")
Back to Journals » The Application of Clinical Genetics » Volume 12
COL2A1 Gene Mutations: Mechanisms of Spondyloepiphyseal Dysplasia Congenita
Authors Nenna R , Turchetti A, Mastrogiorgio G, Midulla F
Received 16 August 2019
Accepted for publication 23 November 2019
Published 4 December 2019 Volume 2019:12 Pages 235—238
DOI https://doi.org/10.2147/TACG.S197205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Raffaella Nenna,1 Arianna Turchetti,1 Gerarda Mastrogiorgio,2 Fabio Midulla1
1Department of Paediatrics, Sapienza University, Rome, Italy; 2Department of Biomedicine and Prevention, University of Rome “Tor Vergata”, Rome, Italy
Correspondence: Raffaella Nenna
Department of Paediatrics, “Sapienza” University of Rome, V.le Regina Elena 324, Rome 00161, Italy
Tel +390649979363
Email [email protected]
Abstract: The COL2A1 gene consists of 54 exons spanning over 31.5 kb and encodes for type II collagen. Type II collagen is the main component of hyaline cartilage extracellular matrix, nucleus pulposus of intervertebral discus, vitreous humor of the eye and inner ear structure. Molecular defects in COL2A1 gene cause a wide variety of rare autosomal-dominant conditions known as type II collagenopathies. A clear genotype–phenotype relationship is not yet known. However, some correlations are described. Spondyloephyseal dysplasia congenita was suggested for a short-trunk dwarfing condition affecting primarily the vertebrae and the proximal epiphyses of the long bones.
Keywords: COL2A1 gene, type II collagen, spondyloepiphyseal dysplasia congenita
COL2A1 gene (12q13.1-q13.2) consists of 54 exons spanning over 31.5 kb and encodes for type II collagen, a 1487-amino acid (134.4 kDa) protein.1
Type II collagen, a large homotrimeric protein, is the main component of hyaline cartilage extracellular matrix (95% of the collagens and approximately 60% of dry weight in adults), nucleus pulposus of intervertebral discus, vitreous humor of the eye and inner ear structure. It plays a fundamental role on the endochondral bone formation and growth. In the cartilage growth plate, it is synthesized by proliferating chondrocytes until they differentiate on hypertrophic chondrocytes.
Type II collagen acts as an autocrine factor of proliferation and differentiation via multiple downstream effectors and a potent suppressor for chondrocyte hypertrophy and apoptosis through the negative regulation of SMAD1 activity.2
Type II collagen molecules have three identical α1-polypeptide chains of 1060 amino acid residues each, with a large uninterrupted triple-helical region and relatively short, non-helical telopeptides (19 amino acid residues in the N-telopeptide and 27 amino acid residues in the C-telopeptide) that do not possess the Gly-X-Y-repeating primary structure found in the triple-helical region. “X” and “Y” positions are frequently occupied by proline and hydroxyproline residues, respectively. N- and C-telopeptide regions allow the initiation of the triple-helical configuration.
Type II collagen molecules spontaneously self-assemble into fibrils, which, together with other macromolecules, form the extracellular scaffold critical for the integrity and biomechanical functions of cartilage.3 The fibrils are cross-linked to form mature type II collagen fibers.
In vivo, the polymerization of collagen molecules into fibrils involves cellular and specific extracellular matrix interactions. Proteoglycans, such as decorin, fibromodulin, and biglycan, bind type II collagen fibrils to stabilize the larger fibril bundles (fibers composed of multiple fibrils).
Molecular defects in COL2A1 gene cause a wide variety of rare autosomal-dominant conditions known as type II collagenopathies.4 So far more than 400 mutations have been described in public databases and previous literature (329 pathogenic variants, 153 variants of uncertain significance). Molecular spectrum of alterations includes point mutations (missense, nonsense, deletion, insertion, insertion-deletion and frame-shift mutation) and complex rearrangements5–7 (Table 1). No mutational hot spots within COL2A1 gene have been identified and the severity of the phenotype can be explained by the nature of mutation and the localization in the protein. We used the reference sequence NM 001844.4 combined with mutalyzer release 2.0.6 (https://mutalyzer.nl/), which is a program suite that examines the sequence variant nomenclature according to HGVS guidelines.
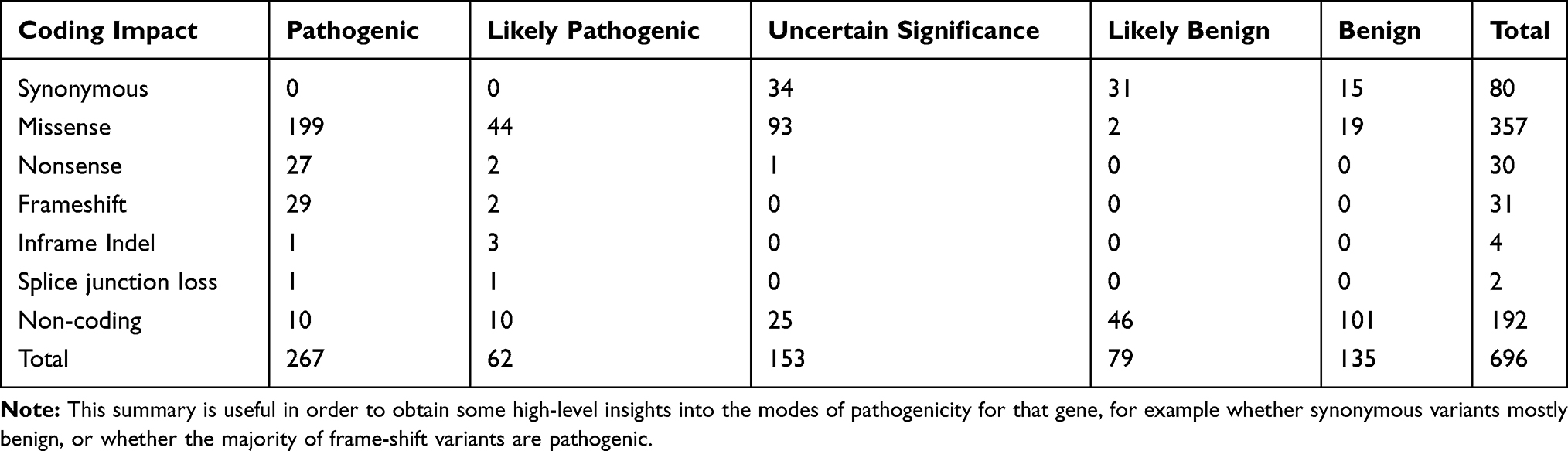 |
Table 1 Summary of the Variants Reported in UniProt, ClinVar & VarSome User Entries for a Given Gene (Version: 18_oct_2019) |
A clear genotype–phenotype relationship is not yet known. However, some correlations are described. Type II collagenopathies dominant inheritance basically depends on two molecular mechanisms: dominant-negative mutants and haploinsufficiency. The most common mutation (over 70%, Table 1) is missense mutation, some of which lead to substitution of glycine residue in the Gly-X-Y repeat, presenting as dominant-negative effect,8 generally seen in the more severe collagen type II disorders. A few truncating and some exon-skipping mutations have been reported to cause more severe type II collagenopathies.9 Missense mutations leading to other amino acids than glycine substitution causes generally milder phenotype due to impairment in protein stability, and subsequent damage in helical structure and proper function of type II collagen.
Haploinsufficiency is a mechanism due to non-sense substitutions or out-of-frame deletions, resulting in premature stop codons which cause reduced synthesis of normal collagen. These mutations are associated with milder phenotypes.
Furthermore, phenotypic variation is likely caused by environmental factors and the polymorphisms in disease-modifying genes and/or regulatory elements.
Type II collagenopathies clinical features show a wide range of severity and complexity.
Moreover, several type II collagenopathies clinical features are shared by other syndromes due to defects in other components of cartilage (eg, otospondylomegaepiphyseal dysplasia caused by COL11A2 mutation, multiple epiphyseal dysplasia principally caused by COMP mutation).10,11
Phenotypic overlap in COL2A1-related disorders and wide inter- and intra-familiar phenotypic variation have been commonly reported.
At one end of the spectrum, achondrogenesis type II (ACG2)/hypochondrogenesis and platyspondylic lethal skeletal dysplasia, Torrance type (PLSDT) are perinatally lethal conditions. They are characterized by micromelia, narrow chest with pulmonary hypoplasia, absent ossification of vertebras bodies and sacrum, Pierre Robin sequence and several visceral anomalies. At the other end of the spectrum are listed some conditions typical of adolescent or adult age: avascular necrosis of the femoral head (ANFH), Legg-Calvè-Perthes disease, early-onset osteoarthritis (OA), Strickler syndrome type1 (STL1), vitreoretinopathy with phalangeal epiphyseal dysplasia (VPED). These conditions are characterized by normal stature and early development of arthrosis or ocular defects. A third group of type II collagenopathies shows at birth or during childhood clinical features of spondyloepiphyseal dysplasia with wide variety of severity. Flat midface and anomalies of eye and inner ear are also common in this group.
Kniest dysplasia, spondyloepimetaphyseal dysplasia (SEMD) Strudwick type and Algerian type, and spondyloepiphyseal dysplasia congenita (SEDC) are characterized by dwarfism caused by delayed ossification of the vertebrae and pubic bones. Frequently odontoid hypoplasia coexists. Long bones are short and kyphoscoliosis develops in childhood.
On the contrary, in spondyloperipheral dysplasia (SPPD), patients only show short stature associated with important lumbar lordosis, in Czech dysplasia (CD) instead hallmarks are broad or prominent knees and wide metatarsals and phalanges in the third or fourth toe.
In 1966, the spondyloephyseal dysplasia congenita was suggested by Spranger and Wiedemann12 for a short-trunk dwarfing condition affecting primarily the vertebrae and the proximal epiphyses of the long bones.
SEDC is a rare disease with a prevalence of 3.4/1,000,000. At birth patients with SEDC are short (mean length 45 cm at term) with flattened vertebras; ossification is absent in pubic bones and distal femoral epiphyses, absent or reduced in cervical and sacral vertebras. The iliac bones are short in length, with acetabular roof more horizontal than normal and without normal flaring of iliac wings. Infants with severe SEDC are often stillborn or premature and die shortly after birth because of hypoventilation. The developing respiratory failure can be temporarily overcome only by delivering intensive ventilator support. Recently13 we describe a case of a child that exceptionally survived to 13.5 years with the placement of airway stenting. The characteristics of the child (small rib cage, severe tracheo-bronchomalacia, hypo-expanded lungs, respiratory muscle failure and marked abdominal distention) in fact, led the child to suffer from a chronic severe obstructive respiratory disease that were constantly monitored and treated by endoscopic surgical procedures.
Frequently patients suffer from atlantoaxial instability due to odontoid hypoplasia that can cause cervical cord compression, especially when repairing during intubation or surgery.14,15
Shortly after birth, platispondyly of the lower thoracic and lumbar vertebral bodies develops. This ultimately leads to wedge-shaped thoracic vertebrae and severe kyphoscoliosis with lumbar lordosis. Tubular bones, except hands, are short with delayed and dysplastic epiphyseal ossification. From this it follows short-trunk dwarfism (mean adult height is 140 cm). Adult patients also show flat face for skeletal hypoplasia with prominent eyes and cleft palate, barrel-chest and pectus carinatum. Their hands are normal but coxa vara, dislocated hips, talipes equinovarus, clubfeet and waddling gate are reported. Hypoplastic abdomen, mitral prolapse have also been reported.
Adult patients suffer from sensorineural (25% to 30% of reported cases) or less frequently mixed hearing loss. Ocular complications such as myopia have been reported in 45% of patients but retinal detachment is less frequent (12%) than in type 1 Strickler syndrome.16
In spondyloephyseal dysplasia congenita over 100 COL2A1 mutations have been described. Most common are in the triple helix (74% glycine substitutions and 10% Arg-to-Cys changes) and are dominantly inherited while only few mutations have been found involving C-peptide region. Furthermore, recently also a recessive pattern has been demonstrated.5
Differently from osteogenesis imperfecta,15 in SEDC patients with glycine substitutions in the triple helical domain, it is not reported any amino-to-carboxyterminal gradient in the radiological phenotype severity.17
Pathophysiological mechanisms and relationship between gene mutations and cartilage/bone defects are largely speculative. Only few data result from studies in mice carrying spontaneous missense mutations in COL2A1 gene18 or in transgenic mice harboring human gene COL2A1 mutations. In transgenic mice mutations with dominant-negative effect19 and mutations causing haploinsufficiency have been generated.20 In these models, the delay of ossification was observed very precociously in fetus development. The grow plates show severe alterations. Proliferative and hypertrophic zones of cartilage were shorter or indistinguishable and deposition of cartilage matrix is notably impaired, collagen fibrils were fewer and less elaborate.
Mutant type II collagen molecules show altered electrophoretic mobility, relatively low thermostability and slow rates of secretion into the extracellular space. They self-assemble in abnormal fibrils which are not able to properly interact with other elements of the extracellular matrix. Proper fibrillar architecture and mechanical characteristics of the interterritorial and pericellular collagenous matrix are critical for a correct columnar arrangement of chondrocytes at the growth plate.21 In transgenic mice, moreover, chondrocytes show greatly extended cisternae of rough endoplasmic reticulum with a retention of procollagen and other molecules (eg, fibronectin). This retention hence causes endoplasmic reticulum stress sufficient to reduce proliferation rate at the growth plates.22,23 Absence or marked reduction in the mRNA expression of chondrocyte markers, including Cdkn1a, Ihh, Fgfr3, COL10A1, and Runx2, has also been reported.
The abnormal chondrocyte differentiation negatively affects linear bone growth altering the normal cells relationships and provision of growth factors during endochondral ossification.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Huerre-Jeanpierre C, Mattei MG, Weil D, et al. Further evidence for the dispersion of the human fibrillary collagen genes. Am J Hum Genet. 1986;38:26–37.
2. Lian C, Wang X, Qiu X, et al. Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin β1–SMAD1 interaction. Bone Res. 2019;7:8. doi:10.1038/s41413-019-0046-y
3. Antipova O, Orgel JPRO. In situ D-periodic molecular structure of type II collagen. J Biol Chem. 2010;285(10):7087–7096. doi:10.1074/jbc.M109.060400
4. Spranger J, Winterpacht A, Zabel B. The type II collagenopathies: a spectrum of chondrodysplasias. Eur J Pediatr. 1994;153(2):56–65. doi:10.1007/bf01959208
5. Barat-Houari M, Sarrabay G, Gatinois V, et al. Mutation update for COL2A1 gene variants associated with type II collagenopathies. Human Mutat. 2016;37:7–15. doi:10.1002/humu.22915
6. Kannu P, Bateman J, Savarirayan R. Clinical phenotypes associated with type II collagen mutations. J Paediatr Child Health. 2012;48(2):E38–E43. doi:10.1111/j.1440-1754.2010.01979.x
7. Xu L, Qiu X, Zhu Z, Yi L, Qiu Y. A novel mutation in COL2A1 leading to spondyloepiphyseal dysplasia congenita in a three-generation family. Eur Spine J. 2014;23(Suppl 2):271–277. doi:10.1007/s00586-014-3292-0
8. Zankl A, Neumann L, Ignatius J, et al. Dominant negative mutations in the C-propeptide of COL2A1 cause platyspondylic lethal skeletal dysplasia, Torrance type, and define a novel subfamily within the type 2 collagenopathies. Am J Med Genet. 2005;133A(1):61–67. doi:10.1002/ajmg.a.30531
9. Nishimura G, Haga N, Kitoh H, et al. The phenotypic spectrum of COL2A1 mutations. Hum Mutat. 2005;26(1):36–43. doi:10.1002/humu.v26:1
10. Chakchouk I, Grati M, Bademci G, et al. Novel mutations confirm that COL11A2 is responsible for autosomal recessive non-syndromic hearing loss DFNB53. Mol Genet Genomics. 2015;290(4):1327–1334. doi:10.1007/s00438-015-0995-9
11. Liu HY, Xiao JF, Huang J, et al. Diagnosis with multiple epiphyseal dysplasia using whole-exome sequencing in a chinese family. Chin Med J (Engl). 2017;130(1):104–107. doi:10.4103/0366-6999.196568
12. Spranger J, Wiedemann HR. Dysplasia spondyloepiphysaria congenita. Helv Paediatr Acta. 1966;21:598.
13. Nenna R, Midulla F, Masi L, et al. Airway stenting in a child with spondyloepiphyseal dysplasia congenita:13-year survival. Int J Pediatr Otorhinolaryngol. 2017;99:13–16. doi:10.1016/j.ijporl.2017.05.008
14. Isik D, Guner S, Avcu S, et al. A case report of a patient with cleft palate carrying the risk of tetraplegia. Cleft Palate Craniofac J. 2011;48(6):773–775. doi:10.1597/09-239
15. Al Kaissi A, Ryabykh S, Pavlova OM, et al. The management of cervical spine abnormalities in children with spondyloepiphyseal dysplasia congenita: observational study. Medicine (Baltimore). 2019;98(1):e13780. doi:10.1097/MD.0000000000013780
16. Turner LM, Steffensen TS, Leroy L, Gilbert-Barness E. Spondyloepiphyseal dysplasia congenita. Fetal Pediatr Pathol. 2010;29(1):57–62. doi:10.3109/15513810903266310
17. Rauch F, Lalic L, Roughley P, Glorieux FH. Genotype-phenotype correlations in nonlethal osteogenesis imperfect caused by mutations in the helical domain of collagen type I. Eur J Hum Genet. 2010;18(6):642–647. doi:10.1038/ejhg.2009.242
18. Terhal PA, Nievelstein RJA, Verver EJ, et al. A study of the clinical and radiological features in a cohort of 93 patients with a COL2A1 mutation causing spondyloepiphyseal dysplasia congenital or a related phenotype. Am J Med Genet. 2015;167A(3):461–475. doi:10.1002/ajmg.a.36922
19. Donahue LR, Chang B, Mohan S, et al. A missense mutation in the mouse COL2A1 gene causes spondyloepiphyseal dysplasia congenita, hearing loss and retinoschisis. J Bone Miner Res. 2003;18(9):1612–1621. doi:10.1359/jbmr.2003.18.9.1612
20. Barbieri O, Astigiano S, Morini M, et al. Depletion of cartilage collagen fibrils in mice carrying a dominant negative COL2A1 transgene affects chondrocyte differentiation. Am J Physiol Cell Physiol. 2003;285(6):C1504–12. doi:10.1152/ajpcell.00579.2002
21. Chung HJ, Jensen DA, Gawron K, et al. R992C (p. R1192C) substitution in collagen II alters the structure of mutant molecules and introduces the unfolded protein response. J Mol Biol. 2009;390(2):306–318. doi:10.1016/j.jmb.2009.05.004
22. Prein C, Warmbold N, Farkas Z, et al. Structural and mechanical properties of the proliferative zone of the developing murine growth plate cartilage assessed by atomic force microscopy. Matrix Biol. 2016;50:1–15. doi:10.1016/j.matbio.2015.10.001
23. Kung LH, Rajpar MH, Preziosi R, et al. Increased classical endoplasmic reticulum stress is sufficient to reduce chondrocyte proliferation rate in the growth plate and decrease bone growth. PLoS One. 2015;10(2):e0117016. doi:10.1371/journal.pone.0117016
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.