PARP inhibition, suggesting that BRCA1/2 mutations may not be the sole predictors of response.11 The combination of EGFR inhibition and PARP inhibition is another area of potential synergistic activity.
Materials and methods
Cell lines
A2780 (EGFR-overexpressing, BRCA1/2 wild-type) cell lines were obtained from the Chinese Academy of Sciences (Shanghai, People’s Republic of China) and maintained in Roswell Park Memorial Institute 1640 (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum and penicillin/streptomycin (Invitrogen). Incubation was carried out at 37°C under 5% CO2 in air.
Efficacy study in vivo
Erlotinib and AZD2281 were purchased from Selleck Chemicals (Houston, TX, USA). BALB/C nude mice (female, aged 6–8 weeks) were obtained from Shanghai SLAC Laboratory Animal Co Ltd (Shanghai, People’s Republic of China) and housed in a pathogen-free environment under controlled conditions (temperature 20°C–26°C, humidity 40%–70%, light–dark cycle 12–12 hours). Chlorinated water and irradiated food were provided ad libitum. The animals were allowed to acclimatize and recover from shipping-related stress for 1 week prior to the study. The health of the mice was monitored daily. The mice were injected subcutaneously with 3×106 A2780 cells that had been resuspended in phosphate-buffered saline (PBS). Erlotinib, solubilized in 6% Captisol (Cydex Inc, Lenexa, KS, USA), was administered orally once daily at a dose of 50 mg/kg for 3 weeks. AZD2281 was reconstituted in 1-methyl-2 pyrrolidone and poly(ethylene glycol) 300, and injected by oral gavage once daily at a dose of 30 mg/kg for 3 weeks. Tumor diameter was measured twice a week using calipers, and tumor volume was calculated as: ab2/2 mm3, where a is the length and b is the width of the tumor.
The protocol was reviewed by the Institutional Animal Care and Use Committee at Central South University. The animal experiments were performed in accordance with the Guidelines for the Accommodation and Care of Laboratory Animals at Central South University.
Mice that developed tumors reaching 150–200 mm3 in size were randomized into four groups with ten mice in each group: vehicle (PBS), 50 mg/kg erlotinib, 30 mg/kg AZD2281, and a combination of erlotinib + AZD2281. sA sister efficacy study was performed, in which mice were randomly divided into three groups to receive vehicle, the combination treatment, or the combination treatment in addition to 5 mg/kg 3-methyladenine (3-MA). At the end of each study, the A2780 xenograft tumor tissues were isolated, placed in fresh sterile Roswell Park Memorial Institute 1640 medium, and then transferred to a new dish for removal of necrotic areas, fatty tissues, blood clots, and connective tissue with forceps and scalpel. Finally, the tumor tissues were washed with PBS and stored in liquid nitrogen.
Western blot analysis
Expression of phospho-EGFR (Tyr1068, p-EGFR), phospho-ERK1/2 (Thr202/Tyr204, p-ERK1/2), phospho-AKT (Ser473, p-AKT), phospho-p53 (Ser46, p-p53), beclin 1, and LC3 in the tumor tissues was examined by Western blotting. The tissues were washed with PBS and lysed on ice in radioimmunoprecipitation assay buffer containing a complete protease inhibitor cocktail (Roche, Basel, Switzerland). The lysates were cleared by centrifugation at 12,000 rpm and 4°C for 15 minutes, and protein was quantified using bicinchoninic acid reagent (Thermo Scientific, Waltham, MA, USA). Protein lysates (30 μg) were run on Bis–Tris gels (Invitrogen) using 3-Morpholinopropanesulfonic acid or 2-(N-morpholino) ethanesulfonic acid buffer (Invitrogen). The proteins were transferred to nitrocellulose membranes by wet transfer or by iBlot (Invitrogen) dry transfer. Membranes were blocked in 10% milk in 1× PBS containing 0.01% Tween 20 (PBST) and were incubated with antibodies in 5% milk or bovine serum albumin in 1× PBST, according to the manufacturer’s instructions. Membranes were washed in 1× PBST and incubated with secondary antibodies (Bio-Rad, Hercules, CA, USA) in 5% milk/1× PBST at room temperature. Membranes were washed with 1× PBST and visualized with enhanced chemiluminescence (Amersham, GE Healthcare, Little Chalfont, UK). All antibodies were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). The results were quantified using Quantity One software (Bio-Rad, version 4.4.0.36).
Immunohistochemical staining
Paraffin tissue sections (4 mm) were dried in a 37°C oven overnight. The slides were deparaffinized in xylene and rehydrated through graded alcohol to water. Endogenous peroxidase was blocked in 3% hydrogen peroxide for 30 minutes. Antigen retrieval was carried out using a microwave oven (800 W for 10 minutes) in 10 mM citrate buffer (pH 6.0). Slides were incubated with individual primary antibodies overnight at 4°C inside a moist chamber. A TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay was purchased from Millipore (Billerica, MA, USA). TUNEL positivity was quantified as mean number of TUNEL-positive cells/total number of glandular cells per high power field (mean of 16 high power fields) ×100. Measurements were performed by three blinded, independent observers for four control and four treated tumors.
Caspase activity assay
The activity of caspase-3, caspase-8, and caspase-9 was evaluated in cytoplasmic extracts using appropriate colorimetric kits (MBL International, Woburn, MA, USA). Briefly, fresh tumors in each group were isolated after the last treatment with erlotinib, AZD2281, or the combination treatment in 3 hours on day 21 of the efficacy study, and tumor lysis containing 200 μg of protein was then incubated with 5 μL of 4 mM pNA-conjugated substrate (DEVD-pNA, IETD-pNA, and LEHD-pNA) at 37°C for 2 hours. The amount of pNA released was measured at 405 nm using a microplate reader (Tecan Spectra, Wetzlar, Germany). Tumors collected from another efficacy study were also analyzed for caspase activity.
Autophagy assay
The fluorescent compound monodansylcadervarine (MDC) is used as a tracer for autophagic vacuoles. In order to investigate whether the combination treatment would induce autophagy in A2780 xenografts, the tumor tissues were collected after treatment with erlotinib, AZD2281, or erlotinib + AZD2281. The autophagic vacuoles were labeled with MDC by incubating with 0.05 mM/L MDC in PBS at 37°C for 2 hours. The tumor tissues were then lysed, washed three times with cold PBS, and immediately measured using a FACScan flow cytometer (Beckman Coulter, Miami, FL, USA). 3-MA, a specific inhibitor of autophagy, was purchased from Sigma–Aldrich (St Louis, MO, USA).
Statistical analysis
The data are presented as the mean ± standard error. The Student’s t-test was used to analyze mean values, and P<0.05 was considered to be statistically significant.
Results
Effects of erlotinib, AZD2281, and erlotinib + AZD2281 in an A2780 xenograft model
Inhibition of tumor growth
Firstly, we validated the efficacy of the combination against tumor growth in the A2780 xenograft model. Both erlotinib and AZD2281 partially, but not significantly, inhibited the growth of A2780 xenografts; however, the combination of erlotinib and AZD2281 was significantly more potent than each single agent used alone in inhibiting the growth of the xenografts, as measured by both tumor size and weight (P<0.01, Figure 1A–C). These in vivo data further demonstrate that the combination of erlotinib and AZD2281 displays augmented anticancer activity. The single agent and combination treatment protocols were well tolerated by the mice, with no weight loss or other signs of acute or delayed toxicity (Figure 1D).
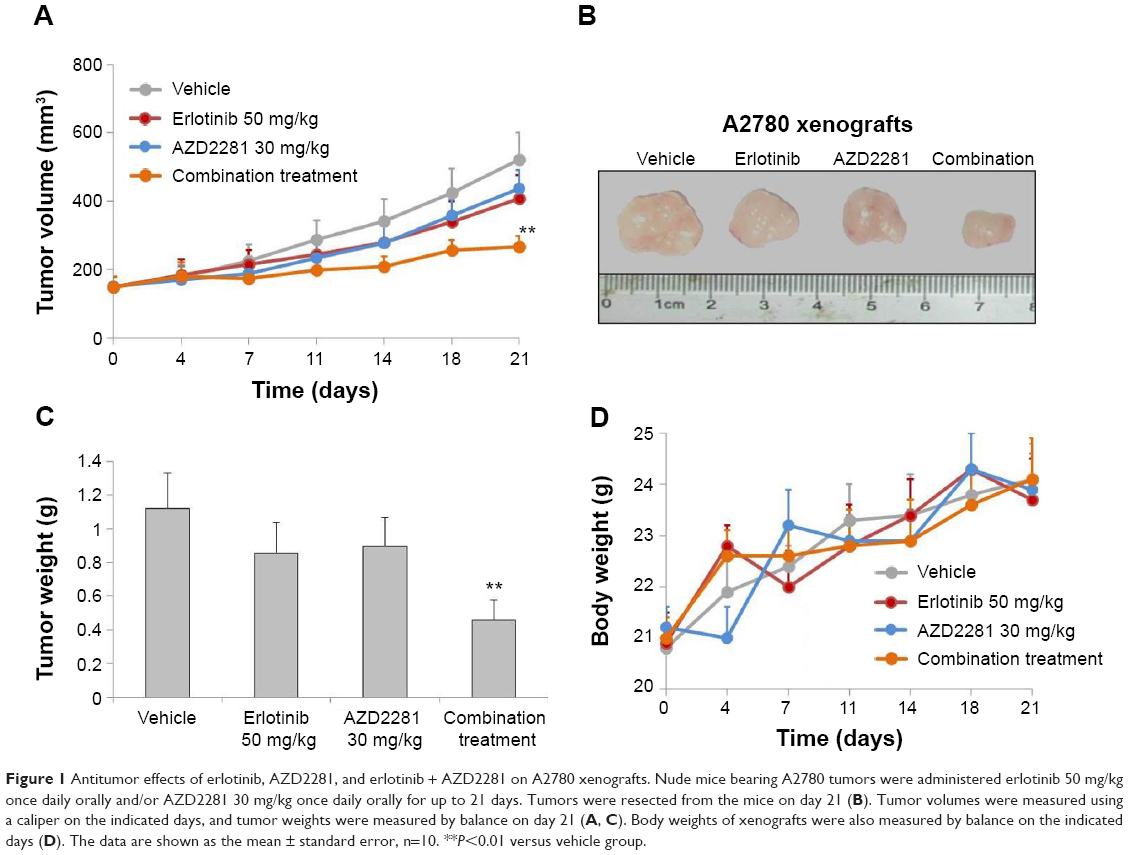 | Figure 1 Antitumor effects of erlotinib, AZD2281, and erlotinib + AZD2281 on A2780 xenografts. Nude mice bearing A2780 tumors were administered erlotinib 50 mg/kg once daily orally and/or AZD2281 30 mg/kg once daily orally for up to 21 days. Tumors were resected from the mice on day 21 (B). Tumor volumes were measured using a caliper on the indicated days, and tumor weights were measured by balance on day 21 (A, C). Body weights of xenografts were also measured by balance on the indicated days (D). The data are shown as the mean ± standard error, n=10. **P<0.01 versus vehicle group. |
PI3K and MEK signaling
To assess the effects of each compound on downstream molecules in the MEK and phosphatidylinositide 3-kinase (PI3K) pathways, we used Western blot analysis to observe phosphorylation status and total protein expression in the A2780 tumor tissues. The results indicated that p-EGFR, p-AKT, and p-ERK1/2 were inhibited by erlotinib. In addition, AZD2281 had no effect on the MEK and PI3K pathways. Interestingly, the erlotinib and AZD2281 combination could not induce a more inhibitory effect on expression of p-AKT, p-ERK1/2, or p-EGFR. In addition, expression of matrix metalloproteinase (MMP)-2 and MMP-9 was not affected by erlotinib, AZD2281, or erlotinib + AZD2281 (Figure 2).
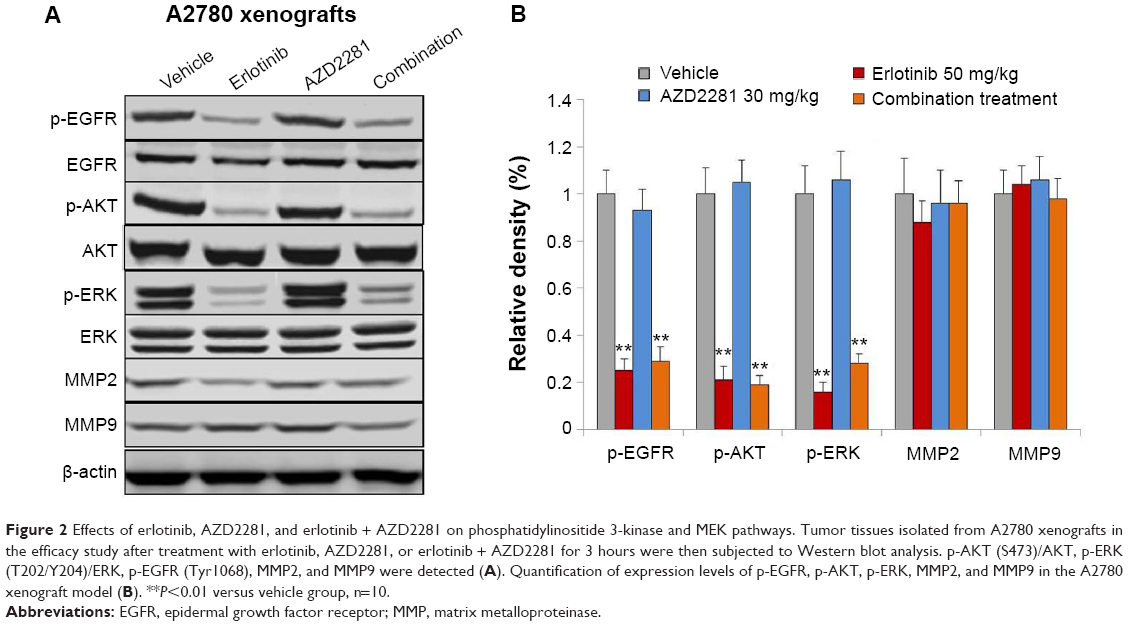 | Figure 2 Effects of erlotinib, AZD2281, and erlotinib + AZD2281 on phosphatidylinositide 3-kinase and MEK pathways. Tumor tissues isolated from A2780 xenografts in the efficacy study after treatment with erlotinib, AZD2281, or erlotinib + AZD2281 for 3 hours were then subjected to Western blot analysis. p-AKT (S473)/AKT, p-ERK (T202/Y204)/ERK, p-EGFR (Tyr1068), MMP2, and MMP9 were detected (A). Quantification of expression levels of p-EGFR, p-AKT, p-ERK, MMP2, and MMP9 in the A2780 xenograft model (B). **P<0.01 versus vehicle group, n=10. |
Apoptosis
In order to determine whether the inhibition of tumor growth caused by erlotinib, AZD2281, and erlotinib + AZD2281 was accompanied by apoptosis, parameters indicating apoptosis were analyzed by TUNEL assay. The results showed that treatment with erlotinib alone could induce TUNEL-positive nuclei in A2780 xenografts, which could be completely reversed by AZD2281 (Figure 3A and B).
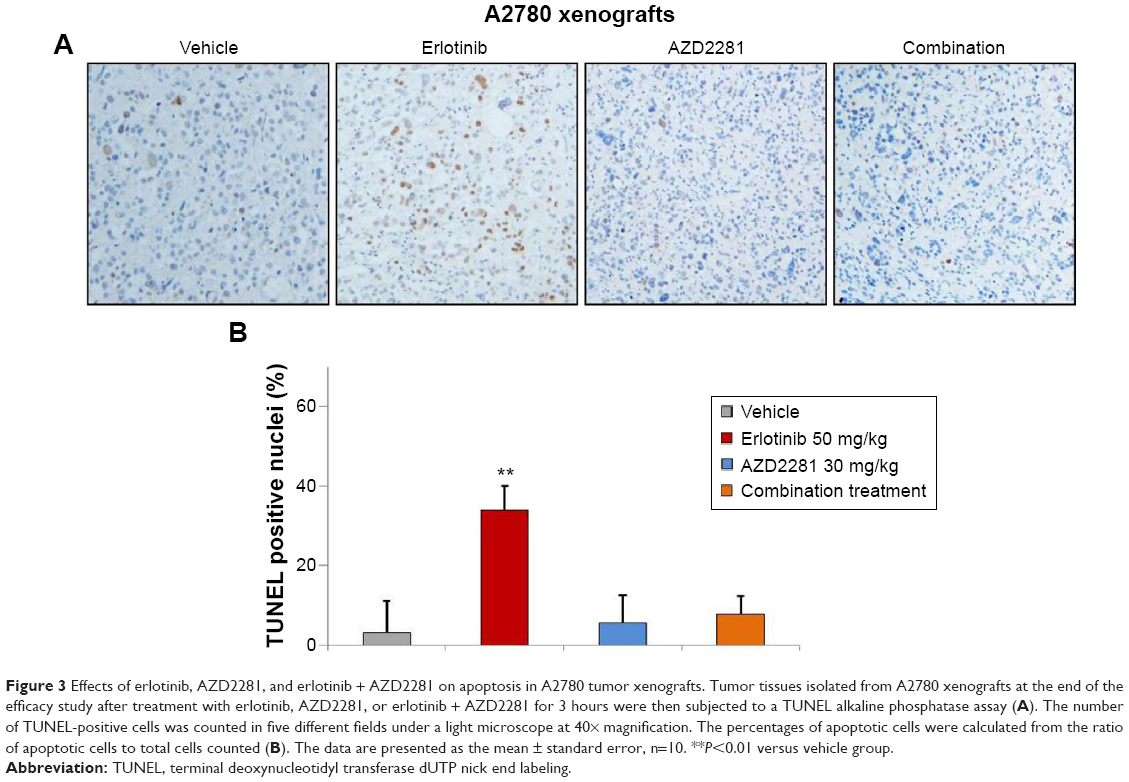 | Figure 3 Effects of erlotinib, AZD2281, and erlotinib + AZD2281 on apoptosis in A2780 tumor xenografts. Tumor tissues isolated from A2780 xenografts at the end of the efficacy study after treatment with erlotinib, AZD2281, or erlotinib + AZD2281 for 3 hours were then subjected to a TUNEL alkaline phosphatase assay (A). The number of TUNEL-positive cells was counted in five different fields under a light microscope at 40× magnification. The percentages of apoptotic cells were calculated from the ratio of apoptotic cells to total cells counted (B). The data are presented as the mean ± standard error, n=10. **P<0.01 versus vehicle group. |
Caspase activity
Next, we evaluated the effects of erlotinib, AZD2281, and erlotinib + AZD2281 on the activity of caspase-3, caspase-8, and caspase-9 in A2780 xenografts using a colorimetric assay. The results showed an approximately five-fold increase in caspase-3 and caspase-9 activity in A2780 xenografts treated with erlotinib. However, erlotinib had no effect on caspase-8 activity, indicating that erlotinib induces apoptosis in A2780 xenografts via the mitochondrial pathway. Not surprisingly, AZD2281 could partially reverse the upregulation of caspase-3 and caspase-9 induced by erlotinib (Figure 4).
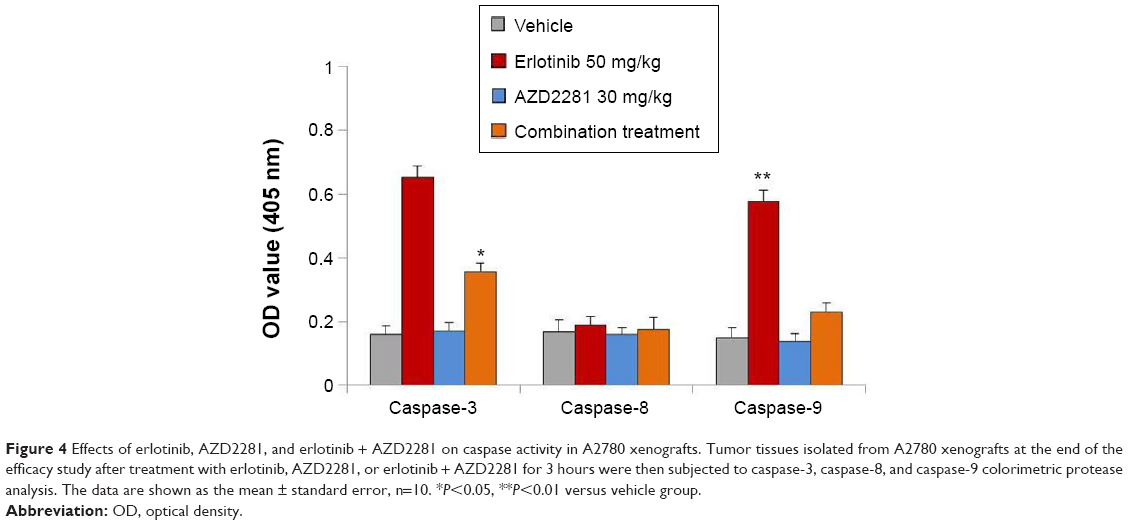 | Figure 4 Effects of erlotinib, AZD2281, and erlotinib + AZD2281 on caspase activity in A2780 xenografts. Tumor tissues isolated from A2780 xenografts at the end of the efficacy study after treatment with erlotinib, AZD2281, or erlotinib + AZD2281 for 3 hours were then subjected to caspase-3, caspase-8, and caspase-9 colorimetric protease analysis. The data are shown as the mean ± standard error, n=10. *P<0.05, **P<0.01 versus vehicle group. |
Autophagy
To explore the involvement of erlotinib, AZD2281, and erlotinib + AZD2281 in the modulation of autophagy, the autophagic ratio was measured using the fluorescent dye MDC, which specifically stains autophagosomes. As shown in Figure 5A, erlotinib or AZD2281 slightly induced autophagy in A2780 xenografts. However, compared with the single treatments, the autophagic ratio was significantly enhanced by the combined use of erlotinib and AZD2281. To further assess whether the combination treatment would induce autophagy, levels of LC3 and Beclin 1 were examined by Western blot analysis. Activation of Beclin 1 was markedly increased, and conversion from LC3-I to LC3-II was also obvious after administration of the combination. We also found that the combination treatment could significantly decrease expression of p-p53 in tumor tissues (Figure 5B). We then performed a sister efficacy study in vivo. As expected, autophagy induced by the combination treatment could be completely suppressed by 3-MA, a specific autophagy inhibitor. In addition, expression of Beclin 1 and conversion from LC3-I to LC3-II were reversed by 3-MA (Figure 5B). We also found that 3-MA attenuated the antitumor activity of the erlotinib + AZD2281 combination in A2780 xenografts (Figure 5C and D). 3-MA had no effect on the activity of caspase-3 or caspase-9 (Figure 5E).
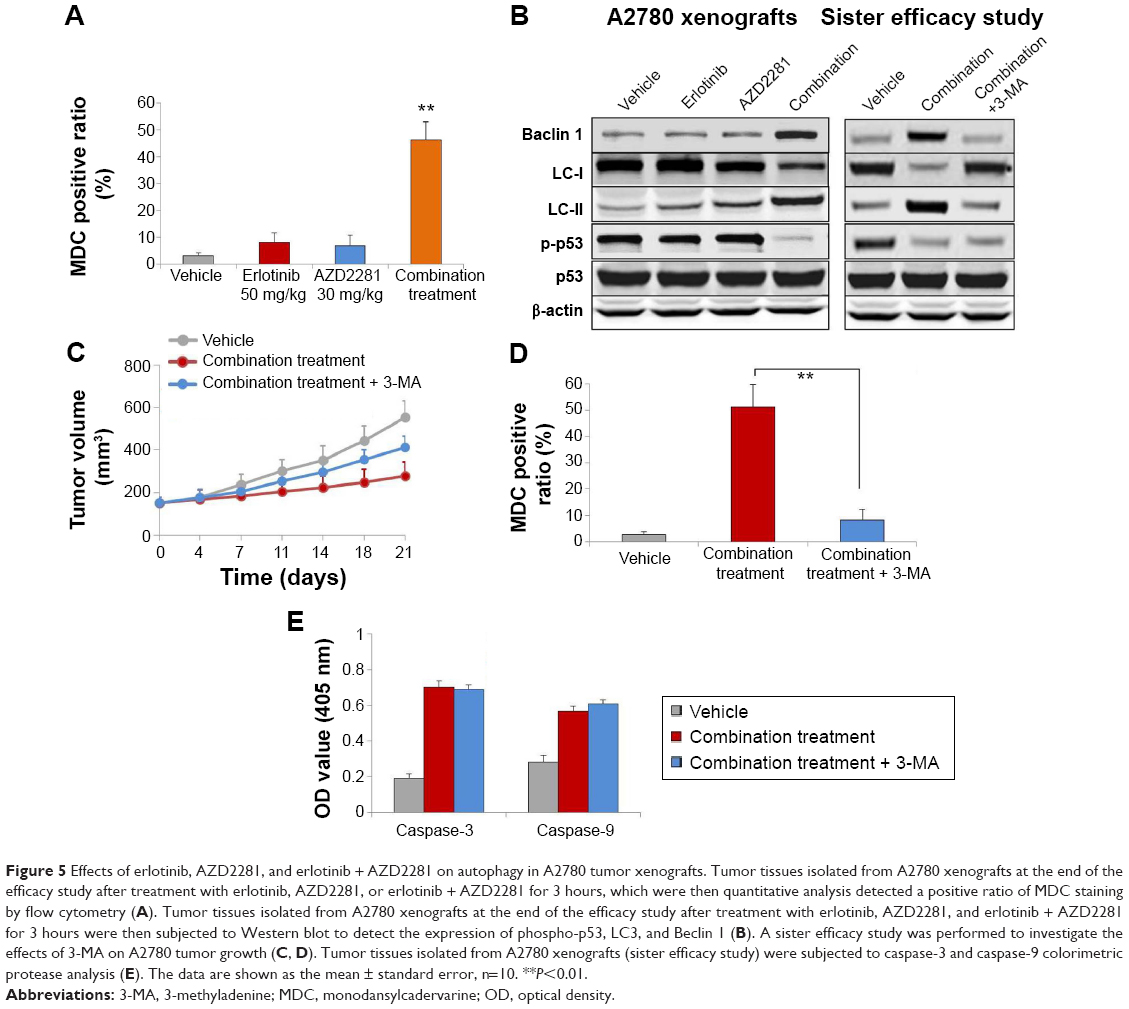 | Figure 5 Effects of erlotinib, AZD2281, and erlotinib + AZD2281 on autophagy in A2780 tumor xenografts. Tumor tissues isolated from A2780 xenografts at the end of the efficacy study after treatment with erlotinib, AZD2281, or erlotinib + AZD2281 for 3 hours, which were then quantitative analysis detected a positive ratio of MDC staining by flow cytometry (A). Tumor tissues isolated from A2780 xenografts at the end of the efficacy study after treatment with erlotinib, AZD2281, and erlotinib + AZD2281 for 3 hours were then subjected to Western blot to detect the expression of phospho-p53, LC3, and Beclin 1 (B). A sister efficacy study was performed to investigate the effects of 3-MA on A2780 tumor growth (C, D). Tumor tissues isolated from A2780 xenografts (sister efficacy study) were subjected to caspase-3 and caspase-9 colorimetric protease analysis (E). The data are shown as the mean ± standard error, n=10. **P<0.01. |
Discussion
Clinical development of PARP inhibitors in advanced ovarian cancer have been shown in hereditary ovarian cancer caused by germline mutations in BRCA1/2.12 However, the presence of a mutation in BRCA was not predictive of response, as also shown in a Canadian multicenter study in which 55 patients with known or unknown BRCA status but high-grade serous histotype received olaparib 400 mg twice daily.9 The antitumor activity in BRCA-mutated patients was 41% compared with 24% in those with wild-type BRCA, indicating the potential of these drugs in the treatment of ovarian cancer beyond BRCA mutation carriers has yet to be defined. Many small-molecule inhibitors and monoclonal antibodies that target multiple crucial cancer characteristics, including vascular endothelial growth factor receptor, EGFR, PARP, PI3K, and phosphatase and tensin homolog, are now entering clinical trials.13 However, some patients are not sensitive to these inhibitors/antibodies, and the emergence of drug resistance has highlighted the continued need for targeted combination therapies. In the current study, we designed a new combinatorial strategy and showed that combination of a selective EGFR inhibitor and a PARP inhibitor was effective in an A2780 (EGFR-overexpressing, BRCA1/2 wild-type) xenograft model in vivo.
Programmed cell death, a critical mechanism for development and homeostasis of multicellular organisms, consists of two main forms, ie, apoptosis and autophagy.14 Apoptosis (type I programmed cell death) is a physiological process of cell suicide, characterized by cell shrinkage, DNA fragmentation, chromatin condensation, membrane blebbing, and formation of apoptotic bodies.15 Autophagy is a conserved degradation/recycling system for long-lived protein and damaged organelles.16 In the present study, we demonstrated that erlotinib can induce apoptosis in A2780 via the mitochondrial pathway. In addition, we provide new evidence that AZD2271 suppresses erlotinib-induced apoptosis by decreasing p-p53 levels. More interesting is that erlotinib or AZD2271 used alone had no significant effect on autophagy in an A2780 xenograft model, but the combination treatment induced notable autophagy by increasing Beclin 1 and LC3-II levels.
The tumor suppressor p53 plays a pivotal role in safeguarding the integrity of the genome and is also a critical mediator of cell death.17 Yan et al reported that p53 promoted tumor necrosis factor alpha-induced apoptosis and autophagy, which indicated apoptosis promoted autophagy. Meanwhile, activation of autophagy participated in the process of apoptosis.18 However, in the present study, inhibition of apoptosis promoted autophagy after the combination treatment. Moreover, expression of p-p53 was downregulated, indicating that p53 may play a critical negative role in regulation of autophagy in A2780 xenografts, subsequently inhibiting apoptosis and promoting autophagy. It is possible that the combination treatment affects the cell cycle or senescence in A2780 xenografts. Further investigations are needed in order to explain the details. In some situations, autophagy and apoptosis seem to be interconnected positively or negatively, introducing the concept of “molecular switches”.19 Undoubtedly, there are multiple connections between the apoptotic and autophagic processes.
In summary, a wide range of novel targeted agents have been developed and are currently under investigation for the treatment of patients with ovarian cancer. Although angiogenesis inhibitors are the most promising therapy for these patients, the present study showed that combination of a selective EGFR inhibitor and a PARP inhibitor was effective in ovarian cancer harboring EGFR overexpression and wild-type BRCA1/2. This combinatorial strategy has provided new insights into the treatment of ovarian cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Heintz AP, Odicino F, Maisonneuve P, et al. Carcinoma of the ovary. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006;1:S161–S192. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics. CA Cancer J Clin. 2014;64(1):9–29. | ||
Bartlett JM, Langdon SP, Simpson BJ, et al. The prognostic value of epidermal growth factor receptor mRNA expression in primary ovarian cancer. Br J Cancer. 1996;73(3):301–306. | ||
Fischer-Colbrie J, Witt A, Heinzl H, et al. EGFR and steroid receptors in ovarian carcinoma: comparison with prognostic parameters and outcome of patients. Anticancer Res. 1997;17(1B):613–619. | ||
Sirotnak FM. Studies with ZD1839 in preclinical models. Semin Oncol. 2003;30(1 Suppl 1):12–20. | ||
Ciardiello F, Caputo R, Bianco R, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000;6(5):2053–2063. | ||
Gordon AN, Finkler N, Edwards RP, et al. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: results from a phase II multicenter study. Int J Gynecol Cancer. 2005;15(5): 785–792. | ||
Konner J, Schilder RJ, DeRosa FA, et al. A phase II study of cetuximab/paclitaxel/carboplatin for the initial treatment of advanced-stage ovarian, primary peritoneal, or fallopian tube cancer. Gynecol Oncol. 2008;110(2):140–145. | ||
Gelmon KA, Hirte HW, Robidoux A, et al. Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serious cancer and triple negative breast cancer. J Clin Oncol. 2010;28(15 Suppl):Abstr 3002. | ||
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | ||
Ledermann JA, Harter P, Gourley C, et al. Phase II randomized placebo-controlled study of olaparib (AZD2281) in patients with platinum-sensitive relapsed serous ovarian cancer (PSR SOC). J Clin Oncol. 2011;29 Suppl:Abstr 5003. | ||
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | ||
De Placido S, Scambia G, Di Vagno G, et al. Topotecan compared with no therapy after response to surgery and carboplatin/paclitaxel in patients with ovarian cancer: Multicenter Italian Trials in Ovarian Cancer (MITO-1) randomized study. J Clin Oncol. 2004;22(13):2635–2642. | ||
Savill J, Fadok VA. Corpse clearance defines the meaning of cell death. Nature. 2000;407(6805):784–788. | ||
Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis. 2005;26(2):263–270. | ||
Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. | ||
Vousden KH, Lane DP. P53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–283. | ||
Cheng Y, Qiu F, Tashiro S, Onodera S, Ikejima T. ERK and JNK mediate TNFalpha-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem Biophys Res Commun. 2008;21:376(3):483–488. | ||
Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11(10):1121–1132. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.