")
Back to Journals » OncoTargets and Therapy » Volume 10
Combination of azacitidine and trichostatin A decreased the tumorigenic potential of lung cancer cells
Authors Yang Y, Yin W, Wu FY, Fan J
Received 6 March 2017
Accepted for publication 3 May 2017
Published 14 June 2017 Volume 2017:10 Pages 2993—2999
DOI https://doi.org/10.2147/OTT.S136218
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ingrid Espinoza
Yang Yang,1,* Wei Yin,2,* Fengying Wu,3,* Jiang Fan1
1Department of Thoracic Surgery, Shanghai Pulmonary Hospital, Tongji University, Shanghai, China; 2Key Laboratory of Oral Biomedical Engineering of Ministry of Education, Hospital of Stomatology, School of Stomatology, Wuhan University, Wuhan, China; 3Oncology Department, Shanghai Pulmonary Hospital, Tongji University, Shanghai, China
*These authors contributed equally to this work
Purpose: This study aims to investigate the possibility of using epigenetic inhibitors against lung cancer.
Methods: The changes in the proliferation of human lung cancer cells, NCI-H1975 and NCI-H1299 cells, treated with various doses of inhibitors of DNA methyltransferase (azacitidine [5-AZA]) or histone deacetylase inhibitors (trichostatin A [TSA]) were determined by cell counting. The cell viability of NCI-H1975 and NCI-H1299 cells treated with 5-AZA and/or TSA was measured by the MTT assay. The changes in expression of the AKT signaling pathway molecules caused by the application of 5-AZA and TSA were analyzed through their protein and mRNA levels. A xenograft model was used to observe the effects of 5-AZA and TSA on tumor growth in vivo.
Results: 5-AZA and TSA inhibited the proliferation and viability of NCI-H1975 and NCI-H1299 cells. Their joint application significantly influenced the expression of key molecules in AKT signaling pathway in vitro, and inhibited the growth of xenograft tumors in vivo. Furthermore, TSA and 5-AZA decreased the tumorigenic ability of NCI-H1975 cells in vivo.
Conclusion: The decreased cell viability and tumorigenic ability, as well as increased anti-oncogene expression following the joint application of 5-AZA and TSA, make these epigenetic inhibitors prospective therapeutic agents for lung cancer.
Keywords: lung cancer, epigenetic inhibitor, azacitidine (5-AZA), trichostatin A
Introduction
Lung cancer, the deadliest type of cancer for both males and females, has the highest prevalence among all cancers. In 2012, 13% of diagnosed cancers were lung cancer, which was responsible for 1.6 million deaths worldwide.1 Epidemiological research indicates that tobacco use, environmental pollution, and genetic factors are the leading pathological causes of lung cancer. Recently, several studies have suggested that smoking can influence DNA methylation in lung tissue. The methylation of the aryl-hydrocarbon receptor repressor (AHRR) gene, which is involved in the metabolism of cigarette smoke components, was 20% lower in current smokers than in individuals who had never smoked.2 Moreover, there is growing evidence that air pollutants which have been officially identified as carcinogenic by the World Health Organization, can induce epigenetic changes which may be implicated in carcinogenesis. PM2.5 exposure, a risk factor for lung cancer, has been demonstrated to influence DNA methylation in a gene-specific manner.3 The previously mentioned findings suggest that epigenetic modifications play a critical role in lung tumorigenesis.
Epigenetics refers to the heritable changes which are not caused by changes in DNA nucleotide sequence. Epigenetic variabilities, including DNA methylation and histone modifications, have been demonstrated to be involved in several human tumors.4 DNA methylation refers to the biochemical process where methyl groups are added onto the cytosine or adenine DNA nucleotides. It is essential for normal development and is associated with a number of key processes including genomic imprinting and suppression of repetitive elements. Histone modifications have important functions in chromatin assembly and their alterations are among the major causes of altered gene expression leading to tumorigenesis.5 Histone deacetylase (HDAC), which catalyzes the removal of acetyl groups on the histone tail and results in a transcriptional inactive heterochromatic state, is the most common histone modification.
Presently, DNA hypermethylation, especially in CpG islands of tumor suppressor gene promoters, has been identified in human tumors including bladder,6 breast,7–9 and lung cancer.10 Furthermore, hypermethylation has been regarded as an early event in lung tumorigenesis.11
Epigenetic changes supply not only a causative factor for tumor development but also a potential strategy for therapeutic intervention, as epigenetic alterations are reversible. DNA methyltransferase (DNMT) inhibitors can induce DNA hypomethylation at specific gene loci resulting in sustained gene reactivation.12 Currently, several inhibitors of DNMT and histone deacetylase (HDAC) are in clinical use and/or clinical testing against various malignancies.13
The cross-talk among different epigenetic activities provides the basis for using a combination of epigenetic agents. In this report, we determined the optimal dosages of DNMT- and HDAC-inhibitors, azacitidine (5-AZA) and trichostatin A (TSA) for inhibiting the growth of lung cancer cells. We then investigated the changes in expression of the AKT signaling pathway molecules caused by 5-AZA and TSA. Our findings shed light on the role of 5-AZA and TSA in lung cancer progression, and provide potential therapeutic agents for the treatment of lung cancer in the near future.
Materials and methods
Ethical approval
This study was designed and conducted in accordance with the amended Declaration of Helsinki. This study was approved by the Institutional Review Board (IRB) of Shanghai Pulmonary Hospital affiliated with Tongji University prior to the commencement of the study. In vivo experiments were also approved by the IRB of Shanghai Pulmonary Hospital affiliated with Tongji University. All experiments were performed following the approved guidelines and guidelines recommended by the Animal Care and Use Committee in Shanghai Pulmonary Hospital affiliated with Tongji University.
Cell culture
Lung cancer cell lines, NCI-H1975 and NCI-H1299, were purchased from American Type Culture Collection (CRL 5908). NCI-H1975 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin. NCI-H1299 cells were cultured in DMEM supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37°C with 5% CO2. All media for cell culture were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA).
Cell growth and viability
NCI-H1975 and NCI-H1299 cells (5×103) were seeded on a 96-well plate. They were treated with 0.1, 1, 5, 10, and 20 μM 5-AZA for 48 h and 72 h and 0.1, 0.3, 0.5, and 1 μM TSA for 24 h and 48 h. Cell counting was used to evaluate the changes in cell growth. Cell viability was estimated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) metabolism assay performed using the commercially available Vybrand® MTT Cell Proliferation Assay Kit (Molecular Probes, Thermo Fisher Scientific). In brief, 0.5 mg/mL MTT was added to each well and incubated at 37°C for 4 h. Next, 100 μL DMSO was used to terminate the reaction. Cell viability was evaluated based on the OD490 data.
Changes in mRNA and protein expression
The optimal doses and treatment times of 5-AZA and TSA were determined based on the results of the cell viability assay. Following treatment with the inhibitors, cells were divided into two parts. One part was used to extract the total protein. The total RNA was extracted from the other part using Trizol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. The changes in expressions of target proteins were analyzed by Western blotting with the corresponding antibodies. The total RNA concentration was quantified using NanoDrop 1000 (Nanodrop, Wilmington, DE, USA). Reverse transcription reaction was performed with 500 ng of the total RNA using the TAKARA Reverse Transcription kit. The mRNA expressions of target genes were determined by real-time polymerase chain reaction (PCR) using SYBR® qPCR Mix and the ABI 7500 Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific). β-actin was used as the internal control. The entire experiment was performed in triplicate.
Xenograft model
Eight-week-old male athymic immunodeficient BALB/c nude mice were purchased from the Shanghai Laboratory Animal Center. TSA (0.3 μM) and 5-AZA (5 μM) treated or untreated NCI-H1975 (5×107) cells were injected subcutaneously into the left flank of the mice.
Statistical analysis
Gene expression at the mRNA level was presented as the mean ± SD. The Student’s t-test was used to compare the expression differences between the control and drug groups. All data were analyzed by the SPSS package for Windows (Version 18.0, SPSS Inc., Chicago, IL, USA). P<0.05 was considered statistically significant.
Results
Effects of 5-AZA and TSA on the proliferation and viability of lung cancer cells
We evaluated the effects of 5-AZA and TSA on human lung cancer cells by analyzing cell proliferation and viability. 5-AZA did not affect the viability of NCI-H1975 cells at day 2. Interestingly, 10 μM of 5-AZA seemed to promote the proliferation of NCI-H1975 cells. However, at day 3, 5-AZA exhibited evident inhibition of NCI-H1975 cell proliferation. TSA inhibited the growth of NCI-H1975 cells at 24 h, and further decreased growth at 48 h. Similar inhibitory effects were also observed in NCI-H1299 cells with the application of 5-AZA and TSA. The decrease in NCI-H1975 cells’ growth was approximately 30% during treatment with 5 μM 5-AZA for 72 h or 0.3 μM TSA for 24 h. Hence, these were set as the optimal doses for all further experiments (Figure 1).
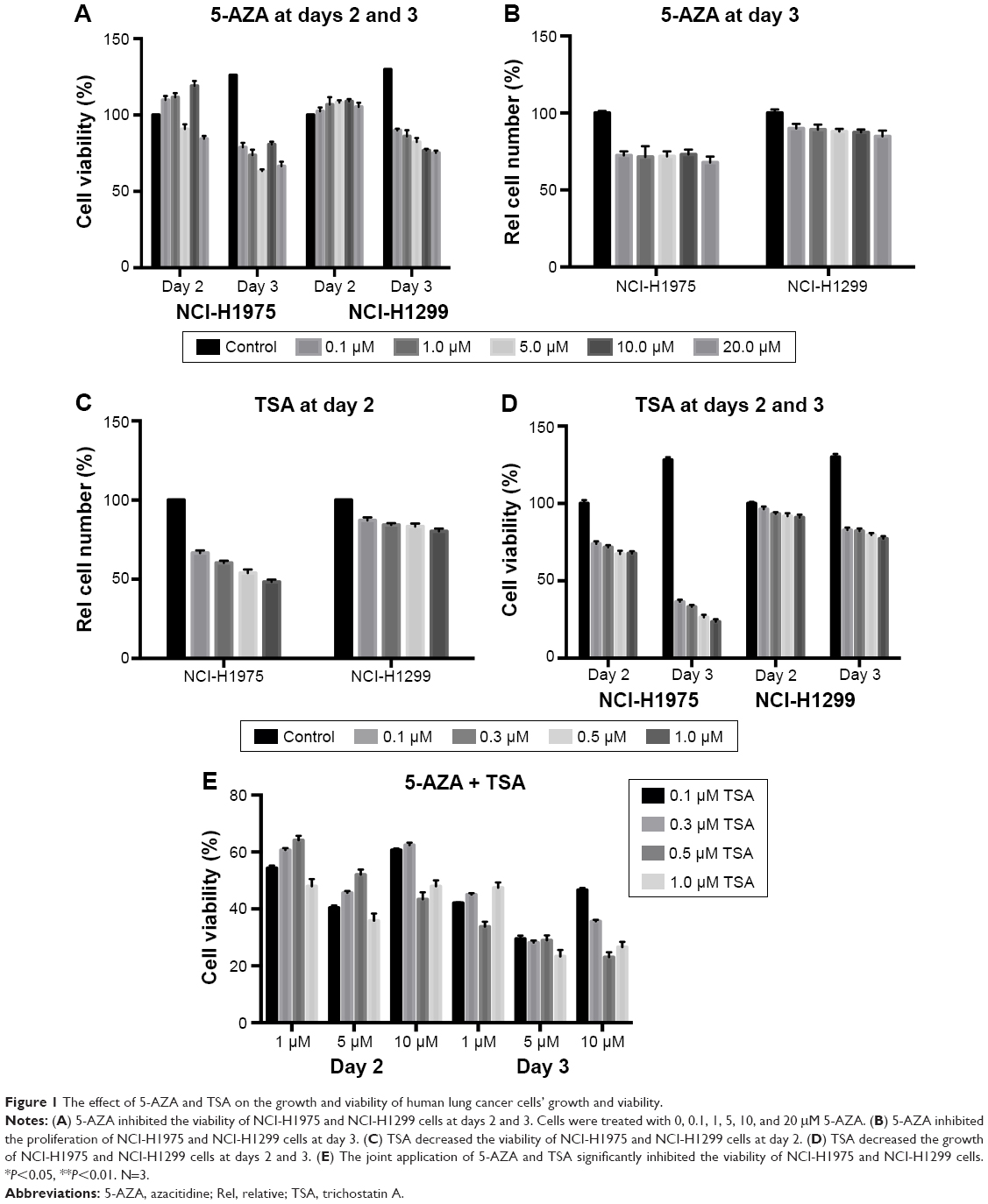 | Figure 1 The effect of 5-AZA and TSA on the growth and viability of human lung cancer cells’ growth and viability. |
Changes in expressions of anti-oncogenes
After determining the appropriate doses of 5-AZA and TSA, we further analyzed the effects of 5-AZA and TSA on lung cancer cells. We screened previously reported molecules targeted by epigenetic modifications which were involved in tumorigenesis. TFF1 and VCAM1 were detected at significantly increased levels compared to those in the control groups. Next, we examined changes in expressions of the upstream signaling molecules which modulate TFF1 and VCAM1 levels. Our data suggested that the expressions of key molecules in the AKT1 signaling pathway were influenced by 5-AZA and TSA. Furthermore, the mRNA expression of TFF1and VCAM1 genes were evidently increased (Figure 2).
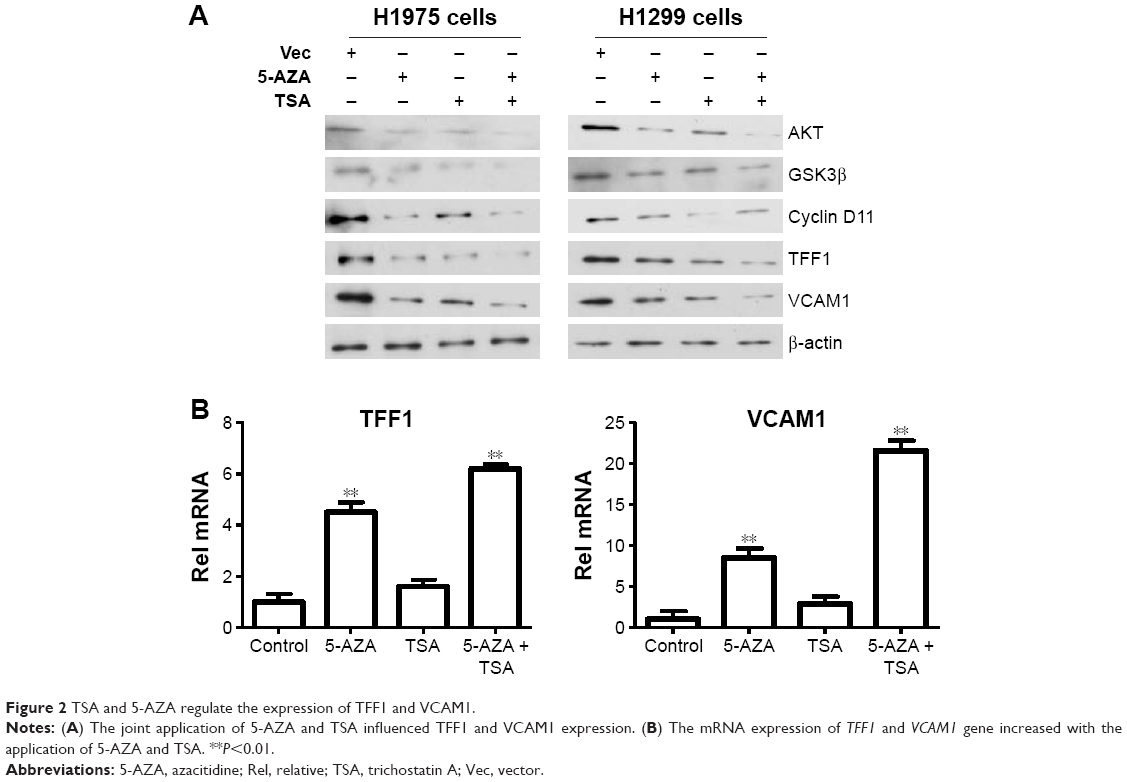 | Figure 2 TSA and 5-AZA regulate the expression of TFF1 and VCAM1. |
TSA and 5-AZA decreased the tumorigenic ability of NCI-H1975 cells in vivo
Next, we observed the effects of TSA and 5-AZA on the tumorigenic ability of NCI-H1975 cells in nude mice. The xenograft model was observed at 8 days post-injection in the control group, while in the TSA- and 5-AZA-treated group, the tumor did not appear until 14 days post-injection. Specimens were harvested at 28 days post-injection. Treatment with TSA and 5-AZA significantly inhibited the tumor volume (Figure 3).
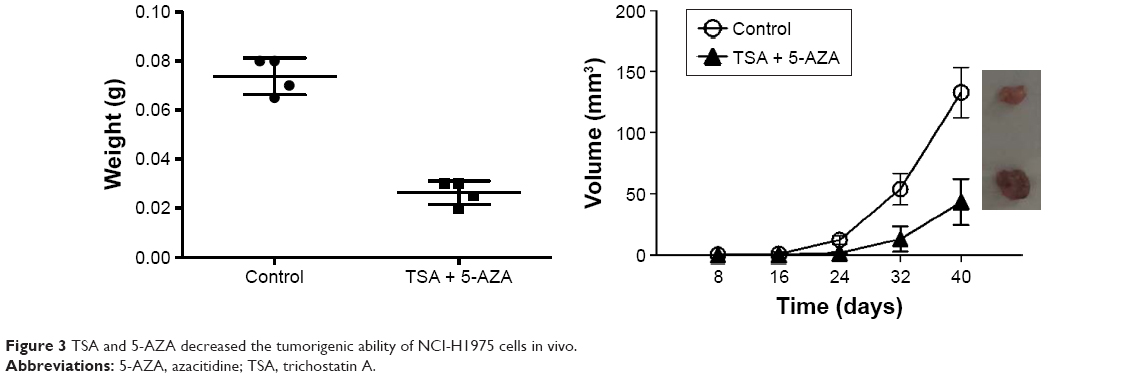 | Figure 3 TSA and 5-AZA decreased the tumorigenic ability of NCI-H1975 cells in vivo. |
Discussion
According to histological analysis, lung cancer is classified as small-cell lung carcinoma and non-small-cell lung carcinoma (NSCLC), which has three main subtypes: adenocarcinoma, squamous-cell carcinoma, and large-cell carcinoma. Various alterations which drive tumorigenesis are involved in the development of lung cancer, which is a molecularly complex and heterogeneous disease. In addition to DNA sequence alterations and copy number alterations, epigenetics also plays a powerful role in cancer development.
Although recent studies have demonstrated that adjuvant chemotherapy improved the survival rate in completely resected NSCLC patients, only a small fraction of the treated individuals ultimately benefited from it.14 Limited understanding of the molecular mechanisms underlying lung carcinogenesis has hindered the application of effective and specific therapeutic reagents. Improvements in our knowledge of molecular alterations at the genetic, epigenetic, and protein level have the potential to influence lung cancer diagnosis and treatment. Gene-specific epigenetic modifications have been reported with high frequency in lung tumors. Therefore, it would be advantageous to investigate the cellular consequences of normal and aberrant epigenetics in lung tumors.
In premalignant and malignant states, promoter methylation is commonly observed in genes involved with crucial functions, including cell cycle control, proliferation, apoptosis, cell adhesion, motility, and DNA repair.15 DNA methylation at CpG dinucleotides in gene promoter regions is a major mechanism of regulating gene expression. A number of novel tumor suppressor genes have been found which were inactivated by promoter hypermethylation. Genome-wide DNA methylation profiling of NSCLC has been used to identify the increased promoter methylation levels.16 More than 30% of lung cancer patients have promoter hypermethylation of the RASSF1A gene.17 Promoter methylation of the ESR1 gene has been associated with stage I NSCLC, which suggests that it may be involved in tumorigenesis.18 HDACs are also overexpressed in lung cancer.19,20 These hypermethylated tumor suppressor genes associated with lung cancer are also frequently hypermethylated in other types of tumors.21
The fact that epigenetic processes can be reverted, provides the rationale for using chromatin re-modeling agents to restore the normal expression of anti-oncogenes. The successful treatment of myelodysplastic syndromes (MDS) with DNMT inhibitors, AZA, and decitabine, warrants increased attention to their epigenetic roles in tumorigenesis and treatment. AZA, a demethylating agent, is the first therapeutic agent demonstrated to have a verified survival benefit for patients with MDS.22 AZA is one of the few epigenetic medicines which have been approved by the US Food and Drug Administration for routine clinical treatment.23 However, in several solid tumor malignancies, the application of AZA showed varying results. Nevertheless, the recent preclinical success of inhibitors of BRD4, an acetyl-lysine chromatin-binding protein, has made epigenetic cancer therapies more promising.24,25 The synergy between DNA methylation and histone modifications may begin in the early stages of tumorigenesis.26,27 Therefore, applying a combination of drugs inhibiting both these processes may work better than application of a single drug. In this study, we observed that 5-AZA and/or TSA treatments could inhibit tumor cell proliferation. The combination of these two drugs was more effective and produced better results than those with single drug treatment. The co-treatment induced significant upregulation of the tested genes. Decreased cell viability and increased anti-oncogene expression aids in the suppression of tumor growth. TFF1 is one of the trefoil factor family members which contain a trefoil domain with cysteine residues and disulfide bridges. It is an epithelial protector and can restitute mucous membranes.28,29 It was reported as an effective marker which distinguished lung carcinoma from breast carcinoma.30 In lung cancer, TFF1 was found to be associated with improved survival of patients.31 Therefore, lung cancer patients may benefit from the increased TFF1 levels caused by using 5-AZA and TSA. However, further study is needed to validate this hypothesis.
In summary, our study suggests that the combined application of 5-AZA and TSA may be a promising therapeutic strategy for lung cancer. Expanding our understanding of how epigenetic events result in the genesis of lung cancer, and the application of this understanding to clinical treatments, will enhance our ability to properly and effectively manage lung cancer and, ultimately, to reduce the heavy global burden of this devastating disease in the future.
Acknowledgments
This work was supported by grants from the National Natural Scientific Foundation of China (81370107, 81602412, and 81501750), the Natural Scientific Foundation of Shanghai Municipal Commission of Health and Family Planning (20134279), the Scientific Research Project of Shanghai Municipal Commission of Health and Family Planning (20164Y0121), and the Scientific Research Project of Science and Technology Commission of Shanghai Municipality (134119b1002). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Global battle against cancer won’t be won with treatment alone – effective prevention measures urgently needed to prevent cancer crisis. Cent Eur J Public Health. 2014;22(1):23–28. | ||
Shenker NS, Ueland PM, Polidoro S, et al. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology. 2013;24(5):712–716. | ||
Hou L, Zhang X, Zheng Y, et al. Altered methylation in tandem repeat element and elemental component levels in inhalable air particles. Environ Mol Mutagen. 2014;55(3):256–265. | ||
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–298. | ||
Pandey M, Sahay S, Tiwari P, Upadhyay DS, Sultana S, Gupta KP. Involvement of EZH2, SUV39H1, G9a and associated molecules in pathogenesis of urethane induced mouse lung tumors: potential targets for cancer control. Toxicol Appl Pharmacol. 2014;280(2):296–304. | ||
Marsit CJ, Karagas MR, Danaee H, et al. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. 2006;27(1):112–116. | ||
Avraham A, Uhlmann R, Shperber A, et al. Serum DNA methylation for monitoring response to neoadjuvant chemotherapy in breast cancer patients. Int J Cancer. 2012;131(7):E1166–E1172. | ||
Buhmeida A, Merdad A, Al-Maghrabi J, et al. RASSF1A methylation is predictive of poor prognosis in female breast cancer in a background of overall low methylation frequency. Anticancer Res. 2011;31(9):2975–2981. | ||
Cho YH, Shen J, Gammon MD, et al. Prognostic significance of gene-specific promoter hypermethylation in breast cancer patients. Breast Cancer Res Treat. 2012;131(1):197–205. | ||
Rauch TA, Zhong X, Wu X, et al. High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc Natl Acad Sci U S A. 2008;105(1):252–257. | ||
Lyn-Cook L, Word B, George N, Lyn-Cook B, Hammons G. Effect of cigarette smoke condensate on gene promoter methylation in human lung cells. Tob Induc Dis. 2014;12(1):15. | ||
Raynal NJ, Si J, Taby RF, et al. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 2012;72(5):1170–1181. | ||
Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. | ||
Hotta K, Matsuo K, Ueoka H, Kiura K, Tabata M, Tanimoto M. Role of adjuvant chemotherapy in patients with resected non-small-cell lung cancer: reappraisal with a meta-analysis of randomized controlled trials. J Clin Oncol. 2004;22(19):3860–3867. | ||
Langevin SM, Kratzke RA, Kelsey KT. Epigenetics of lung cancer. Transl Res. 2015;165(1):74–90. | ||
Karlsson A, Jonsson M, Lauss M, et al. Genome-wide DNA methylation analysis of lung carcinoma reveals one neuorendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin Cancer Res. 2014;20(23):6127–6140. | ||
Ko E, Lee BB, Kim Y, et al. Association of RASSF1A and p63 with poor recurrence-free survival in node-negative stage I–II non-small cell lung cancer. Clin Cancer Res. 2013;19(5):1204–1212. | ||
Lin Q, Geng J, Ma K, et al. RASSF1A, APC, ESR1, ABCB1 and HOXC9, but not p16INK4A, DAPK1, PTEN and MT1G genes were frequently methylated in the stage I non-small cell lung cancer in China. J Cancer Res Clin Oncol. 2009;5(12):1675–1684. | ||
Nakagawa M, Oda Y, Eguchi T, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18(4):769–774. | ||
Bartling B, Hofmann HS, Boettger T, et al. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer. 2005;49(2):145–154. | ||
Herceg Z, Vaissière T. Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics. 2011;6(7):804–819. | ||
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232. | ||
Barrow TM, Michels KB. Epigenetic epidemiology of cancer. Biochem Biophys Res Commun. 2014;455(1–2):70–83. | ||
Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. | ||
Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904-917. | ||
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. | ||
Capobianco E, Mora A, La Sala D, et al. Separate and combined effects of DNMT and HDAC inhibitors in treating human multi-drug resistant osteosarcoma HosDXR150 cell line. PLoS One. 2014;9(4):e95596. | ||
Kjellev S. The trefoil factor family – small peptides with multiple functionalities. Cell Mol Life Sci. 2009;66(8):1350–1369. | ||
Davidson B, Stavnes HT, Risberg B, et al. Gene expression signatures differentiate adenocarcinoma of lung and breast origin in effusions. Hum Pathol. 2012;43(5):684–694. | ||
Yang M, Nonaka D. A study of immunohistochemical differential expression in pulmonary and mammary carcinomas. Mod Pathol. 2010;23(5):654–661. | ||
Dietrich D, Hasinger O, Liebenberg V, Field JK, Kristiansen G, Soltermann A. DNA methylation of the homeobox genes PITX2 and SHOX2 predicts outcome in non-small-cell lung cancer patients. Diagn Mol Pathol. 2012;21(2):93–104. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.