")
Back to Journals » International Medical Case Reports Journal » Volume 14
Coexistence of Acquired Hemophilia and Antiphospholipid Serology in Monoclonal Gammopathy Patient
Authors Belfeki N, Hamrouni S, Strazzulla A, Diamantis S
Received 26 November 2020
Accepted for publication 6 March 2021
Published 28 April 2021 Volume 2021:14 Pages 261—264
DOI https://doi.org/10.2147/IMCRJ.S293931
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Nabil Belfeki, Sarra Hamrouni, Alessio Strazzulla, Sylvain Diamantis
Department of Internal Medicine, Groupe Hospitalier Sud-Ile de France, Melun, 77000, France
Correspondence: Nabil Belfeki
Department of Internal Medicine, Groupe Hospitalier Sud Ile de France, 77000, Melun Tel +33 1 81 74 17 04
Fax +33 1 81 74 18 12
Email [email protected]
Abstract: Acquired hemophilia is a rare coagulopathy with hemorrhage into the skin, muscle, or soft tissues and mucous membranes and caused by inhibitor antibodies, mainly against FVIII. We report a case of acquired hemophilia presenting with diffuse cutaneous hemorrhage and hemothorax. The patient was found to have acquired an FVIII inhibitor and a high titer of anti β 2 glycoprotein 1 IgG and IgM, and anticardiolipin IgM in the context of IgA kappa-type monoclonal gammopathy. He received 3 injections of recombinant factor VII (rFVIIa) and blood transfusion. He was started on steroids and oral cyclophosphamide for 6 weeks. Thromboprophylaxis with aspirin at 100 mg/day was started 3 months after discharge. Antiphospholipid antibodies remained positive after 3 months as well as prolonged aPTT, factor VIII raised at 100%, and the inhibitor was not detected. The association between acquired hemophilia and antiphospholipid antibodies is rare and its distinction is mandatory because clinical presentation ranges from massive hemorrhage to thrombosis.
Keywords: acquired hemophilia, factor VIII deficiency, antiphospholipid antibodies, hemorrhage, treatment
Introduction
Acquired hemophilia is a rare coagulopathy with hemorrhage into the skin, muscle, or soft tissues and mucous membranes and is caused by inhibitor antibodies, mainly against FVIII. This disorder can be idiopathic but also associated with other conditions. Antiphospholipid antibodies are antibodies directed against components of the cell coat called phospholipids and are associated with predisposition for blood clot.1 Gammopathy of unknown significance causes coagulation abnormalities in approximately 6% to 7% of patients.2 We herein present the case of an association between acquired hemophilia and antiphospholipid antibodies in a patient with monoclonal gammopathy of unknown significance.
Case Report
A 77-year-old male with a past history of arterial hypertension, type 2 diabetes, and chronic renal insufficiency presented to the emergency room for diffuse ecchymosis all over his body after a mechanic fall (Figure 1). There was no history of any drug intake or family past history of bleeding. Physical examination showed mucocutaneous pallor and a diffuse hematoma taking all the left hemi-body. Blood pressure was at 110/70 mmHg and heart beats 100 per minute. The pulmonary auscultation revealed a murmur abolition on the left hemithorax. Thoraco-abdomino-pelvic CT scan showed non-compressive left hemothorax and a CT angiogram did not find the source of bleeding in the thorax. There are no underlying parenchymal tumoral process, no lymph nodes or spleen enlargement. Laboratory investigations showed an aregenerative normochromic normocytic anemia at 5 g/dl (normal range 12–14 g/dl), white cells 4000/L (normal range 4000–10,000/L), platelet count 450 000/L (normal range 150 000–450,000/L), C reactive protein 130 mg/dl (normal range < 5 mg/dl), creatinine 167 μmol/l (normal range 88–155μmol/l), and calcium 2.4 mmol/L (normal range 2.2–2.6 mmol/L). Haptoglobin level was 3 g/L (normal range 0.5–2.2 g/L), reticulocytes 20,000/L, ferritin 365 µg/L (normal range 24–336 µg/L), and anti-globulin test was negative. Hemostasis assessment showed prolonged activated partial thromboplastin time (aPTT) (113 seconds) with failure to correct (73 seconds) with 1:1 mixing studies (Rosner index 33.5), prothrombin time 83% (normal range 85–100%), fibrinogen 4.5 g/L (normal range 2–4 g/L), and D-Dimer 500 ng/mL (normal range < 500 ng/mL). Peripheral blood smear was normal. Repeat aPTT was performed to confirm the results. Chromogenic quantification of the coagulation factors revealed the presence of 5% FVIII activity measured by FVIII assay; in this assay, the percentage of FVIII in plasma is determined by the degree of correction obtained when patient plasma is added to human plasma that has been depleted of FVIII but contains normal concentrations of all other coagulation factors. Other factors were within the normal range. The anti-human FVIII inhibitor concentration as quantified by the Bethesda assay was 26 Bethesda units (BU).3 The PFA-100 (Platelet Function Assay) and the von Willebrand factor were normal. Serum electrophoresis showed monoclonal immunoglobulin IgA kappa type of 4 g/L. Bone marrow aspiration showed 3% of non-dystrophic plasma cell infiltration. The autoimmune work-up showed negative antinuclear antibodies, positive anti β2 glycoprotein 1 antibodies IgG serotype at 25 U/mL (normal range < 7 U/mL) and IgM serotype at 143 U/mL (normal range < 7 U/mL), and positive anticardiolipin antibodies IgM serotype at 175 UMPL/mL (normal range 15–40 U/mL) and anticardiolipin IgG serotype at 13 U GPL/mL (normal range 15–40 UGPL/mL). Hepatitis B, C serology and HIV serology were negative as well as cytomegalovirus and Epstein-Barr serology. The diagnosis of acquired hemophilia associated with antiphospholipid antibodies in a patient with monoclonal gammopathy of unknown significance was retained. The patient received 3 injections of recombinant factor VII (rFVIIa) and blood transfusion. Besides, he was started on oral steroids (60 mg/d with progressive tapering) and oral cyclophosphamide (2 mg/kg/day) for 6 weeks. We observe an increase in FVIII levels and a disappearance of anti FVIII inhibitor concentration 1 month after starting immunosuppressive treatment. The patient was discharged after 1 month in hospital. He was reevaluated at an outpatient clinic. Antiphospholipid antibodies remained positive after 3 months as well as prolonged aPTT, factor VIII raised at 100%, and the inhibitor was persistently not detected. The patient recovered progressively without treatment side effects. Neither bleeding nor thrombotic events were reported. Three months after discharge, we started thromboprophylaxis with aspirin at 100 mg/day.
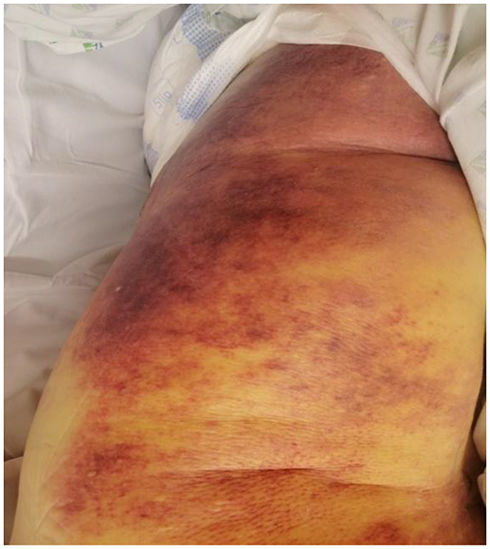 |
Figure 1 Diffuse cutaneous hemorrhage over the thigh. |
Discussion
We report the case of an elderly patient presenting with diffuse subcutaneous hematoma and hemothorax after a fall revealing a coexistence of acquired hemophilia and positive antiphospholipid antibodies. Acquired hemophilia is a very rare (1.5 per million per year) and life-threatening coagulopathy causing death of 7.9% to 22% of patients.4 According to the European Acquired Hemophilia (EACH2) registry, bleeding episodes are spontaneous in 77% of cases and cause at major time diffuse cutaneous and internal hemorrhage. Contrary to hereditary hemophilia, hemarthrosis is rare.5 This condition results from antibodies targeting coagulation factor usually factor VIII and specifically its C2 domain. Several large registers have established that idiopathic AHA is the most frequent type accounting for up to 50% of the cases.6 Underlying malignancy is present in 10% to 20% in the most important registers. The majority are related to solid tumors, especially carcinoma of the prostate and lung, each of which accounts for about 25% of cases of AHA. Of the hematological malignancies, which are less frequently associated with AHA than are solid tumors, lymphoid neoplasms were the most frequent.7 In a recent literature review, the main locations of bleeding were the skin and muscle; gastrointestinal and retroperitoneal bleeding and hematuria were other noted symptoms. Hemothorax, as illustrated in this case, was reported in only one report.8 Coexistence of factor VIII inhibitor and lupus anticoagulant was reported.6,9 Both will cause prolongation of aPTT with different mechanism of inhibition but different clinical presentation. This would be of great interest for physician managing patients with acute hemorrhage and prolonged aPTT and highlight the importance of the dosage of factor VIII as first step, and think of acquired cause in the elderly patient with past history of coagulopathy. The coexistence of antiphospholipid antibodies with a biological tendency to systemic thrombosis is not well established and complicate the patient management. Hemorrhagic complications in primary antiphospholipid syndrome are rare and reported in patients with lupus anticoagulant-hypoprothrombinemia syndrome or catastrophic antiphospholipid syndrome because of an association of an acquired factor II deficiency in the first case and low platelet count in the second.10 In the current observation, neither low prothrombin time nor low platelet count was found. The exact significance of high titles of antiphospholipid antibodies has to be defined but could mitigate the bleeding episodes caused by acquired factor VIII deficiency and vice versa. We hypothesize an association of two autoimmune diseases with a common etiopathogenesis due to endothelial injury. The treatment consists of hemostatic management and eradication of the inhibitors. Few data are available to guide the management of acquired hemophilia-related bleeding and the eradication of disease-causing antibodies. According to North American recommendations, the patient received a by-passing therapy consisting of recombinant FVII activated (rFVIIa) and blood transfusion support. The management of patients with acute hemorrhage with acquired hemophilia has two targets: hemostatic control and clearance of inhibitors. Bypass agents are the first-line treatments for bleeding control, including recombinant activated factor VII (rFVIIa), activated prothrombin complex concentrate, or recombinant porcine FVIII in bleeding patients.5,11 According to the international recommendations on the diagnosis and treatment of acquired hemophilia A, desmopressin is not recommended for the treatment of clinically relevant bleeding in patients with AHA. The use of recombinant or plasma-derived human FVIII concentrates only if by passing agents or rpFVIII are unavailable or ineffective and the inhibitor titer is low 11. Autoantibody eradication can be achieved with immunosuppressive therapy, including prednisone at a dose of 1 mg/kg/d for a maximum of 4–6 weeks (followed by a tapered withdrawal), cyclophosphamide at a dose of 1.5–2 mg/kg/day per os for 6 weeks, rituximab at a dose of 375 mg/m2 weekly for a maximum of 4 cycles, or mycophenolate mofetil at a dose of 1g/day for 1 week, followed by 2g/day.14 In our case, we used 3 injections of recombinant factor VII (rFVIIa) with favorable outcome. The eradication of the inhibitors required immunosuppressants. Of the reported cases, seven received steroids alone or were associated with other immunosuppressants, such as rituximab, vincristine or cyclophosphamide.12 Our patient received oral cyclophosphamide and antibodies control showed a success of eradication at the end of treatment.
Ethics and Consent Statements
A written informed consent has been signed by the patient to have the case details with published images. No institutional approval was required to publish the case details.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gupta D, Chatterjee T, Sharma A, et al. Rare case of acquired haemophilia and lupus anticoagulant. Indian J Hematol Blood Transfus. 2014;30(3):197–200. doi:10.1007/s12288-012-0204-5
2. Auwerda JJA, Sonneveld P, de Maat MP, et al. Prothrombotic coagulation abnormalities in patients with paraprotein-producing B-cell disorders. Clin Lymphoma Myeloma. 2007;7(7):462–466. doi:10.3816/CLM.2007.n.027
3. Sahud MA. Factor VIII inhibitors. Laboratory diagnosis of inhibitors. Semin Thromb Hemost. 2000;26:195–203. doi:10.1055/s-2000-9823
4. Sallah S, Wan JY. Inhibitors against factor VIII in patients with cancer. Analysis of 41 patients. Cancer. 2001;91(6):1067–1074. doi:10.1002/1097-0142(20010315)91:6<1067::AID-CNCR1101>3.0.CO;2-4
5. Baudo F, Collins P, Huth-Kühne A, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012;120(1):39–46. doi:10.1182/blood-2012-02-408930
6. Brings HA, Waas JK, McCrae KR, et al. Successful management of life-threatening hemorrhage in a patient with synchronous lupus anticoagulant and factor VIII inhibitor. J Vasc Surg. 2002;36(4):853–855. doi:10.1067/mva.2002.127336
7. Bastida JM, Cano-Mozo MT, Lopez-Cadenas F, Vallejo VE. Hemorrhagic pericardial effusion as the debut of acquired hemophilia in a chronic lymphocytic leukemia patient: a case report, and a review of acquired hemophilia A-related hematological malignancies. Medicine (Baltimore). 2017;96(47):e8669. doi:10.1097/MD.0000000000008669
8. Barbara DW, McKenzie KM, Parikh SA, et al. Successful perioperative management of severe bleeding from undiagnosed acquired factor VIII inhibitors. J Cardiothorac Vasc Anesth. 2015;29(3):731–734. doi:10.1053/j.jvca.2013.10.015
9. Taher A, Abiad R, Uthman I. Coexistence of lupus anticoagulant and acquired haemophilia in a patient with monoclonal gammopathy of unknown significance. Lupus. 2003;12(11):854–856. doi:10.1191/0961203303lu463cr
10. Bel Feki N, Zayet S, Ben Ghorbel I, et al. Lupus anticoagulant-hypoprothrombinemia syndrome presenting with coexisting cerebral venous thrombosis and subdural hemorrhage. J Mal Vasc. 2016;41:403–406. doi:10.1016/j.jmv.2016.07.003
11. Kruse-Jarres R, Kempton CL, Baudo F, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92(7):695–705. doi:10.1002/ajh.24777
12. Páramo L, Enciso Olivera LJ, Noreña I, et al. First case of acquired hemophilia B in a patient with HIV infection: case report and literature review. Cureus. 2019;11(3):e4179. doi:10.7759/cureus.4179
13. Tiede A, Collins P, Knoebl P, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica. 2020;105(7):1791–1801. doi:10.3324/haematol.2019.230771
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.