")
Back to Journals » OncoTargets and Therapy » Volume 13
Coexisting of COX7A2L–ALK, LINC01210–ALK, ATP13A4–ALK and Acquired SLCO2A1–ALK in a Lung Adenocarcinoma with Rearrangements Loss During the Treatment of Crizotinib and Ceritinib: A Case Report
Authors Cai C, Long Y, Li Y, Huang M
Received 14 April 2020
Accepted for publication 3 August 2020
Published 20 August 2020 Volume 2020:13 Pages 8313—8316
DOI https://doi.org/10.2147/OTT.S258067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Chengzhi Cai, Yaling Long, Yanying Li, Meijuan Huang
Department of Thoracic Oncology, Cancer Center, West China Hospital, Sichuan University, Chengdu, People’s Republic of China
Correspondence: Meijuan Huang
Department of Thoracic Oncology, Cancer Center, West China Hospital, Sichuan University, No. 37 Guoxue Alley, Wuhou District, Chengdu City, Sichuan Province 610041, People’s Republic of China
Tel +86 189 80602026
Email [email protected]
Abstract: ALK rearrangements account for ∼ 5% of non-small-cell lung cancer (NSCLC). Numerous rearrangement partners have been discovered. Here, we describe a 53-year-old nonsmoker with NSCLC, in whom we identified four novel rearrangements. The patient was diagnosed as adenocarcinoma in the right middle lobe of lung, with metastases in subcarinal lymph node, ipsilateral lung, pleura and contralateral rib (cT4N2M1, stage IV). Next-generation sequencing (NGS) identified three baseline ALK fusions: COX7A2L–ALK (C[intragenic]:A20), LINC01210–ALK (L[intergenic]:A20) and ATP13A4–ALK (A9:A19). The patient exhibited 12 months of progression-free survival (PFS) and a partial response (PR) to first-line crizotinib therapy. We then discovered a new SLCO2A1–ALK fusion (S[intergenic]:A18) and a missense mutation C1156Y after resistance developed. Sequential ceritinib resulted in further 8 months of PFS, after which NGS results demonstrated the loss of ATP13A4–ALK and SLCO2A1–ALK. This is the first description a NSCLC patient harbors four ALK fusions and was sensitive to tyrosine kinase inhibitors (TKIs). Acquisition and loss of ALK fusions after ALK inhibitors may account for resistance.
Keywords: NSCLC, intergenic ALK, intragenic ALK, non-EML4–ALK, target therapy
Introduction
ALK rearrangements resulting in overexpression of the anaplastic lymphoma kinase (ALK) protein are the second most common type of genetic aberrations which occurs in non-small-cell lung cancer (NSCLC). These rearrangements are found in ~5% of affected patients.1 The gene encoding echinoderm microtubule associated protein like 4 (EML4) is a common ALK fusion partner, but at least 20 other partners have also been reported.2 Patients with ALK rearrangements usually benefit from tyrosine-kinase inhibitor (TKI) treatment. However, the inhibitor sensitivity of recombinant ALK proteins can vary depending on the nature of the fusion partners.3 Furthermore, multiple fusion events can coexist in individual patients,4 thereby complicating the tumor response to ALK inhibitors. The development of ALK-specific TKIs has revolutionized the treatment of ALK-rearranged NSCLC. Crizotinib is a first-generation ALK inhibitor that is superior to chemotherapy in terms of tumor control and progression-free survival (PFS).5 However, resistance to such inhibitors inevitably develops, with missense mutations encoding amino acid substitutions such as L1196M, C1156Y, G1202R, and I1171N being the most frequent causes of inhibitor failure. The occurrence of these mutations in the ALK gene reduces the effectiveness of combined inhibitor and protein therapies, necessitating the development of new treatments. Thus, second-generation inhibitors such as ceritinib, alectinib, and brigatinib, and third-generation inhibitors such as lorlatinib, have now been developed.
Case Description
A 53-year-old man presented at our hospital complaining of tightness in the chest. Imaging analyses identified a mass in the right middle lobe of the lung, with ipsilateral thickened pleura and enlarged subcarinal lymph nodes. Lesions in the ipsilateral superior lobe and left eighth rib were considered to be metastatic (Figure 1A). Thoracentesis confirmed that the medium amount of pleural effusion was hydrothorax with blood. After exfoliative cytology and immunohistochemistry of the hydrothorax (using Ventana ALK D5F3), the patient was diagnosed with ALK-positive lung adenocarcinoma (cT4N2M1, stage IV). Next-generation sequencing (NGS) of genomic DNA isolated from exfoliated adenocarcinoma cells revealed three novel ALK rearrangements: COX7A2L–ALK (C[intragenic]:A20, abundance 41.2%), in which an intron sequence from COX7A2L was joined to exon 20 of ALK; LINC01210–ALK (L[intergenic]:A20, abundance 34.7%), in which an intergenic sequence near to LINC01210 was joined to exon 20 of ALK; and ATP13A4–ALK (A9:A19, abundance 21.5%), in which exon 9 of ATP13A4 was joined to exon 19 of ALK. No aberrations were discovered in other common driver genes. The patient’s Eastern Cooperative Oncology Group (ECOG) performance status (PS) score was 0. Then patient received crizotinib (250 mg, twice daily) as a first-line treatment. According to the Response Evaluation Criteria in Solid Tumors version 1.1, the patient was evaluated after 2 months of treatment and found to have achieved a partial response (PR), as evidenced by shrinkage of the lung lesions and elimination of lymph-node lesions (Figure 1B). During crizotinib treatment, the only adverse event was level 1 hypoleukemia, from which the patient recovered following symptomatic treatment. After 12 months PFS, hydrothorax and a subcutaneous node were discovered. Thoracentesis was performed again to obtain exfoliated cancer cells. NGS identified a new rearrangement, SLCO2A1–ALK (S[intergenic]:A18, abundance 49.9%), and a missense mutation encoding C1156Y. Resistance to crizotinib was also accompanied by an increased abundance of the COX7A2L–ALK (53.6%), LINC01210–ALK (52.4%) and ATP13A4–ALK (33.8%) fusions. Therefore, ceritinib was administered orally once daily (450 mg), on the basis of resistance mutation sensitivity. The results of follow-up computed tomography (CT) scans suggested that the patient experienced 7 months of PFS, with the best evaluation of PR (Figure 1C). Progression occurred in the lung, and subcutaneous lesions with the recurrence of hydrothorax were observed later. The patient underwent a third thoracentesis for hydrothorax exfoliative cytology and NGS. The ATP13A4–ALK and SLCO2A1–ALK fusions were no longer detectable. The remaining fusions included COX7A2L–ALK (33.8%) and LINC01210-ALK (13.3%). The missense mutation C1156Y was also still present.
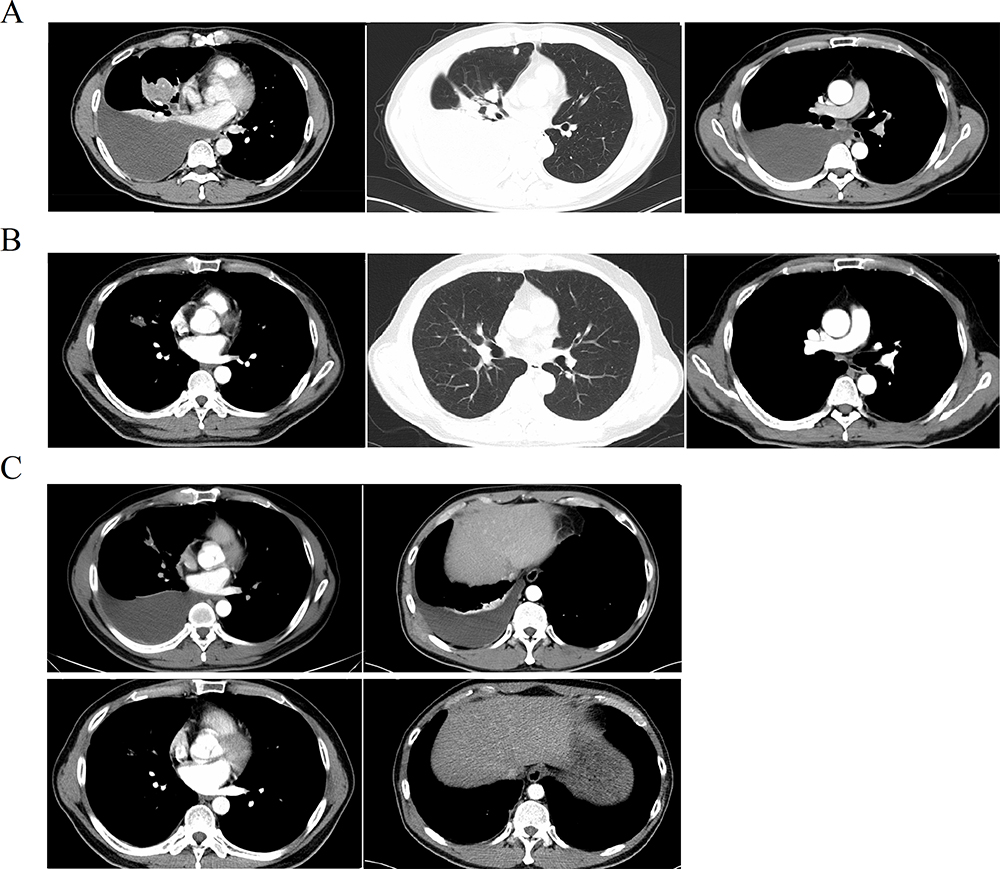 |
Figure 1 Image study and therapeutic evaluation. (A) CT scan showed primary lung lesion in right middle lobe and metastasis to subcarinal lymph node, ipsilateral lung and left eighth rib in baseline; (B) PR assessment during crizotinib because of the shrinking of lung lesions and elimination of lymph node; (C) progression when resistant to crizotinib (hydrothorax and subcutaneous metastasis) and the partial response (PR) to ceritinib in hydrothorax elimination and subcutaneous lesion control. |
Discussion
We have presented here the first known case of a NSCLC patient in whom four ALK gene fusions coexisted. These rearrangements were COX7A2L–ALK (C[intragenic]:A20), LINC01210–ALK (L[intergenic]:A20), ATP13A4–ALK (A9:A19) and SLCO2A1–ALK (S[intergenic]:A18). To the best of our knowledge, these events are rare and constitute the first report. The abundance of each rearrangement changed over the course of treatment with targeted therapies (Table 1). At baseline, we detected three fusions (COX7A2L–ALK, LINC01210–ALK and ATP13A4–ALK), and the patient received crizotinib. NGS at progression additionally detected an SLCO2A1–ALK fusion. The patient then received second-line ceritinib until progression, at which point ATP13A4–ALK and SLCO2A1–ALK were no longer detected, indicating that resistance had developed again.
 |
Table 1 Abundance of ALK Mutations at Different Stages of the Patient’s Treatment |
Among the four rearrangement partners, ATP13A4, which is located on chromosome 3, was the only gene to contribute exon sequences. The protein encoded by ATP13A4 gene is a member of the subfamily of P5-type ATPases, and is involved in cardiac conduction and the transport of glucose and other sugars, bile salts, organic acids, metal ions and amine compounds.6 By contrast, intergenic and intragenic ALK fusions are rare mutations in which the rearrangement partners do not contribute exon sequences to the gene fusions. Intergenic fragments are gene spacer sequences that are located near to the partner gene, whereas intragenic fragments are derived from intron regions of the partner gene. It is unknown whether these intergenic and intragenic fusions are tumor drivers. Further analysis of the fragment functions and mRNA sequences may provide additional information.
The development of ALK inhibitors has resulted in considerable improvements in survival among patients diagnosed with NSCLC with ALK-rearrangement. However, these patients inevitably acquire treatment resistance. Along with missense mutations, the acquisition and loss of ALK rearrangements may also be involved in inhibitor resistance. In this case, an SLCO2A1–ALK (S[intergenic]:A18) fusion was detected by NGS following the development of resistance to crizotinib. Some evidence suggests that rare fusions may contribute to crizotinib resistance.7 The development of rearrangements encoding TKI-insensitive ALK might, therefore, lead to acquired resistance. Notably, resistance to epidermal growth factor receptor (EGFR) TKIs has been observed in patients with NSCLC and EGFR mutations who acquired EML4–ALK fusions.8 Loss of ALK rearrangements can also lead to resistance.9 Additionally, no new missense mutations were detected when the patient was assessed for progression following ceritinib treatment. In this regard, we believed the disappearance of the ATP13A4–ALK and SLCO2A1–ALK rearrangements is in association with the development of resistance to ceritinib. The maintenance of the crizotinib-resistant C1156Y mutation indicated that retained COX7A2L–ALK and LINC01210–ALK might have been involved in tumor growth and sensitivity to crizotinib.
Conclusion
ATP13A4–ALK is a therapeutic target for ALK-rearrangement NSCLC. Intergenic and intragenic ALK fusions might be involved in tumor growth and sensitivity to ALK TKIs. Both acquisition and loss of ALK fusions after inhibitor treatment might lead to resistance.
Abbreviations
NSCLC, non-small-cell lung cancer; TKI, tyrosine kinase inhibitor; NGS, next-generation sequencing; ALK, anaplastic lymphoma kinase; EML4, echinoderm microtubule associated protein like 4; PFS, progression-free survival; ECOG, Eastern Cooperative Oncology Group; PS, performance status; PR, partial response; CT, computed tomography; N.D., not detected; EGFR, epidermal growth factor receptor.
Ethics Approval and Consent to Participate
This study was approved by the ethics committee of the Sichuan University Affiliated West China Hospital. Additionally, written informed consent has been provided by the patient to have the case details and any accompanying images published.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ramalingam SS, Owonikoko TK, Khuri FR. Lung cancer: new biological insights and recent therapeutic advances. CA Cancer J Clin. 2011;61:91–112. doi:10.3322/caac.20102
2. Zhu VW, Schrock AB, Bosemani T, et al. Dramatic response to alectinib in a lung cancer patient with a novel VKORC1L1-ALK fusion and an acquired ALK T1151K mutation. Lung Cancer (Auckl). 2018;9:111–116. doi:10.2147/LCTT.S186804
3. Du X, Shao Y, Qin HF, et al. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer. 2018;9(4):423–430. doi:10.1111/1759-7714.12613
4. Yin J, Zhang Y, Peng F, et al. Reporting on two novel fusions, DYSF-ALK and ITGAV-ALK, coexisting in one patient with adenocarcinoma of lung, sensitive to crizotinib. J Thorac Oncol. 2018;(3):e43–e45. doi:10.1016/j.jtho.2017.10.025
5. Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi:10.1056/NEJMoa1408440
6. Vallipuram J, Grenville J, Crawford DA. The E646D-ATP13A4 mutation associated with autism reveals a defect in calcium regulation. Cell Mol Neurobio. 2010;30(2):233–246. doi:10.1007/s10571-009-9445-8
7. Du X, Shao Y, Gao H, et al. CMTR1-ALK: an ALK fusion in a patient with no response to ALK inhibitor crizotinib. Cancer Biol Ther. 2018;19(11):1–5. doi:10.1080/15384047.2018.1480282
8. Xu H, Shen J, Xiang J, et al. Characterization of acquired receptor tyrosine-kinase fusions as mechanisms of resistance to EGFR tyrosine-kinase inhibitors. Cancer Manag Res. 2019;11:6343–6351. doi:10.2147/CMAR.S197337
9. Lin JJ, Zhu VW, Yoda S, et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J Clin Oncol. 2018;36(12):1199–1206. doi:10.1200/JCO.2017.76.2294
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.