")
Back to Journals » Research and Reviews in Parkinsonism » Volume 8
Cognitive deficits in Parkinson’s disease: current perspectives
Authors Cosgrove J, Alty JE
Received 27 October 2017
Accepted for publication 14 November 2017
Published 9 January 2018 Volume 2018:8 Pages 1—11
DOI https://doi.org/10.2147/JPRLS.S125064
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Peter Hedera
Jeremy Cosgrove, Jane Elizabeth Alty
Department of Neurology, Leeds Teaching Hospitals NHS Trust, Leeds, UK
Abstract: Cognitive dysfunction is a common and significant non-motor symptom of Parkinson’s disease (PD). PD mild cognitive impairment (PD-MCI) is evident in approximately one-quarter of patients at the time of PD diagnosis, and half of PD patients have progressed to PD dementia (PDD) after 10 years. The transition to PDD from PD-MCI is not linear and may depend on the facets of cognition affected. Despite increased understanding of pathological, neurotransmitter and genetic drivers, there are no proven pharmacological treatments for PD-MCI and those licensed for PDD are of modest benefit only. Biomarkers to predict those most at risk of developing PDD are under investigation and are likely to be essential so that early and individualized treatment can be provided.
Keywords: Parkinson’s disease, mild cognitive impairment, dementia
Introduction
Modern understanding has changed the perception of Parkinson’s disease (PD) from a pure movement (or motor) disorder to a multisystem disease. PD comprises motor and non-motor symptoms (NMSs) and involves changes to dopaminergic, noradrenergic, serotonergic and cholinergic neurotransmitter systems within the brain. A range of NMSs are described in PD, and among the most common is cognitive impairment. A spectrum of cognitive deficit is associated with PD, from subjective cognitive decline (PD-SCD) to mild cognitive impairment (PD-MCI) and dementia (PDD). Longitudinal cohort studies have demonstrated that approximately half of those diagnosed with PD for 10 years develop PDD,1–3 while the point prevalence of dementia among those with PD is between 35% and 30%.4 In context, PDD accounts for 3%–4% of dementia of all types within the general population, a much smaller percentage than Alzheimer’s disease (AD; 50%–70%) or vascular dementia (15%–25%).5 There is good evidence that PD cognitive impairment reduces quality of life, increases mortality and intensifies caregiver burden.6–8
The construct of PD-MCI was formally defined in 2012 by the International Parkinson and Movement Disorder Society (MDS),9 and it has since been shown that approximately one-third of people have PD-MCI at the time they are diagnosed with PD.10,11 The risk of progression from PD-MCI to PDD is not uniform – PD-MCI may remain static or even reverse rather than always inexorably progress to PDD. Understanding the complex pathological and genetic factors governing transition between normal cognition, PD-SCD, PD-MCI and PDD is an active area of research. One major aim is to identify biomarkers that can accurately predict those PD patients most at risk of developing PDD, as this would allow targeted studies of such people and early delivery of disease-modifying medication, when this becomes available.
Cognitive subtypes in PD
Subjective cognitive decline
SCD is a relatively new term, introduced over the past few years to describe cognitive impairment identified by the patient, their family or health care professionals in the context of normal performance on cognitive tests. There is established evidence that in the general population SCD is a risk factor for amnestic MCI and AD.12 There are currently no established criteria to define this syndrome in PD and limited published research. Small studies have shown that PD-SCD patients are at increased risk of developing PD-MCI compared to those without SCD,13,14 but further research is required.
PD mild cognitive impairment
The 2012 MDS PD-MCI diagnostic criteria9 were published to create a unifying set of diagnostic measures to standardize practice across clinical trials and improve understanding of PD-MCI. The key difference between PD-MCI and PDD is that cognitive deficits are not sufficient to significantly impair activities of daily living in the former. In order to align with the 2007 MDS PDD diagnostic criteria,15 the 2012 MDS PD-MCI criteria contain two diagnostic categories: a “level 1” category diagnosis allows an approved test of global cognitive function to demonstrate deficits, whereas a “level 2” category diagnosis is more rigorous, requiring two neuropsychological tests of the five core cognitive domains most often affected in PD, which are attention and working memory, executive function, language, memory and visuospatial function.9 Deficits need to be identified in at least two tests to make a level 2 diagnosis. The abnormal tests can both be within the same cognitive domain or within different cognitive domains, allowing subtyping of level 2 PD-MCI into single or multiple cognitive domains.
The definition of “impairment” in cognitive tests is not precisely defined by the MDS criteria, and large-scale validation is underway.16 The results will help resolve a number of problems identified and refine them appropriately. For example, there is uncertainty regarding the optimum cut off for impairment values when compared to normative means, which has a marked influence on the number of PD subjects classified as having PD-MCI.11,17 The validity of approved global cognitive screening tests to make a level 1 diagnosis of PD-MCI also needs to be established. To achieve a sensitivity and specificity of 80%, the best of three approved tests in one study (the Montreal Cognitive Assessment [MoCA]) had a diagnostic accuracy of 57% and 68%, respectively.18
The literature looking at the outcome of PD-MCI in longitudinal studies suggests that it is a risk factor for PDD. In one study, PD-MCI was strongly associated with PDD development during 4 years of follow-up after controlling for age, disease stage, education and gender (odds ratio 5.1).19 Three-year follow-up of 182 participants in the Norwegian ParkWest Study cohort showed that 10 of 37 subjects with PD-MCI at baseline had converted to PDD, compared to one of 145 with normal cognition, a relative risk of 39.2.20 However, this study also found that 22% of PD-MCI subjects at baseline reverted to normal cognition, implying that PD-MCI can be reversible. The PD-MCI converters were significantly older and had significantly worse scores on tests of executive function at baseline compared to the PD-MCI reverters.20 More information is needed to understand the transition between normal cognition, PD-MCI and PDD. PD-MCI and PDD appear to share several risk factors such as level of education,21 severity of motor deficit22,23 and male gender.24
The CamPaIGN cohort study assessed PD patients’ cognition at baseline and conversion to dementia at 3.5 and 5.2 years of follow-up; it showed that deficits in semantic fluency and visuospatial function at baseline were risk factors for dementia development but deficits in executive function were not.22,23 This finding, among others, led to the “dual syndrome hypothesis”25,26 which proposes that those with PD-MCI characterized by executive dysfunction, principally driven by changes in dopaminergic pathways, are less likely to transition to PDD. In contrast, those PD-MCI patients with deficits in memory and visuospatial function (Figure 1), caused predominantly by deficits in acetylcholine (ACh), are more prone to rapid cognitive decline and PDD. In newly diagnosed PD, there is some evidence that functional magnetic resonance imaging (fMRI) changes and genetic risk factors support the dual syndrome hypothesis,27 but further longitudinal follow-up, for example, via the ICICLE-PD study,11 is required to test this theory in more detail.
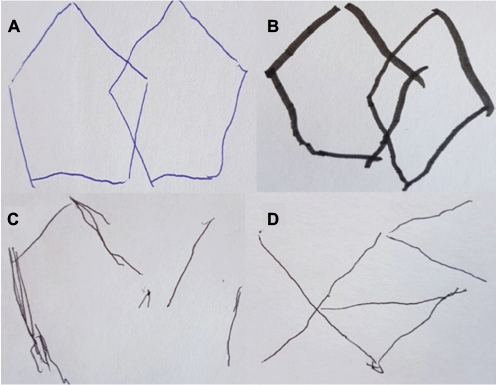 | Figure 1 Visuospatial function is affected in Parkinson’s cognitive impairment. Notes: Progressive difficulty copying interlocking pentagons. Based on level 1 MDS diagnostic criteria, patient “A” has normal cognition, patient “B” has PD-MCI and patients “C” and “D” have PDD. Abbreviations: MDS, Movement Disorder Society; PD, Parkinson’s disease; PDD, PD dementia; PD-MCI, PD mild cognitive impairment. |
PD dementia
The importance of cognitive decline in PD was highlighted when several longitudinal studies demonstrated that approximately 50% of patients have developed dementia 10 years after the initial diagnosis of PD.1–3 The Sydney Multicenter Study – the longest published follow-up of a newly diagnosed PD cohort to date – found that 83% of those alive at 20 years had developed PDD and 75% developed PDD before death,28 suggesting that dementia may be an inevitable consequence of PD if survival allows. However, the 17% of subjects in the study without dementia at 20 years could represent a distinct subgroup with some protection from PDD.
Disentangling the effect of age, age at the onset of PD and duration of disease in cohort studies is complex, but it appears that age, rather than age at the onset of PD, conveys an increased risk of developing PDD (Box 1).29 Those diagnosed with PD earlier in life live for longer before developing dementia,30 and four clinical milestones, including dementia, have been shown to herald the onset of a terminal phase of PD.31
 | Box 1 Summary of risk factors for PDD |
The postural instability gait disorder (PIGD) motor phenotype is associated with a more rapid cognitive decline than the tremor-dominant motor phenotype,23,32 and in some studies, PDD is almost exclusively a condition occurring in PIGD.33 It has been argued that ACh deficit may link cognitive decline – particularly deficits in attention – and PIGD due to degeneration of the nucleus basalis of Meynert (nbM) and the pedunculopontine nucleus (PPN), respectively.34,35
Mechanisms causing cognitive deficits
Understanding the drivers of cognitive deficit in PD is complex and hindered in part by the absence of good animal models. The pathological changes seen are important, but other factors such as alterations to neurotransmitter systems and genetic influences are also significant. Disentangling these factors and their relevance to each individual patient is an unmet need.
Pathological studies
The major pathological driver of cognitive decline in PD is believed to be the presence of Lewy bodies and Lewy neurites within the limbic system and neocortex, although AD-related pathology – tau neurofibrillary tangles (NFTs) and amyloid beta plaques (Aβ) – is also important. The core constituent of Lewy bodies and neurites is abnormal alpha synuclein (α-syn), which causes neurotoxicity via a range of proposed mechanisms including impairment of axonal transport, oxidative stress, mitochondrial changes and synaptic dysfunction.36 The Braak pathological stage37 – a six-stage caudo-rostral propagation of Lewy pathology from the medulla oblongata to the neocortex – has been linked to cognitive function,31,38 but not all brains with evidence of Lewy pathology within the neocortex are associated with a history of cognitive impairment. Some have argued that α-syn may be a marker of neural protection rather than cause neural death,39 or alternately that those with neocortical Lewy pathology may have died prior to developing cognitive dysfunction.36 Other pathological markers of cognitive decline must exist, because it is possible, although rare, for people with a pathological diagnosis of PD and a clinical diagnosis of PDD to have no evidence of cortical Lewy pathology.37
The presence of combined Lewy pathology, NFT and Aβ is a better neuropathological correlate of PDD than any single pathology in isolation,40 and animal models suggest that the trio may be synergistic.41 Furthermore, 40%–50% of pathologically proven PDD cases also fulfill pathological diagnostic criteria for AD.36 Coexisting PDD and AD pathology is more likely to be seen in elderly brains because of the association between increasing age and presence of NFT and Aβ.42 Cerebrovascular disease and hippocampal sclerosis may also be important pathological markers of cognitive deficit in PD, but further work is needed to establish their role in more detail. Pathological studies of subjects with PD-MCI are currently lacking.
Neurotransmitter dysfunction
Dopamine
The relationship between dopaminergic activity in the mesolimbic and mesocortical pathways, and executive function and attention, is complicated. It has long been demonstrated that PD patients have deficits in facets of executive function and some studies have shown an association between deficit severity and motor symptoms.43 The profile of deficits in executive function in PD is linked to dopaminergic stimulation of frontostriatal cortical loops.43 An improvement in some tests of executive function (such as spatial working memory and planning44) and attention flexibility45 when PD patients are tested “on” compared to “off” has been shown. However, the relationship between dopamine and cognitive performance is not linear because other tests of executive function (such as reversal learning46 and motor sequencing learning47) were shown to deteriorate in the “on” state.
The “dopamine overdose hypothesis” was proposed to explain these findings and is based on the sequential degeneration of dopaminergic neurons within the substantia nigra pars compacta (SNpc). The ventral lateral tier, whose dopaminergic projections primarily connect to the dorsal putamen, is most severely affected.48 Executive functions such as flexibility and response inhibition, as well as working memory, are mediated by the dorsal putamen and are improved by dopaminergic stimulation early in the disease course. However, the dorsal SNpc tier, whose dopaminergic projections primarily connect to the ventral striatum, is less affected early in the disease course (Figure 2) and cognitive functions mediated by this pathway, such as reversal learning and motor sequence learning, are effectively “overdosed” by dopaminergic stimulation at this stage.25 Longitudinal positron emission tomography (PET) scanning studies have demonstrated that the dorsal–ventral gradient of reduced dopamine storing within the putamen persists as disease progresses but reduces in prominence over time.49
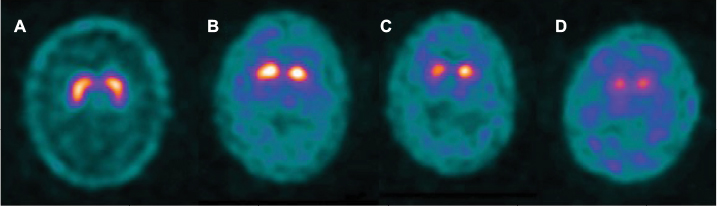 | Figure 2 The dopamine overdose hypothesis. Notes: Progressive dorsal–ventral loss of striatal dopaminergic neurons is demonstrated on Dopamine transporter imaging using 123I-N-3-fluoropropyl-2beta-carbomethoxy-3beta-4-iodophenyl tropane single-photon emission computed tomography (123I-FP-CIT SPECT): substantia nigral projections to the dorsal putamen are lost early in PD (A), then the ventral putamen (B) and caudate in later stages (C and D). Dorsal putamen domains (executive function and working memory) are improved by dopaminergic stimulation early in the disease course, whereas the cognitive functions associated with the relatively preserved ventral striatum (reversal learning and motor sequence learning) are “overdosed” by dopaminergic stimulation at this stage. Abbreviation: PD, Parkinson’s disease. |
In addition to its influence on the dorsal and ventral striatum, there is evidence that excessive dopaminergic stimulation can have deleterious consequences on the dorsolateral prefrontal cortex (DLPFC), which receives dopaminergic stimulation from neurons located in the ventral tegmental area via the mesocortical pathway.50,51 Establishing the effect of dopamine levels on the functioning of the DLPFC has occurred indirectly, by studying PD patients who have been stratified according to COMT gene polymorphism.23 A methionine to valine polymorphism at residue 158 of the COMT gene exists, and each valine substitution is associated with a fourfold increase in the efficiency of the enzyme COMT and therefore a more rapid removal of dopamine.52 Moreover, because dopamine transporters are relatively absent in the DLPFC, levels of COMT are the major factor in regulating dopamine levels.26 In the CamPaIGN study, it was shown that in “early PD” (<1.6 years disease duration) the methionine homozygotes performed worse than the “later” PD group (>1.6 years since diagnosis) in a test of executive function.23 This suggests that methionine homozygosity, and therefore relative inefficiency of COMT, results in overdosing of the DLPFC leading to impaired executive function in the early stages of PD. As the disease progresses, and dopaminergic stimulation of the mesocortical pathway is reduced, the inefficiency of COMT metabolism results in higher, more optimum, levels of DLPFC dopamine. Further advancement of disease then reduces available DLPFC still further leading to impaired executive function due to dopamine deficiency. Each individual with PD will have a different starting point on this “inverted U shape” pattern (poorer performance, better performance, poorer performance) and the overall effect of dopamine on executive function at a particular time will be related to disease progression, the striatal structure used in a cognitive test and genetic factors including COMT polymorphism.23,50 Interestingly, there is some emerging evidence from animal studies that adenosine A2A antagonism may influence facets of cognition, including working memory and attention, via changes to dopamine levels in the prefrontal cortex.53
Noradrenaline and serotonin
In PD, there is a loss of noradrenergic neurons from the locus coeruleus and serotonergic neurons from the dorsal and median raphe nuclei.54 These nuclei have extensive mesocortical connections, and there is growing evidence of noradrenergic and serotonergic influence on cognition in PD. The selective noradrenaline reuptake inhibitor (SNRI) atomoxetine caused a small but statistically significant improvement in cognition compared to placebo in one study, although this was a secondary outcome measure.55 Subsequently, it has been shown that a single dose of atomoxetine can reduce impulsivity in PD,56 and fMRI suggests that this medication can restore connectivity between the pre-supplementary motor cortex and inferior frontal gyrus.57 A single dose of citalopram, a selective serotonin reuptake inhibitor (SSRI), has also been shown to influence impulsivity compared to placebo in selected PD patients with more severe motor disease.58 Recent work has shown that it is possible to predict whether atomoxetine and citalopram will affect impulsivity, as measured by response inhibition, using a combination of clinical, structural and functional imaging.59 There is some evidence of downregulation of serotonin receptors associated with cognition in PD, although no association with executive function was found.60
Acetylcholine
The basal forebrain nuclei, including the nbM, are the major source of ACh to the cortex, whereas the PPN is the major source of cholinergic projections to the thalamus, as well as projecting to the spinal cord, the cerebellum and numerous brainstem nuclei.34,61 ACh is believed to be particularly important in regulating attention via projections from the basal forebrain nuclei to the DLPFC in animals62 and humans.63 The exact mechanism by which ACh modulates attention is unknown although it is established that ACh levels within the prefrontal cortex are transiently increased during tasks requiring attention.64
The nbM is significantly depleted in postmortem studies of PD patients when compared to controls,65 and the PPN loses about 50% of its laterally placed large neurons in PD, whereas it remains relatively intact in AD.66 According to the Braak staging model of PD,37 the two major ACh-producing nuclei are infiltrated with Lewy pathology at a similar time to the SNpc. PET and single-photon emission computed tomography (SPECT) studies of the cholinergic system in PD are compatible with the pathological changes seen at postmortem.67 Baseline deficits of attention in a cohort of PD patients were associated with a more rapid cognitive decline.35 In addition, medications that reduce ACh levels in the brain are linked to PD deficits. For example, in a PD cohort of 235, it was shown that those taking anti-cholinergics at baseline had a higher median reduction in Mini-Mental State Examination (MMSE) at 8 years (6.5 points) than those not who were not (1 point). Regression analysis adjusting for age, depression and baseline cognition suggested that duration and load of anti-cholinergic drugs were both significantly associated with decline on MMSE score.68 Conversely, cholinesterase inhibitors (ChEIs) – drugs that indirectly increase the availability of ACh – are the only proven treatment to enhance cognitive function in PDD according to meta-analysis.69
Genetics
The contribution of genetic changes to cognitive deficits in PD can broadly be divided into the study of monogenic forms of PD, the study of candidate genes in idiopathic – or sporadic – PD and emerging gene-wide association studies (GWAS).
Gene-wide association studies
The first published GWAS of 443 PD subjects did not find any significant single-nucleotide polymorphism (SNP) associations with cognitive function but was likely to be underpowered.70 Subsequent studies of similar sizes have also failed to find significant associations between SNPs found in GWAS of AD71 or type 2 diabetes72 and PD cognitive impairment. The complexity of cognitive dysfunction in PD means that large, well-powered GWAS are likely to be needed to understand the variance of candidate genes and hopefully this work will be forthcoming.
Monogenic PD
Analysis of monogenic forms of PD has provided information about their influence on cognition, although longitudinal data on dementia frequency in such cases are suboptimal.73 Leucine-rich repeat kinase 2 (LRRK 2) mutations are the most common autosomal-dominant (AuD) form of PD. Clinical features are similar to sporadic PD,74 and studies have generally shown that this mutation is neutral75 or protective76 from cognitive impairment. In contrast, several mutations in SCNA, as well as SCNA triplication,77 suggest that this AuD PD variant is associated with a severe and rapid cognitive change. Pathologically, SCNA mutations are characterized by diffuse Lewy body deposition throughout the cortex and brainstem.78
Parkin mutations account for 50% of autosomal recessive (AR) early-onset PD.79 Lewy bodies are absent or sparse, and cell loss is limited to the ventral substantia nigra and locus coeruleus.80 This restriction of pathology is associated with a very low incidence of cognitive dysfunction; a review of published data on Parkin-associated PD found that dementia occurred in <3% of cases and was more common in heterozygotes (7%) than homozygotes (1%), suggesting a protective effect.81
Sporadic PD
Homozygous mutation in the gene coding for glucocerebrosidase, known as GBA, causes Gaucher’s disease, a multisystem lysosomal storage disease. Heterozygous mutation of GBA is believed to be most frequent genetic risk factor for sporadic PD, occurring in ~3%–4% of cases.82 Those with GBA-associated PD have a younger onset and higher risk of dementia development – the point prevalence of dementia in GBA-associated PD is ~50%, twice that of sporadic PD.83 Progression to dementia in GBA-associated PD is significantly quicker than sporadic PD,84 but the reason for this is not yet fully understood. Pathologically, GBA-associated PD has similar findings to sporadic PD, although glucocerebrosidase is present in the majority of Lewy bodies.85 The mechanism by which glucocerebrosidase causes α-syn accumulation and aggregation is believed to involve changes to lysosomal, mitochondrial and endoplasmic reticulum function.86
COMT polymorphism, as discussed earlier, influences performance on executive function via fronto-striatal dopaminergic pathways but is not believed to be a risk factor for dementia development.23 Polymorphisms of gene coding for apolipoprotein E, APOE, are known to be associated with AD development. For example, those with the E4/E4 genotype are eight times more likely to develop AD than those with the most common E3/E3 genotype, although 50% of those with AD do not carry an E4 allele.87 Given the pathological association between NFT, Aβ and PDD,40 there has been interest in the influence of APOE in PD cognitive impairment and meta-analysis found an increased odds ratio of 1.6 for dementia development in those with at least one E4 allele.88 However, the authors comment that publication bias, study heterogeneity and relatively small numbers (295 PD and 163 PDD) need to be considered when interpreting these data.88 Two longitudinal studies of APOE in newly diagnosed PD subjects have shown no association with dementia development over 523 and 9.7 years,89 whereas a study of subjects with average disease duration of 7 years at trial onset showed that APOE E4 carriers had more rapid cognitive decline.90 It has been suggested the influence of APOE on dementia progression in PD may be greater with advancing age.91
Tau proteins are involved in the stabilization of microtubules, which maintain cell structure and perform other key roles. Microtubule-associated protein tau (MAPT), located on chromosome 17, codes for tau proteins via alternative splicing. An inversion in MAPT, H2, is present in ~25% of people with European ancestry.91 The H1/H1 genotype is a risk factor for PD development (odds ratio 1.4).92 The CamPaIGN study suggested that H1/H1 genotype was strongly associated with dementia development with an odds ratio of 12.1 at 5 years,23 but other studies have not found an association.90,92 The influence of MAPT H1 on cognition might be greatest at disease onset91 – supported by fMRI imaging27 – because the association between PDD and H1/H1 genotype appears to reduce over time.1
Biomarkers
Over the last decade, there has been an enormous drive to find biomarkers that can accurately identify, at the early stages of PD, those individuals who are at highest risk of developing PDD. Biomarker development has largely focused on imaging modalities and cerebrospinal fluid (CSF) analysis, although there are a few studies using electrophysiological techniques and combinations of biomarkers have also been studied. Recent review papers discuss this topic in more depth.93,94
Imaging
Advanced magnetic resonance imaging (MRI) techniques have enabled researchers to demonstrate that both structural and functional connectivity of certain brain areas are altered in PD cognitive impairment. Structural MRI studies of PD-MCI found a loss of volume within the hippocampus and the frontal, parietal and posterior cortices.95,96 Longitudinal studies of patients with PD-MCI have shown that these areas progressively lose volume and this correlates with cognitive deficit.95,97 By the time PDD is diagnosed, there is significant thinning of the parietal, occipital, frontal and temporal regions, plus further atrophy of the hippocampus, parahippocampus, insular and cingulate.98,99
Diffusion tensor imaging (DTI) has revealed evidence of reduced structural connectivity in similar regions – in particular, the hippocampus and frontal and parietal white matter tracts – and this is associated with a decline in executive functions and verbal and visuospatial memory tests.100,101 fMRI studies have demonstrated progressive impaired connectivity of various brain regions in PD, especially posteriorly, and this correlates with reduction in cognitive performance.102 The frontal and corticostriatal tract networks are particularly disrupted in PDD, and the level of cognitive decline is associated with the degree of impaired connectivity.103
PET imaging has provided useful in vivo information on the activity of certain neurotransmitters and the presence of abnormal proteins that are important in the pathogenesis of PD cognitive impairment. For example, cholinergic PET molecular imaging has shown that loss of cholinergic activity occurs throughout the cortex early in the course of PD but is much more severe, especially in the forebrain, in PDD.67 It is unclear at this stage if cholinergic PET imaging is sensitive to PD-MCI, but reduced levels of acetylcholinesterase were correlated with worse performance on tests of memory, executive functioning and attention in PD subjects without a diagnosis of cognitive impairment.67,104
In contrast to AD, where PET imaging of Aβ plaques is nearly always abnormal, there are abnormalities in less than 20% of PDD patients on PET.105 PD-MCI patients with Aβ on their PET scan are at increased risk of further cognitive decline.106 It has also been shown that the distribution of Aβ deposition is important, with cognitive impairment worse in PD cases with Aβ deposition in striatum and cortex, compared to the cortex alone.107 Likewise, a recent PET imaging study of tau deposition in PDD showed that cortical deposition, especially in the inferior temporal lobes, may potentially be a biomarker.108
It is perhaps not surprising that dopaminergic imaging has only revealed subtle differences between PD and PDD when one considers the long preclinical period of neurodegeneration that has occurred before a motor diagnosis is made. Nevertheless, there is some evidence that reduced dopamine transporter levels in the caudate are associated with impaired executive function109 and that more advanced nigrostriatal degeneration occurs in those who develop PDD.110
Cerebrospinal fluid
Several studies have assessed CSF proteins in PD, and it has been consistently shown that there is an association between low CSF levels of Aβ42 and cognitive impairment in PD.94 Longitudinal studies have shown that newly diagnosed PD patients with lower levels of CSF Aβ42 show a more rapid cognitive decline.111 The findings for tau are less clear – some studies found an association between cognitive decline and increased levels of tau in CSF but others have not. The relevance of α-syn CSF levels is also inconclusive; associations with cognitive impairment have been found with both high112 and low113 levels.
Treatment
Non-pharmacological
A systematic review of eight studies comprising a total of 158 PD patients suggested that physical exercise has beneficial effects on global cognition (measured with the MoCA, for example)114 and in particular there is evidence of improvement in executive function.115,116 Speculated mechanisms include enhanced perfusion and angiogenesis.117 However, published studies are heterogeneous in relation to intensity, mode and duration of exercise, and more research is needed.
Cognitive training (CTr) is a structured teaching or practice designed to target a specific cognitive domain. Research suggests that it is efficacious in healthy elderly adults118 and those with MCI.119 CTr in PD was reviewed in a meta-analysis comprising seven studies and 272 patients;120 overall, a small but statistically significant benefit was found compared to control conditions. Subanalysis suggested that CTr benefited processing speed and executive function but no other facets of cognition, including memory and visuospatial function. No significant difference in MMSE or MoCA scores was found.
Pharmacological
ChEIs are licensed for use in the treatment of PDD primarily based on the results of two large, randomized controlled trials (RCTs). The EDON study compared donepezil 5 mg daily, 10 mg daily and placebo over 24 weeks in 550 participants,121 and the EXPRESS study compared rivastigmine at doses of between 3 and 12 mg per day (highest well-tolerated dose maintained) with placebo in a 2:1 ratio over 24 weeks in 541 participants.122 There is better evidence overall to support the use of rivastigmine than donepezil although the benefits of both are modest – for example, the EXPRESS study showed a single-point difference in MMSE score (secondary outcome measure) at 24 weeks between groups.
The N-methyl-d-aspartate (NMDA) receptor antagonist, memantine, a drug that blocks activity of the excitatory neurotransmitter glutamate, has been studied in PDD. Three RCTs have been performed on a total of 299 subjects, each comparing memantine 20 mg per day with placebo. Meta-analysis concluded that memantine has a mild beneficial effect on global impression of change assessments but not on cognitive function as measured using MMSE.69
No successful treatments for PD-MCI have yet been identified in RCTs. A 24-week RCT crossover study of rivastigmine in PD-MCI was performed in 28 patients, of whom 26 completed the trial and 23 tolerated medication for both phases.123 Only a trend toward significance was demonstrated in the primary outcome – global impression of change (p 0.096) – and secondary outcome measures of cognition, including MoCA, were not significantly different. The small number of participants in the study may have influenced results, and it has been suggested that the heterogeneous nature of PD-MCI may also have contributed: perhaps only those with PD-MCI characterized by posterior cortical dysfunction, mediated primarily by ACh deficiency, will benefit from ChEI,124 as extrapolated from the dual syndrome hypothesis.
The monoamine oxidase B inhibitor (MAO-B), rasagiline, was studied in 170 PD patients in a 24-week, placebo-controlled RCT, and no significant effect on global impression of change or MoCA was detected.125 As discussed earlier, there is accumulating evidence that SNRIs55,56 and SSRIs58 might positively influence cognition in PD but large-scale RCTs are awaited. A number of other active clinical trials are underway, including the use of dual 5-HT6/5-HT2 antagonists, and these were summarized in a recent review.126 As with other neurodegenerative conditions such as AD, disease-modifying agents are a huge unmet need in PD. Many potential avenues are being explored in animal models, including the use of immunotherapies to target α-syn,127 tau and amyloid.128
Conclusion
Cognitive deficits in PD are common, even at the point of diagnosis, and have a significant negative impact on patients and their carers. The influence of neuronal toxicity by α-syn and AD-related pathology, neurotransmitter changes, genetic risks, age and other factors on cognition is different in each PD patient. Despite major research advances in the last 2 decades, the current pharmacological treatment of cognitive deficits in PD is unsatisfactory; benefits are modest and only drugs originally designed for use in AD are licensed. A greater understanding of PD cognitive impairment is likely to come via the identification of biomarkers to identify accurately those most at risk. In turn, this will hopefully lead to a move away from a reactive, “one-size-fits-all” treatment approach toward proactive, individualized and targeted therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Williams-Gray CH, Mason SL, Evans JR, et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry. 2013;84(11):1258–1264. | ||
Perez F, Helmer C, Foubert-Samier A, Auriacombe S, Dartigues JF, Tison F. Risk of dementia in an elderly population of Parkinson’s disease patients: a 15-year population-based study. Alzheimers Dement. 2012;8(6):463–469. | ||
Auyeung M, Tsoi TH, Mok V, et al. Ten year survival and outcomes in a prospective cohort of new onset Chinese Parkinson’s disease patients. J Neurol Neurosurg Psychiatry. 2012;83(6):607–611. | ||
Aarsland D, Zaccai J, Brayne C. A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov Disord. 2005;20(10):1255–1263. | ||
Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111–128. | ||
Macleod AD, Taylor KS, Counsell CE. Mortality in Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2014;29(13):1615–1622. | ||
Leroi I, McDonald K, Pantula H, Harbishettar V. Cognitive impairment in Parkinson disease: impact on quality of life, disability, and caregiver burden. J Geriatr Psychiatry Neurol. 2012;25(4):208–214. | ||
Szeto JY, Mowszowski L, Gilat M, Walton CC, Naismith SL, Lewis SJ. Mild cognitive impairment in Parkinson’s disease: impact on caregiver outcomes. J Parkinsons Dis. 2016;6(3):589–596. | ||
Litvan I, Goldman J, Tröster A, et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force Guidelines. Mov Disord. 2012;27(3):349–356. | ||
Broeders M, de Bie RM, Velseboer DC, Speelman JD, Muslimovic D, Schmand B. Evolution of mild cognitive impairment in Parkinson disease. Neurology. 2013;81(4):346–352. | ||
Yarnall AJ, Breen DP, Duncan GW, et al. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology. 2014;82(4):308–316. | ||
Jessen F, Amariglio RE, van Boxtel M, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement. 2014;10(6):844–852. | ||
Erro R, Santangelo G, Barone P, et al. Do subjective memory complaints herald the onset of mild cognitive impairment in Parkinson disease? J Geriatr Psychiatry Neurol. 2014;27(4):276–281. | ||
Hong JY, Sunwoo MK, Chung SJ, et al. Subjective cognitive decline predicts future deterioration in cognitively normal patients with Parkinson’s disease. Neurobiol Aging. 2014;35(7):1739–1743. | ||
Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22(12):1689–1707. quiz 1837. | ||
Geurtsen GJ, Hoogland J, Goldman JG, et al. Parkinson’s disease mild cognitive impairment: application and validation of the criteria. J Parkinson Dis. 2014;4(2):131–137. | ||
Szeto JYY, Mowszowski L, Gilat M, Walton CC, Naismith SL, Lewis SJG. Assessing the utility of the Movement Disorder Society Task Force Level 1 diagnostic criteria for mild cognitive impairment in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21(1):31–35. | ||
Marras C, Armstrong MJ, Meaney CA, et al. Measuring mild cognitive impairment in patients with Parkinson’s disease. Mov Disord. 2013;28(5):626–633. | ||
Janvin C, Larsen J, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson’s disease: progression to dementia. Mov Disord. 2006;21(9):1343–1349. | ||
Pedersen KF, Larsen JP, Tysnes OB, Alves G. Prognosis of mild cognitive impairment in early Parkinson disease: the Norwegian ParkWest study. JAMA Neurol. 2013;70(5):580–586. | ||
Elgh E, Domellof M, Linder J, Edstrom M, Stenlund H, Forsgren L. Cognitive function in early Parkinson’s disease: a population-based study. Eur J Neurol. 2009;16(12):1278–1284. | ||
Williams-Gray CH, Foltynie T, Brayne CE, Robbins TW, Barker RA. Evolution of cognitive dysfunction in an incident Parkinson’s disease cohort. Brain. 2007;130(pt 7):1787–1798. | ||
Williams-Gray CH, Evans JR, Goris A, et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain. 2009;132(pt 11):2958–2969. | ||
Aarsland D, Bronnick K, Williams-Gray C, et al. Mild cognitive impairment in Parkinson disease: a multicenter pooled analysis. Neurology. 2010;75(12):1062–1069. | ||
Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol. 2010;9(12):1200–1213. | ||
Kehagia AA, Barker RA, Robbins TW. Cognitive impairment in Parkinson’s disease: the dual syndrome hypothesis. Neurodegener Dis. 2013;11(2):79–92. | ||
Nombela C, Rowe JB, Winder-Rhodes SE, et al. Genetic impact on cognition and brain function in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain. 2014;137(pt 10):2743–2758. | ||
Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23(6):837–844. | ||
Aarsland D, Kvaløy J, Andersen K, et al. The effect of age of onset of PD on risk of dementia. J Neurol. 2007;254(1):38–45. | ||
Reid WG, Hely MA, Morris JG, Loy C, Halliday GM. Dementia in Parkinson’s disease: a 20-year neuropsychological study (Sydney Multicentre Study). J Neurol Neurosurg Psychiatry. 2011;82(9):1033–1037. | ||
Kempster P, O’Sullivan S, Holton J, Revesz T, Lees A. Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain. 2010;133(6):1755–1762. | ||
Burn DJ, Rowan EN, Allan LM, Molloy S, O’Brien JT, McKeith IG. Motor subtype and cognitive decline in Parkinson’s disease, Parkinson’s disease with dementia, and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry. 2006;77(5):585–589. | ||
Alves G, Larsen J, Emre M, Wentzel-Larsen T, Aarsland D. Changes in motor subtype and risk for incident dementia in Parkinson’s disease. Mov Disord. 2006;21(8):1123–1130. | ||
Yarnall A, Rochester L, Burn DJ. The interplay of cholinergic function, attention, and falls in Parkinson’s disease. Mov Disord. 2011;26(14):2496–2503. | ||
Taylor JP, Rowan EN, Lett D, O’Brien JT, McKeith IG, Burn DJ. Poor attentional function predicts cognitive decline in patients with non-demented Parkinson’s disease independent of motor phenotype. J Neurol Neurosurg Psychiatry. 2008;79(12):1318–1323. | ||
Irwin D, Lee V, Trojanowski J. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci. 2013;14(9):626–636. | ||
Braak H, Del Tredici K, Rüb U, de Vos R, Jansen Steur E, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. | ||
Braak H, Rüb U, Jansen Steur E, Del Tredici K, de Vos R. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64(8):1404–1410. | ||
Parkkinen L, Pirttilä T, Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008;115(4):399–407. | ||
Compta Y, Parkkinen L, O’Sullivan S, et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain. 2011;134(5):1493–1505. | ||
Clinton L, Blurton-Jones M, Myczek K, Trojanowski J, LaFerla F. Synergistic interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30(21):7281–7289. | ||
Matthews F, Brayne C, Lowe J, McKeith I, Wharton S, Ince P. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med. 2009;6(11):e1000180. | ||
Owen AM, James M, Leigh PN, et al. Fronto-striatal cognitive deficits at different stages of Parkinson’s disease. Brain. 1992;115(pt 6):1727–1751. | ||
Lange KW, Robbins TW, Marsden CD, James M, Owen AM, Paul GM. L-dopa withdrawal in Parkinson’s disease selectively impairs cognitive performance in tests sensitive to frontal lobe dysfunction. Psychopharmacology. 1992;107(2–3):394–404. | ||
Cools R, Barker RA, Sahakian BJ, Robbins TW. L-Dopa medication remediates cognitive inflexibility, but increases impulsivity in patients with Parkinson’s disease. Neuropsychologia. 2003;41(11):1431–1441. | ||
Swainson R, Rogers RD, Sahakian BJ, Summers BA, Polkey CE, Robbins TW. Probabilistic learning and reversal deficits in patients with Parkinson’s disease or frontal or temporal lobe lesions: possible adverse effects of dopaminergic medication. Neuropsychologia. 2000;38(5):596–612. | ||
Kwak Y, Muller ML, Bohnen NI, Dayalu P, Seidler RD. Effect of dopaminergic medications on the time course of explicit motor sequence learning in Parkinson’s disease. J Neurophysiol. 2010;103(2):942–949. | ||
Fearnley J, Lees A. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(5):2283–2301. | ||
Nandhagopal R, Kuramoto L, Schulzer M, et al. Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study. Brain. 2009;132(11):2970–2979. | ||
Vaillancourt DE, Schonfeld D, Kwak Y, Bohnen NI, Seidler R. Dopamine overdose hypothesis: evidence and clinical implications. Mov Disord. 2013;28(14):1920–1929. | ||
Lewis SJ, Barker RA. Understanding the dopaminergic deficits in Parkinson’s disease: insights into disease heterogeneity. J Clin Neurosci. 2009;16(5):620–625. | ||
Chen J, Lipska BK, Halim N, et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75(5):807–821. | ||
Ko WK, Camus SM, Li Q, et al. An evaluation of istradefylline treatment on Parkinsonian motor and cognitive deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated macaque models. Neuropharmacology. 2016;110(pt A):48–58. | ||
Halliday GM, Leverenz JB, Schneider JS, Adler CH. The neurobiological basis of cognitive impairment in Parkinson’s disease. Mov Disord. 2014;29(5):634–650. | ||
Weintraub D, Mavandadi S, Mamikonyan E, et al. Atomoxetine for depression and other neuropsychiatric symptoms in Parkinson disease. Neurology. 2010;75(5):448–455. | ||
Kehagia AA, Housden CR, Regenthal R, et al. Targeting impulsivity in Parkinson’s disease using atomoxetine. Brain. 2014;137(7):1986–1997. | ||
Rae CL, Nombela C, Rodriguez PV, et al. Atomoxetine restores the response inhibition network in Parkinson’s disease. Brain. 2016;139(pt 8):2235–2248. | ||
Ye Z, Altena E, Nombela C, et al. Selective serotonin reuptake inhibition modulates response inhibition in Parkinson’s disease. Brain. 2014;137(pt 4):1145–1155. | ||
Ye Z, Rae CL, Nombela C, et al. Predicting beneficial effects of atomoxetine and citalopram on response inhibition in Parkinson’s disease with clinical and neuroimaging measures. Hum Brain Mapp. 2016;37(3):1026–1037. | ||
Varrone A, Svenningsson P, Marklund P, et al. 5-HT1B receptor imaging and cognition: a positron emission tomography study in control subjects and Parkinson’s disease patients. Synapse. 2015;69(7):365–374. | ||
Muller ML, Bohnen NI. Cholinergic dysfunction in Parkinson’s disease. Curr Neurol Neurosci Rep. 2013;13(9):377. | ||
Totah NK, Kim YB, Homayoun H, Moghaddam B. Anterior cingulate neurons represent errors and preparatory attention within the same behavioral sequence. J Neurosci. 2009;29(20):6418–6426. | ||
Bloem B, Poorthuis RB, Mansvelder HD. Cholinergic modulation of the medial prefrontal cortex: the role of nicotinic receptors in attention and regulation of neuronal activity. Front Neural Circuits. 2014;8:17. | ||
Howe WM, Berry AS, Francois J, et al. Prefrontal cholinergic mechanisms instigating shifts from monitoring for cues to cue-guided performance: converging electrochemical and fMRI evidence from rats and humans. J Neurosci. 2013;33(20):8742–8752. | ||
Nakano I, Hirano A. Parkinson’s disease: neuron loss in the nucleus basalis without concomitant Alzheimer’s disease. Ann Neurol. 1984;15(5):415–418. | ||
Zweig RM, Jankel WR, Hedreen JC, Mayeux R, Price DL. The pedunculopontine nucleus in Parkinson’s disease. Ann Neurol. 1989;26(1):41–46. | ||
Bohnen NI, Kaufer DI, Ivanco LS, et al. Cortical cholinergic function is more severely affected in Parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol. 2003;60(12):1745–1748. | ||
Ehrt U, Broich K, Larsen JP, Ballard C, Aarsland D. Use of drugs with anticholinergic effect and impact on cognition in Parkinson’s disease: a cohort study. J Neurol Neurosurg Psychiatry. 2010;81(2):160–165. | ||
Wang HF, Yu JT, Tang SW, et al. Efficacy and safety of cholinesterase inhibitors and memantine in cognitive impairment in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies: systematic review with meta-analysis and trial sequential analysis. J Neurol Neurosurg Psychiatry. 2015;86(2):135–143. | ||
Chung SJ, Armasu SM, Biernacka JM, et al. Genomic determinants of motor and cognitive outcomes in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(7):881–886. | ||
Wang YQ, Tang BS, Yang Y, et al. Relationship between Alzheimer’s disease GWAS-linked top hits and risk of Parkinson’s disease with or without cognitive decline: a Chinese population-based study. Neurobiol Aging. 2016;39: 217.e9–11. | ||
Chung SJ, Kim MJ, Kim J, et al. Association of type 2 diabetes GWAS loci and the risk of Parkinson’s and Alzheimer’s diseases. Parkinsonism Relat Disord. 2015;21(12):1435–1440. | ||
Kasten M, Kertelge L, Bruggemann N, et al. Nonmotor symptoms in genetic Parkinson disease. Arch Neurol. 2010;67(6):670–676. | ||
Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–590. | ||
Shanker V, Groves M, Heiman G, et al. Mood and cognition in leucine-rich repeat kinase 2 G2019S Parkinson’s disease. Mov Disord. 2011;26(10):1875–1880. | ||
Srivatsal S, Cholerton B, Leverenz JB, et al. Cognitive profile of LRRK2-related Parkinson’s disease. Mov Disord. 2015;30(5):728–733. | ||
Farrer M, Kachergus J, Forno L, et al. Comparison of kindreds with Parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55(2):174–179. | ||
Waxman EA, Giasson BI. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta. 2009;1792(7):616–624. | ||
Lücking CB, Dürr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the Parkin gene. N Engl J Med. 2000;342(21):1560–1567. | ||
Mori H, Kondo T, Yokochi M, et al. Pathologic and biochemical studies of juvenile Parkinsonism linked to chromosome 6q. Neurology. 1998;51(3):890–892. | ||
Grunewald A, Kasten M, Ziegler A, Klein C. Next-generation phenotyping using the parkin example: time to catch up with genetics. JAMA Neurol. 2013;70(9):1186–1191. | ||
Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–1661. | ||
Seto-Salvia N, Pagonabarraga J, Houlden H, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov Disord. 2012;27(3):393–399. | ||
Winder-Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain. 2013;136(pt 2):392–399. | ||
Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010;120(5):641–649. | ||
Migdalska-Richards A, Schapira AH. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem. 2016;139(suppl 1):77–90. | ||
Loy CT, Schofield PR, Turner AM, Kwok JBJ. Genetics of dementia. Lancet. 2014;383(9919):828–840. | ||
Huang X, Chen P, Kaufer DI, Troster AI, Poole C. Apolipoprotein E and dementia in Parkinson disease: a meta-analysis. Arch Neurol. 2006;63(2):189–193. | ||
Kurz MW, Dekomien G, Nilsen OB, Larsen JP, Aarsland D, Alves G. APOE alleles in Parkinson disease and their relationship to cognitive decline: a population-based, longitudinal study. J Geriatr Psychiatry Neurol. 2009;22(3):166–170. | ||
Morley JF, Xie SX, Hurtig HI, et al. Genetic influences on cognitive decline in Parkinson’s disease. Mov Disord. 2012;27(4):512–518. | ||
Collins LM, Williams-Gray CH. The genetic basis of cognitive impairment and dementia in Parkinson’s disease. Front Psychiatry. 2016; 7:89. | ||
Mata IF, Leverenz JB, Weintraub D, et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 2014;71(11):1405–1412. | ||
Aarsland D, Creese B, Politis M, et al. Cognitive decline in Parkinson disease. Nat Rev Neurol. 2017;13(4):217–231. | ||
Lin CH, Wu RM. Biomarkers of cognitive decline in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21(5):431–443. | ||
Mak E, Su L, Williams GB, et al. Baseline and longitudinal grey matter changes in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain. 2015;138(pt 10):2974–2986. | ||
Pereira JB, Aarsland D, Ginestet CE, et al. Aberrant cerebral network topology and mild cognitive impairment in early Parkinson’s disease. Hum Brain Mapp. 2015;36(8):2980–2995. | ||
Hanganu A, Bedetti C, Degroot C, et al. Mild cognitive impairment is linked with faster rate of cortical thinning in patients with Parkinson’s disease longitudinally. Brain. 2014;137(pt 4):1120–1129. | ||
Zarei M, Ibarretxe-Bilbao N, Compta Y, et al. Cortical thinning is associated with disease stages and dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2013;84(8):875–881. | ||
Rektorova I, Biundo R, Marecek R, Weis L, Aarsland D, Antonini A. Grey matter changes in cognitively impaired Parkinson’s disease patients. PLoS One. 2014;9(1):e85595. | ||
Agosta F, Canu E, Stefanova E, et al. Mild cognitive impairment in Parkinson’s disease is associated with a distributed pattern of brain white matter damage. Hum Brain Mapp. 2014;35(5):1921–1929. | ||
Chen B, Fan GG, Liu H, Wang S. Changes in anatomical and functional connectivity of Parkinson’s disease patients according to cognitive status. Eur J Radiol. 2015;84(7):1318–1324. | ||
Olde Dubbelink KT, Schoonheim MM, Deijen JB, Twisk JW, Barkhof F, Berendse HW. Functional connectivity and cognitive decline over 3 years in Parkinson disease. Neurology. 2014;83(22):2046–2053. | ||
Rektorova I, Krajcovicova L, Marecek R, Mikl M. Default mode network and extrastriate visual resting state network in patients with Parkinson’s disease dementia. Neurodegener Dis. 2012;10(1–4):232–237. | ||
Bohnen NI, Müller MLTM, Kotagal V, et al. Heterogeneity of cholinergic denervation in Parkinson’s disease without dementia. J Cereb Blood Flow Metab. 2012;32(8):1609–1617. | ||
Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry. 2008;79(12):1331–1338. | ||
Gomperts S, Locascio J, Rentz D, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology. 2013;80(1):85–91. | ||
Shah N, Frey KA, Muller ML, et al. Striatal and cortical beta-amyloidopathy and cognition in Parkinson’s disease. Mov Disord. 2016;31(1):111–117. | ||
Gomperts SN, Locascio JJ, Makaretz SJ, et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol. 2016;73(11):1334–1341. | ||
Marquie M, Locascio JJ, Rentz DM, et al. Striatal and extrastriatal dopamine transporter levels relate to cognition in Lewy body diseases: an (11)C altropane positron emission tomography study. Alzheimers Res Ther. 2014;6(5–8):52. | ||
Song IU, Kim YD, Cho HJ, Chung SW, Chung YA. An FP-CIT PET comparison of the differences in dopaminergic neuronal loss between idiopathic Parkinson disease with dementia and without dementia. Alzheimer Dis Assoc Disord. 2013;27(1):51–55. | ||
Backstrom DC, Eriksson Domellof M, Linder J, et al. Cerebrospinal fluid patterns and the risk of future dementia in early, incident Parkinson disease. JAMA Neurol. 2015;72(10):1175–1182. | ||
Stewart T, Liu C, Ginghina C, et al. Cerebrospinal fluid alpha-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am J Pathol. 2014;184(4):966–975. | ||
Skogseth RE, Bronnick K, Pereira JB, et al. Associations between cerebrospinal fluid biomarkers and cognition in early untreated Parkinson’s disease. J Parkinsons Dis. 2015;5(4):783–792. | ||
Murray DK, Sacheli MA, Eng JJ, Stoessl AJ. The effects of exercise on cognition in Parkinson’s disease: a systematic review. Transl Neurodegener. 2014;3:5–5. | ||
Cruise KE, Bucks RS, Loftus AM, Newton RU, Pegoraro R, Thomas MG. Exercise and Parkinson’s: benefits for cognition and quality of life. Acta Neurol Scand. 2011;123(1):13–19. | ||
Tanaka K, Quadros AC Jr, Santos RF, Stella F, Gobbi LT, Gobbi S. Benefits of physical exercise on executive functions in older people with Parkinson’s disease. Brain Cogn. 2009;69(2):435–441. | ||
Reynolds GO, Otto MW, Ellis TD, Cronin-Golomb A. The therapeutic potential of exercise to improve mood, cognition, and sleep in Parkinson’s disease. Mov Disord. 2016;31(1):23–38. | ||
Lampit A, Hallock H, Valenzuela M. Computerized cognitive training in cognitively healthy older adults: a systematic review and meta-analysis of effect modifiers. PLoS Med. 2014;11(11):e1001756. | ||
Coyle H, Traynor V, Solowij N. Computerized and virtual reality cognitive training for individuals at high risk of cognitive decline: systematic review of the literature. Am J Geriatr Psychiatry. 2015;23(4):335–359. | ||
Leung IHK, Walton CC, Hallock H, Lewis SJG, Valenzuela M, Lampit A. Cognitive training in Parkinson disease: a systematic review and meta-analysis. Neurology. 2015;85(21):1843–1851. | ||
Dubois B, Tolosa E, Katzenschlager R, et al. Donepezil in Parkinson’s disease dementia: a randomized, double-blind efficacy and safety study. Mov Disord. 2012;27(10):1230–1238. | ||
Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med. 2004; 351(24):2509–2518. | ||
Mamikonyan E, Xie SX, Melvin E, Weintraub D. Rivastigmine for mild cognitive impairment in Parkinson disease: a placebo-controlled study. Mov Disord. 2015;30(7):912–918. | ||
Burn DJ. The treatment of mild cognitive impairment associated with Parkinson’s disease. Mov Disord. 2015;30(7):881–882. | ||
Weintraub D, Hauser RA, Elm JJ, Pagan F, Davis MD, Choudhry A. Rasagiline for mild cognitive impairment in Parkinson’s disease: a placebo-controlled trial. Mov Disord. 2016;31(5):709–714. | ||
Goldman JG, Weintraub D. Advances in the treatment of cognitive impairment in Parkinson’s disease. Mov Disord. 2015;30(11):1471–1489. | ||
Lindstrom V, Ihse E, Fagerqvist T, et al. Immunotherapy targeting alpha-synuclein, with relevance for future treatment of Parkinson’s disease and other Lewy body disorders. Immunotherapy. 2014;6(2):141–153. | ||
Valera E, Spencer B, Masliah E. Immunotherapeutic approaches targeting amyloid-beta, alpha-synuclein, and Tau for the treatment of neurodegenerative disorders. Neurotherapeutics. 2016;13(1):179–189. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.