")
Back to Journals » International Journal of Nanomedicine » Volume 13
Codelivery of doxorubicin and JIP1 siRNA with novel EphA2-targeted PEGylated cationic nanoliposomes to overcome osteosarcoma multidrug resistance
Authors Haghiralsadat F, Amoabediny G, Naderinezhad S, Zandieh-Doulabi B , Forouzanfar T, Helder MN
Received 25 August 2017
Accepted for publication 17 March 2018
Published 3 July 2018 Volume 2018:13 Pages 3853—3866
DOI https://doi.org/10.2147/IJN.S150017
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Fateme Haghiralsadat,1–3 Ghasem Amoabediny,3–5 Samira Naderinezhad,4 Behrouz Zandieh-Doulabi,6 Tymour Forouzanfar,5 Marco N Helder5
1Department of Life Science Engineering, Faculty of New Sciences and Technologies, University of Tehran, Tehran, Iran; 2Department of Orthopaedic Surgery, VU University Medical Center, MOVE Research Institute Amsterdam, Amsterdam, the Netherlands; 3Department of Nano Biotechnology, Research Center for New Technologies in Life Science Engineering, 4Department of Biotechnology and Pharmaceutical Engineering, School of Chemical Engineering, College of Engineering, University of Tehran, Tehran, Iran; 5Department of Oral and Maxillofacial Surgery, VU University Medical Center, MOVE Research Institute Amsterdam, 6Department of Oral Cell Biology and Functional Anatomy, Academic Center for Dentistry Amsterdam (ACTA), Vrije Universiteit Amsterdam and University of Amsterdam, MOVE Research Institute, Amsterdam, the Netherlands
Purpose: Osteosarcoma (OS) mostly affects children and young adults, and has only a 20%–30% 5-year survival rate when metastasized. We aimed to create dual-targeted (extracellular against EphA2 and intracellular against JNK-interacting protein 1 [JIP1]), doxorubicin (DOX)-loaded liposomes to treat OS metastatic disease.
Materials and methods: Cationic liposomes contained N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP), cholesterol, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and distearoyl-phosphatidylethanolamine–methyl-poly(ethylene glycol) (DSPE–mPEG) conjugate. EphA2 targeting was accomplished by conjugating YSA peptide to DSPE–mPEG. Vesicles were subsequently loaded with DOX and JIP1 siRNA.
Results: Characteristics assessment showed that 1) size of the bilayered particles was 109 nm; 2) DOX loading efficiency was 87%; 3) siRNA could be successfully loaded at a liposome:siRNA ratio of >24:1; and 4) the zeta potential was 18.47 mV. Tumor-mimicking pH conditions exhibited 80% siRNA and 50.7% DOX sustained release from the particles. Stability studies ensured the protection of siRNA against degradation in serum. OS cell lines showed increased and more pericellular/nuclear localizations when using targeted vesicles. Nontargeted and targeted codelivery caused 70.5% and 78.6% cytotoxicity in OS cells, respectively (free DOX: 50%). Targeted codelivery resulted in 42% reduction in the siRNA target, JIP1 mRNA, and 46% decrease in JIP1 levels.
Conclusion: Our dual-targeted, DOX-loaded liposomes enhance toxicity toward OS cells and may be effective for the treatment of metastatic OS.
Keywords: MAP kinase 8 interacting protein 1, MAPK8IP1, functionalization, cationic liposome, intracellular targeting, extracellular targeting
Introduction
Osteosarcoma (OS) is the most common primary malignant bone tumor in children and adolescents, with a worldwide incidence of 4 million patients/year and a peak incidence between the age range of 15–19 years. At present, the gold standard for the treatment of OS consists of multiagent neoadjuvant chemotherapy, followed by radical excision of the tumor (and metastases when feasible), and subsequently multiagent adjuvant chemotherapy. Five-year survival rates are 65%–70% in patients with localized disease, whereas in patients with metastatic or recurrent disease, survival rates drop to ~20%.1–4 The major cause for the limited efficacy of current therapies is the development of multidrug resistance (MDR) to cytotoxic therapy, which is thought to occur by the selection of resistant cell clones after a treatment cycle.5 These resistant clones can then lead to relapse or metastatic disease progression in a later stage of the disease.6 New treatments, aimed to subvert therapy resistance and reestablish OS sensitivity to existing therapies, are urgently needed.
Previously, we used an siRNA approach to target the intracellular kinase MAPK8IP1 (JNK-interacting protein 1 [JIP1]) and found that JIP1 siRNA was able to restore chemosensitivity to doxorubicin (DOX) in OS cells.7 siRNAs consist of a synthetic nucleic acid of ~21–25 base pairs complementary to one specific mRNA8 and, by interaction, target this mRNA for degradation. This high specificity makes siRNAs potent therapeutic agents for tumor targeting, but current downsides are their poor cellular uptake and instability in plasma.9
To overcome these disadvantages, encapsulation in liposomes is a valid option. In previous works, we developed a thermo- and pH-sensitive liposomal formulation for localized drug release.10,11 Liposomal DOX is a potent nanoscale chemotherapeutic drug, effective in a wide range of human neoplasms, and has been developed to overcome anthracycline-induced cardiomyopathy, improve toxicity against cancer, and reduce systemic drug exposure. Moreover, we functionalized the nanocarriers by coupling a YSA peptide, a ligand for the EphA2 receptor, which is highly upregulated on OS, to the PEGylated liposomes to specifically target the liposomes to OS cells.12
In this study, we tested the hypothesis that simultaneous delivery of DOX and siRNA via this functionalized carrier could efficiently reinstall chemosensitivity to DOX in OS. We extensively characterized our final formulation combining the different moieties and evaluated its synergistic anti-OS effect in vitro.
Materials and methods
Chemicals and biological material
PD-10 desalting columns were purchased from GE Healthcare Bio-sciences Corp. (Piscataway, NJ, USA). Sephadex G-50 was obtained from Pharmacia Biotech (Uppsala, Sweden). DOX hydrochloride (Ebewe Pharma, Unterach am Attersee, Austria) was suspended in water. N-[1-(2,3-Dioleoyloxy)propyl]-N, N, N-trimethylammonium methyl-sulfate (DOTAP) (chloride salt) and cholesterol were purchased from Avanti Polar Lipids (Avanti Polar Lipids Inc, Alabaster, AL, USA) and Sigma-Aldrich Co. (St Louis, MO, USA), respectively. Both 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and distearoyl-phosphatidylethanolamine–methyl-poly(ethylene glycol) (DSPE–mPEG) 2000 were obtained from Lipoid GmbH (Ludwigshafen, Germany). YSA peptide was synthesized by the Department of Oral Biochemistry, ACTA Amsterdam, and then conjugated with DSPE–PEG-2000-NHS to link it to the surface of a liposome. Sodium cholate and triton X-100 were purchased from Sigma-Aldrich Co. All other reagents and chemicals were of analytical grade.
Preparation of positively charged liposomal DOX
We modified our preliminary procedures described in detail in our previous work to prepare the appropriate vesicle for codelivery system.12,13 We synthesized the cationic liposome (L-DOX) by mixing DOTAP (7.89 mg), cholesterol (4.40 mg), DPPC (19.84 mg), and DSPE–PEG-2000 (6.44 mg) in a 20:30:70:5.4 molar ratio. YSA-L-DOX was prepared by covalent coupling of YSA peptide (0.617 mg) to the DSPE–PEG-2000 compound, resulting in a final 20:30:70:5.4:0.6 molar ratio. In brief, total lipids were dissolved in CHCl3 and dried at 55°C to form a film by rotary evaporation instrument and placed in 4°C to overnight for further drying. The dried thin film of liposomes was then hydrated with 3.30 mL (NH4)2SO4 (pH=4) for 45 min at 55°C, followed by transmembrane pH gradient method for hydration to improve the zeta potential and entrapment efficiency (Naderinezhad et al, unpublished data, 2017).
Residual (NH4)2SO4 buffer was removed with PBS (pH=8.87, 25°C) during 2 h by size exclusion chromatography on PD-10 desalting columns. Liposome extrusion was gradually performed using 200 and 100 nm polycarbonate membranes at 25°C using extruder device (Northern Lipids Inc., Vancouver, BC, Canada). DOX, diluted with PBS (pH=4.5) to 0.5 mg/mL, was slowly loaded into the liposomes (45 min at 60°C) after the extrusion step. Untrapped DOX was removed using Sephadex G-50 gel filtration in PBS (pH 7.4, 4°C). The DOX entrapment efficiency inside the vesicles was determined by UV–Vis spectrophotometry (482 nm) after digesting the vesicles with iso-propyl alcohol. The schematic representation of experiments, mechanism of action of the nanocarrier, and the biological outcome assessments are presented in Figure 1.
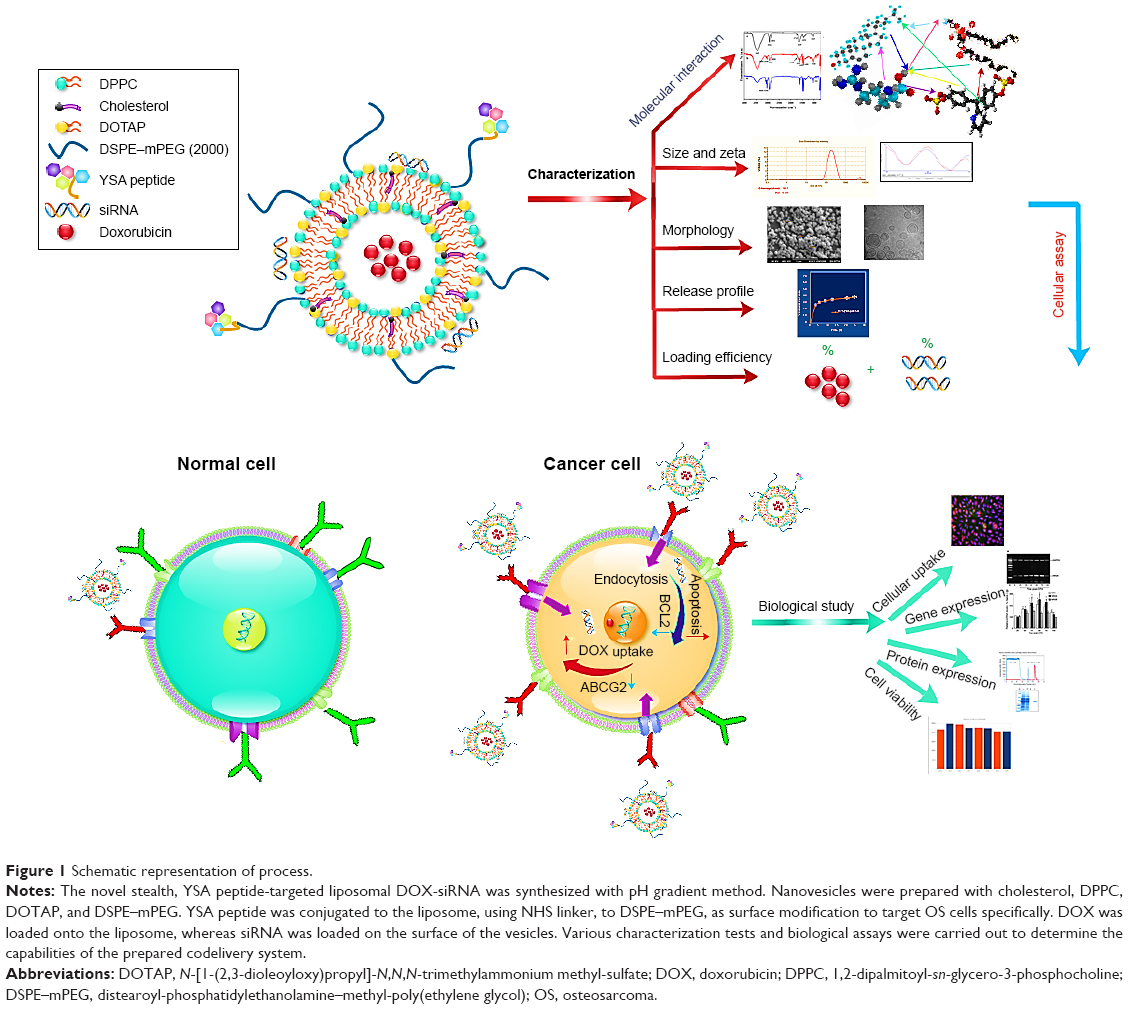 | Figure 1 Schematic representation of process. |
siRNA incubation
siRNA was synthesized by Eurogentec Co. (Liège, Belgium), and sense and antisense sequences were as follows: sense strand: 5′-GUA-CGA-GGC-CUA-CAA-CAU-G99-3′; antisense strand: 5′-CAU-GUU-GUA-GGC-CUC-GUA-C99-3′. The 5′ end was labeled with fluorescein amidite to allow tracking using florescence microscopy. To create the siRNA-loaded lipoplexes (L-siRNA-DOX and YSA-L-siRNA-DOX), the siRNA solution (stock solution with RNase-free H2O) was added to L-DOX and YSA-L-DOX, respectively. The prepared samples were subsequently vortexed and incubated for 30 min at 25°C to load the siRNA into the vesicles.
Determination of siRNA compilation with liposome
The loading efficiency of siRNA in L-siRNA-DOX and YSA-L-siRNA-DOX was investigated using an agarose gel retardation assay based on ethidium bromide (EtBr) dye displacement.
The various liposome/nucleotide acid ratios of complexes (80:1–1:1) were loaded into the agarose/EtBr gels and run in tris-Borate-ethylene-diamine-tetra-acetic acid buffer (4.5 mM of Tris base, 1 mM of sodium EDTA, 4.5 mM of boric acid, pH=8.3) at 90 V and 50 mA for 30 min. Images were analyzed using an UV transilluminator and a digital imaging system (Thermo Fisher Scientific, Waltham, MA, USA) to determine the approximate loading efficiency.
Size and charge determination
The measurement of size diameter (in volume and numeral mode) and polydispersity index (PDI) of the nanovesicles was carried out by dynamic-light scattering using the Particle Sizer Analyzer (Brookhaven Instruments Corp., New York, NY, USA) after diluting the sample with buffer. Zeta potential was determined with the same instrument.
In vitro DOX/siRNA release assay
Liposomal DOX release assessment was performed by the dialysis of 1 mL of the liposome preparations using a 12 kDa cutoff dialysis tube, at 37°C in PBS of either pH 7.4 or pH 5.4, under continuous stirring. The amount of DOX release into vesicle was detected spectrophotometry at 480 nm at predetermined time intervals by taking out of 1 mL of the dialysis medium and replacing it by the same volume of fresh PBS.
To measure the siRNA release pattern, L-siRNA-DOX was incubated in 10 cc of culture medium (DMEM) at pH 5.4 and 7.4 in 96-well plate. The 96-well plate was then placed in a shaker incubator for 1–48 h at 37°C. The concentration of released siRNA in sample was measured using a spectrophotometer at 260 nm followed by centrifuging at 5,000 rpm.
Microscopy
The morphology of YSA-L-siRNA-DOX was examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). For SEM analysis, 5 μL of suspension was poured on the glass plate to form a thin layer film and air dried. The sample was sputter coated with gold, and images were obtained using SEM (model EM3200; KYKY Technology Development Ltd, Beijing, China). For digital TEM imaging, a small sample was brought on a 200 Cu-coated grid, and images were acquired by Cryogenic TEM (FEI TEcnai 20 Sphera microscope, Hillsboro, OR, USA).
Molecular interaction
Droplets of liposome samples were placed on glass, air dried, and kept between two KBr pellets. The fourier-transform infrared spectroscopy (FTIR) spectra were obtained using the FTIR spectrometer (Model 8300; Shimadzu Corporation, Tokyo, Japan).
In vitro intracellular siRNA and DOX accumulation
To visualize cellular localization of the liposomal siRNA/DOX complexes under targeting and nontargeting conditions, an uptake assay was performed. In brief, 104 SaOs-2 cells per well were incubated for 24 h to allow to adhere, followed by 3 or 6 h incubation with YSA-L, YSA-L-siRNA, YSA-L-DOX L-siRNA-DOX, and YSA-L-siRNA-DOX. Afterward, cells were washed with PBS, fixed with 4% formalin, and visualized using the florescent microscopy imaging (Olympus Corporation, Tokyo, Japan). Fluorescein amidite-labeled siRNA (green fluorescence channel) and DOX (red fluorescence channel) and 6,4-diamidino-2-phenylindole (Thermo Fisher Scientific) were used for cell nuclei staining (red fluorescence channel), and tetramethylindocarbocyanine perchlorate (Sigma-Aldrich Co.) marker was added to liposomal formulation to visualize labeled liposomes.
Cell culture and cytotoxicity
SaOs-2 and MG-63 cell lines were provided by the Pasteur Institute (Tehran, Iran) and cultured with DMEM and 10% FBS to reach 70%–80% confluence.
Human primary bone chips (Human 63 and Human 65) were prepared from patients undergoing knee replacement after receiving written informed consent and cultured in RPMI-1640 medium (Thermo Fisher Scientific). All cells, seeded in 96-well plates, were incubated for 24 h to adhere overnight. Then, cells were treated with empty liposome (L) and YSA-liposome (YSA-L) as well as free siRNA, free DOX, liposomal DOX-siRNA (L-siRNA-DOX), and YSA-targeted siRNA-loaded liposomal DOX (YSA-L-siRNA-DOX). Afterward, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was carried out to determine cell viability by the adding MTT solution to the medium in each well and continuing incubation for 3 h. Viability percentage was determined using the EPOCH Microplate Spectrophotometer (BioTek, Winooski, VT, USA) at 570 nm after solubilizing formazan crystals with DMSO.
The Medical Ethics Review Committee of VU University Medical Center confirmed that the WMO Medical Research Involving Human Subjects Act does not apply to this research under decision 2016.105 and is exempt from approval.
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) evaluation
Cells were seeded at a density of 105 cell/well for 24 h before experiment. Then, cells were treated with free DOX, YSA-L, YSA-L-siRNA, YSA-L-DOX, L-siRNA-DOX, and YSA-L-siRNA-DOX for 48 h. The cells were then trypsinized and washed with cold PBS. Samples were centrifuged and suspended in a culture medium. Finally, cells were stained for 5 min with Annexin V-FITC and PI to distinguish apoptotic cells after prior incubation in the dark on ice for 20 min. Apoptosis was studied with flow cytometry (Annexin V-FITC at wavelength 525 nm).
Real-time polymerase chain reaction (PCR) assay
The expression of J1P1 in OS cell lines and primary bone cells was measured by quantitative reverse transcription polymerase chain reaction (qRT-PCR) and using protocol explained elsewhere. In brief, cells were treated with YSA-L, DOX, YSA-L-DOX, siRNA, YSA-L-siRNA, and YSA-L-siRNA-DOX, and total RNA was extracted using the Ambion kit (Thermo Fisher Scientific). The cDNA was synthesized using the RevertAid™ First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), and RT-qPCR was performed using SYBR Green Master Mix (Takara, Dalian, China) by Fast Real-Time PCR System (Rotor-gene Q, Qiagen, Stockach, Germany) with the following steps: 1) denaturation at 95°C for 10 min and 2) 40 cycles amplification at 95°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 30 s. The expression levels of the JIP1 mRNA were normalized to GAPDH mRNA expression levels.
Western blot analysis
SaOs-2 cells were seeded and treated for 24 h either in the absence of liposomes or in the presence of YSA-L and YSA-L-siRNA. Harvested cells were lysed on ice in NP40 buffer with protease inhibitor cocktails and phosphatase. The cell lysates were centrifuged, supernatants were collected, and the concentration of protein was determined using the Bradford protein assay by Qubit Protein Assay Kit Qubit (Thermo Fisher Scientific). Then, 40 μg of protein was loaded on 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis gels. Next, proteins were transferred to nitrocellulose membranes using the Western blot system (Bio-Rad Laboratories Inc., Hercules, CA, USA) under 60 mA for 1.5 h. The membranes were soaked in blocking buffer containing 5% bovine serum albumin for 1 h followed by further shaking at 25°C for 2 h to block nonspecific binding and then incubated overnight at 4°C with primary antibody diluted in tris buffered saline–Tween buffer with the ratio of 1:1,000.
Finally, the membrane was washed with tris buffered saline–Tween for three times and incubated with 1:2,000 diluted goat–anti-rabbit immunoglobulin G-horse radish peroxidase conjugated secondary antibodies for 2 h at room temperature. The blots were then incubated with the electrochemoluminescency Western Blot Detection Reagents (Thermo Fisher Scientific), according to the manufacturer’s instructions. We used β-actin antibody for the normalization of MAPK8IP1 protein levels.
Statistical analysis
Statistical analyses were performed to determine the statistical significance by one-way analysis of variance and Student’s t-test. All tests were performed on triplicate sample data.
Results
Characterization of nanostructure
Size and charge determination
Vesicle size and zeta potential of nanoliposome were measured by Zetasizer, and data are reported in Table 1. The size slightly increased with siRNA loading (average 20.4%). The incorporation of YSA peptide increased the size of L-DOX particles from 88.7 to 90.5 (2.03%) and of L-siRNA-DOX from 106.9 to 108.9 nm (1.87%). The zeta potential was also significantly decreased with siRNA loading (57.42%) and functionalizing with YSA peptide agent (71.2%). All formulations are homogeneously distributed with a PDI of <0.3. Nanoparticles are monodispersing with irrespective changes made by incorporation with YSA.
 | Table 1 The study of liposome formulation when added targeting function (YSA) and siRNA loading and comparison in terms of size, zeta potential, and loading efficiency |
As listed in Table 1, the average loading efficiency of DOX into L-DOX was 86.97%. Incorporation of YSA peptide and/or siRNA did not significantly alter loading efficiency. Figure 2 shows the gel retardation assay of the YSA-L-DOX formulation loaded with siRNA using different ratios of liposome:siRNA, from 1:1 to 80:1 (Figure 2A and B). Unloaded siRNA and siRNA from liposomal formulations prepared using liposome:siRNA ratios <24:1 migrated during electrophoresis, whereas siRNA loaded at a ratio of >24:1 showed successful complexation with the liposomes, as evidenced by its exclusive retention of siRNA signal in the gel slots. A similar trend was observed for the L-siRNA-DOX. Note that the wells showed more signal due to the intrinsic fluorescence of DOX.
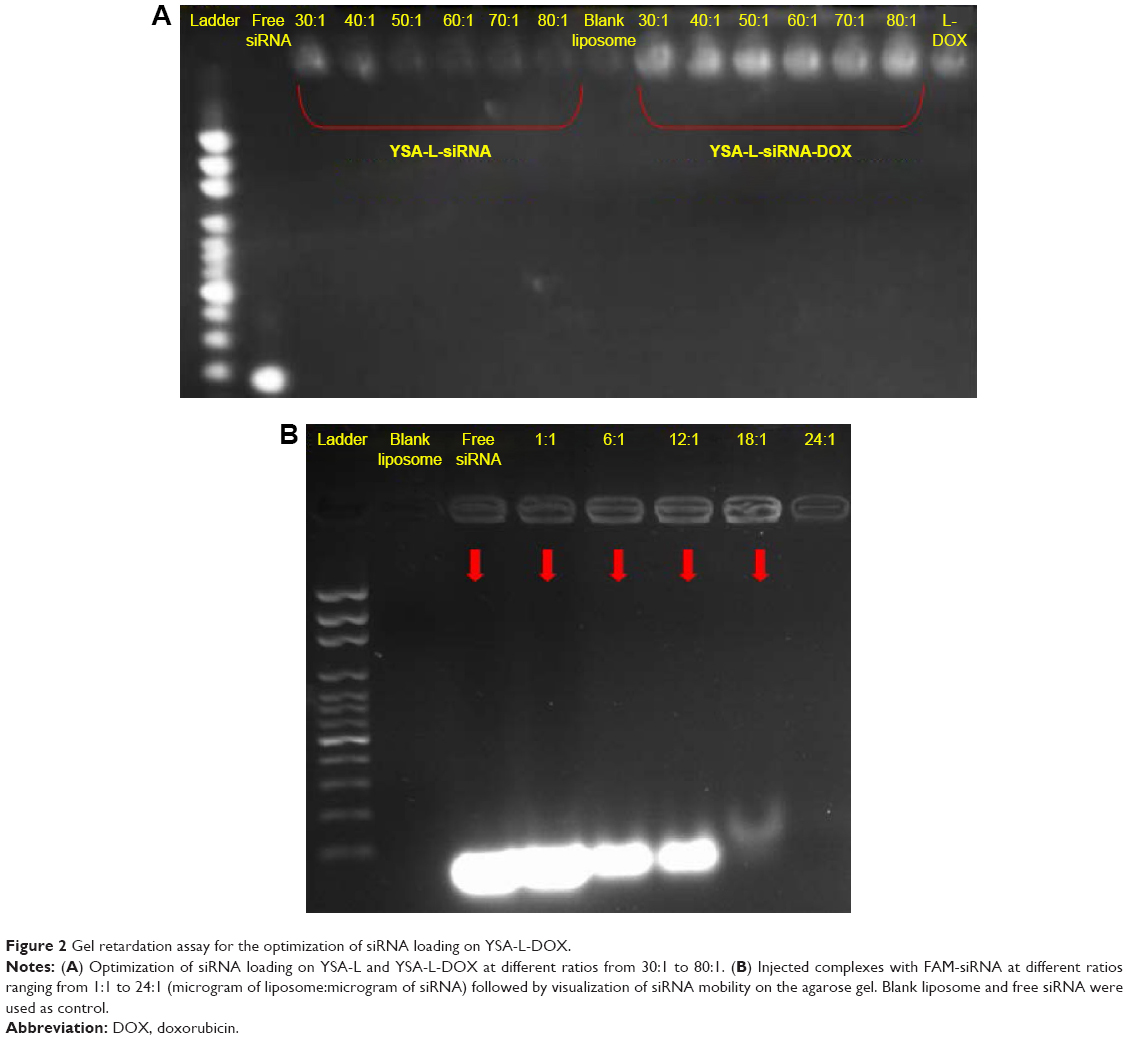 | Figure 2 Gel retardation assay for the optimization of siRNA loading on YSA-L-DOX. |
Drug release pattern
The release of siRNA and DOX from the targeted liposomes displayed a pH-sensitive pattern (Figure 3). Targeted nanoliposome–siRNA–DOX complexes were kept in a medium (PBS/DMEM) at various pH values (5.4 and 7.4). Both DOX and siRNA release from YSA-L-siRNA-DOX in the medium were time dependent and pH dependent. A similar trend was observed for the nontargeted formulation. As shown in Figure 3, the gene and drug release increased with decreasing pH (pH 5.4 mimics the pH in cancerous tissues). After 48 h, the cumulative siRNA release from YSA-L-siRNA-DOX reached to 80% at pH 5.4 and only 21% at pH 7.4. The DOX release acts in the same manner, so that 50.7% and 29.6% are released at pH 5.4 and 7.4, respectively.
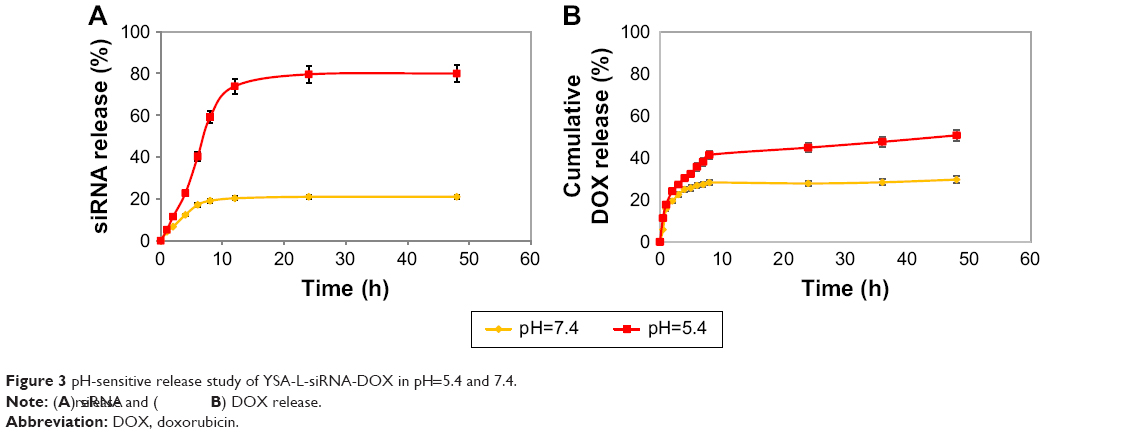 | Figure 3 pH-sensitive release study of YSA-L-siRNA-DOX in pH=5.4 and 7.4. |
Morphology
In this study, homogeneous YSA-L-siRNA-DOX nanoparticles were prepared using the pH gradient method, followed by sonication of the nanoparticle suspension. Figure 4A and B shows the SEM and TEM images of YSA-L-siRNA-DOX, which clearly indicated that nanoparticles are monodispersed with spherical–elliptical structure. As expected, the average diameter of nanoparticle was ~100 nm, which confirms the sizes measured using dynamic light scattering. The SEM images show that the drug- and gene-loaded vesicles were well-defined entities with smooth surface and only few aggregations. The TEM photograph showed the presence of single-layered but primarily bilayered vesicles.
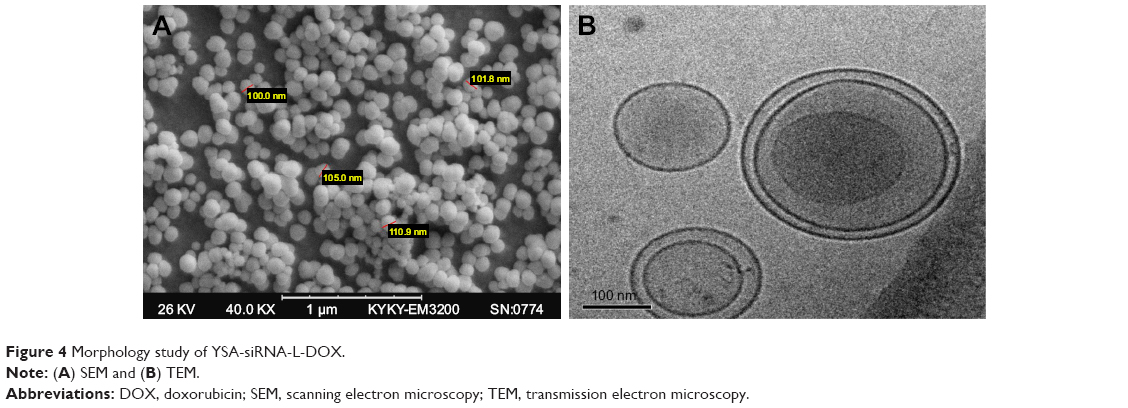 | Figure 4 Morphology study of YSA-siRNA-L-DOX. |
FTIR spectrum
We studied the FTIR spectrum of L-siRNA-DOX and YSA-L-siRNA-DOX, and the results are indicated in Figure 5. In our previous work (Haghiralsadat et al, unpublished data, 2017),14 we illustrated the FTIR analysis of YSA, DOX, and L to show integrity in our formulation and the impact of physical forces on loading DOX and siRNA in liposome. As shown in Figure 4, all characteristic peaks are repeated in FTIR spectrum of YSA-L-siRNA-DOX, no additional peaks were observed, and no new chemical bindings were created, ie, siRNA and DOX were loaded without any direct chemical interaction.
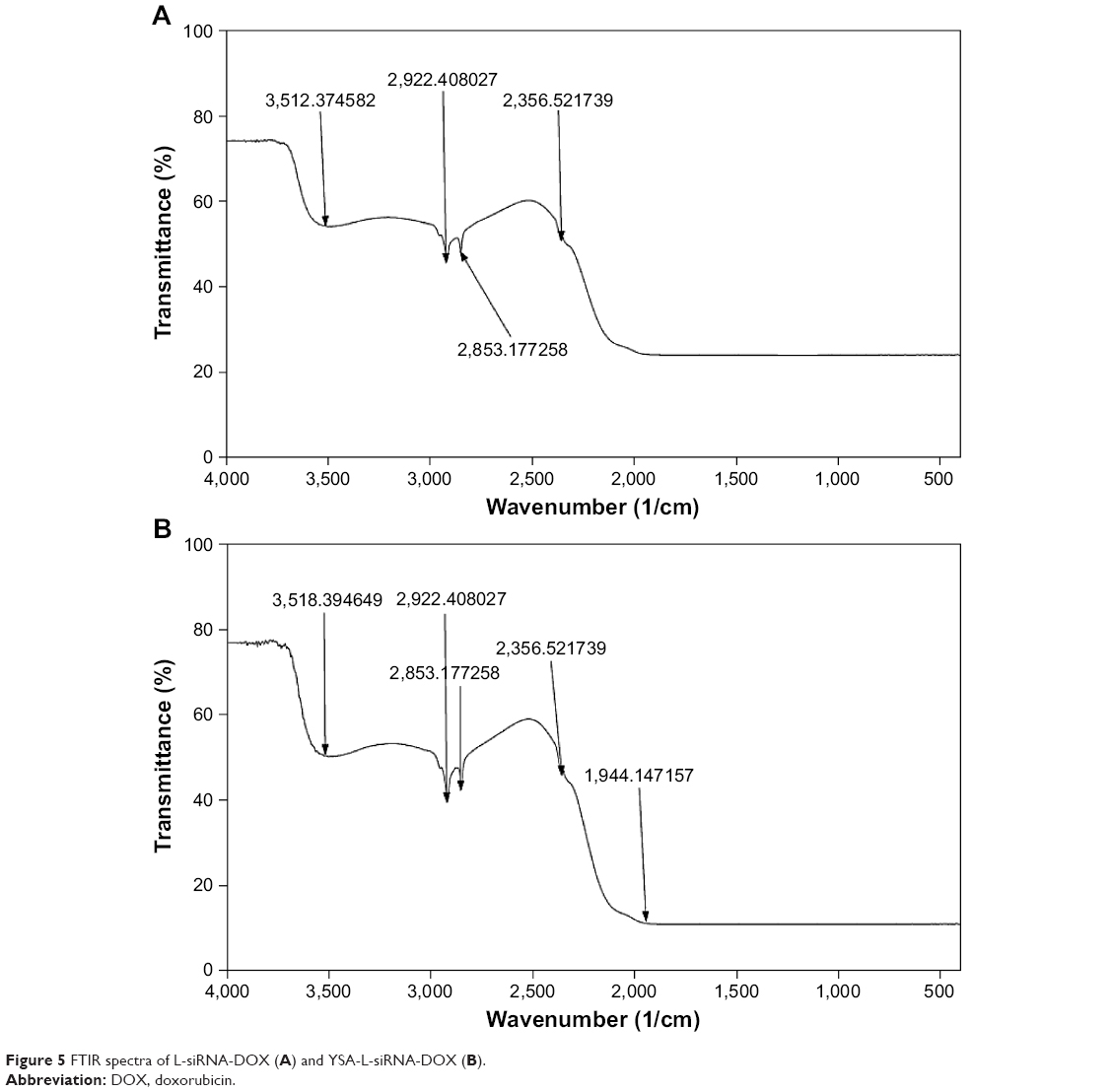 | Figure 5 FTIR spectra of L-siRNA-DOX (A) and YSA-L-siRNA-DOX (B). |
Therapeutic effect
Cellular transfection
The ability of liposomal vesicles to deliver the chemotherapeutic drug DOX and the chemosensitizer JIP1 siRNA to OS cells was studied in SaOs-2 cells (Figure 6). We carried out four types of experiments. First, we showed cellular transfection of carriers targeted to EphA2 with the YSA peptide, loaded with a single therapeutic agent (YSA-L-DOX or YSA-L-siRNA). Second, we assessed the difference in delivery efficiency using the nontargeted and targeted codelivery system (L-siRNA-DOX and YSA-L-siRNA-DOX, respectively) in Figure 6B. It can be deduced that more substantial transfection was achieved when therapeutic agents were loaded simultaneously in the YSA-functionalized carrier compared to its nontargeted counterpart. In the third transfection series, we assessed the time-dependent (3 and 6 h) uptake and intracellular locations of the compounds. As shown in Figure 6C, after 3 h, both DOX and siRNA were detected within the cytoplasm as well as the cytoplasm of the cells, whereas both compounds accumulated primarily in the nucleus after 6 h.
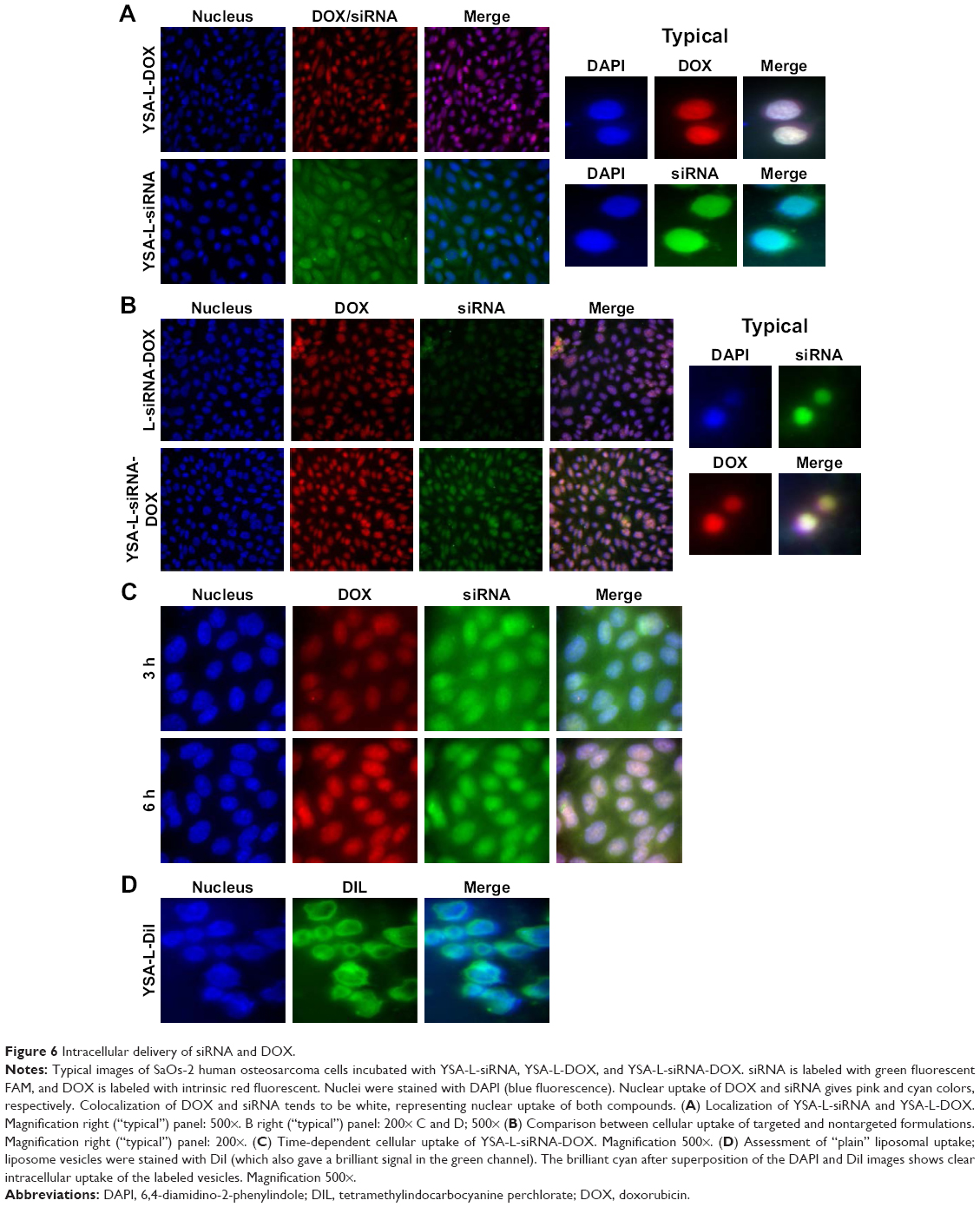 | Figure 6 Intracellular delivery of siRNA and DOX. |
Finally, to address whether the observed florescence intensity for siRNA or DOX was due to active release of DOX and siRNA intracellularly, or to prior released drug/siRNA outside the cells, we formulated our liposome with 0.1 mol% Dil. The intracellular, perinuclear signal confirmed the successful cellular uptake of the nanoliposomes.
Cytotoxicity
Cytotoxicity effects of various formulations were assessed with MTT assays in the MG-63 and SaOs-2 human OS cell lines and two human primary bone cell cultures (Figure 7). It was found that treatment with either blank liposomes or only siRNA (1 μg/mL) for 48 h did not or only marginally inhibit cell growth in all cell cultures compared to control conditions, whereas free DOX reduced cell viability to ~50%, again showing no difference between the OS and the primary bone cells.
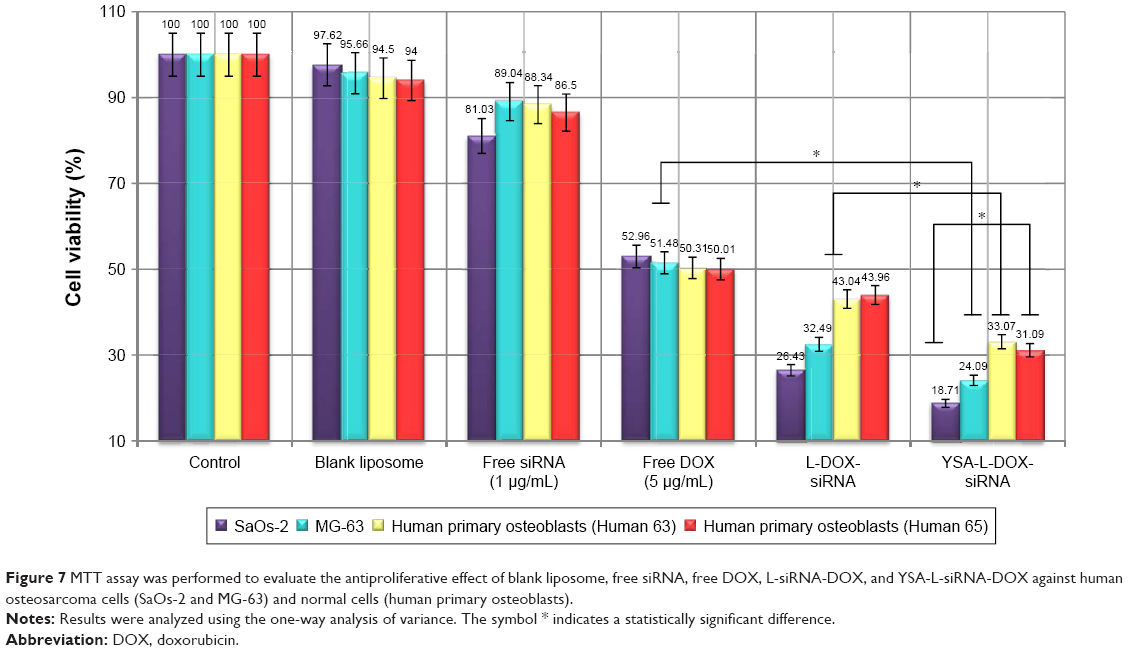 | Figure 7 MTT assay was performed to evaluate the antiproliferative effect of blank liposome, free siRNA, free DOX, L-siRNA-DOX, and YSA-L-siRNA-DOX against human osteosarcoma cells (SaOs-2 and MG-63) and normal cells (human primary osteoblasts). |
Liposomal codelivery of DOX and siRNA markedly increased the cytotoxicity. Nontargeted and targeted codelivery resulted in only 29.5% and 21.4% remaining viability in OS cells, respectively, whereas the primary bone cell cultures showed less-affected viability levels (43.5% and 32.1%). Both the nontargeted and YSA-targeted liposomal formulations showed an 1.5-fold increased cell kill in OS cells compared to primary bone cells. Moreover, the YSA-functionalized formulation (YSA-L-siRNA-DOX) decreased the viability of the OS cells 1.4-fold compared to its nontargeted counterpart (L-siRNA-DOX).
Taken together, the prepared formulation exhibited great potential for application in selective delivery of DOX and siRNA to OS cells.
Apoptosis assay
Apoptosis was investigated in SaOs-2 cells incubated with YSA-L, YSA-L-siRNA, DOX, YSA-L-DOX, L-siRNA-DOX, and YSA-L-siRNA-DOX for 2 days (Figure 8). Cationic YSA-L and YSA-L-siRNA slightly induced apoptosis. Targeted codelivery of DOX and siRNA enhanced apoptosis by 2.16- and 7.75-fold compared to the targeted single delivery of DOX and siRNA, respectively. The delivery of both compounds increased the apoptosis 1.24-fold compared to its nontargeted formulation. Delivery of DOX by the targeted carrier induced the apoptosis of DOX by 1.34 times compared to free DOX.
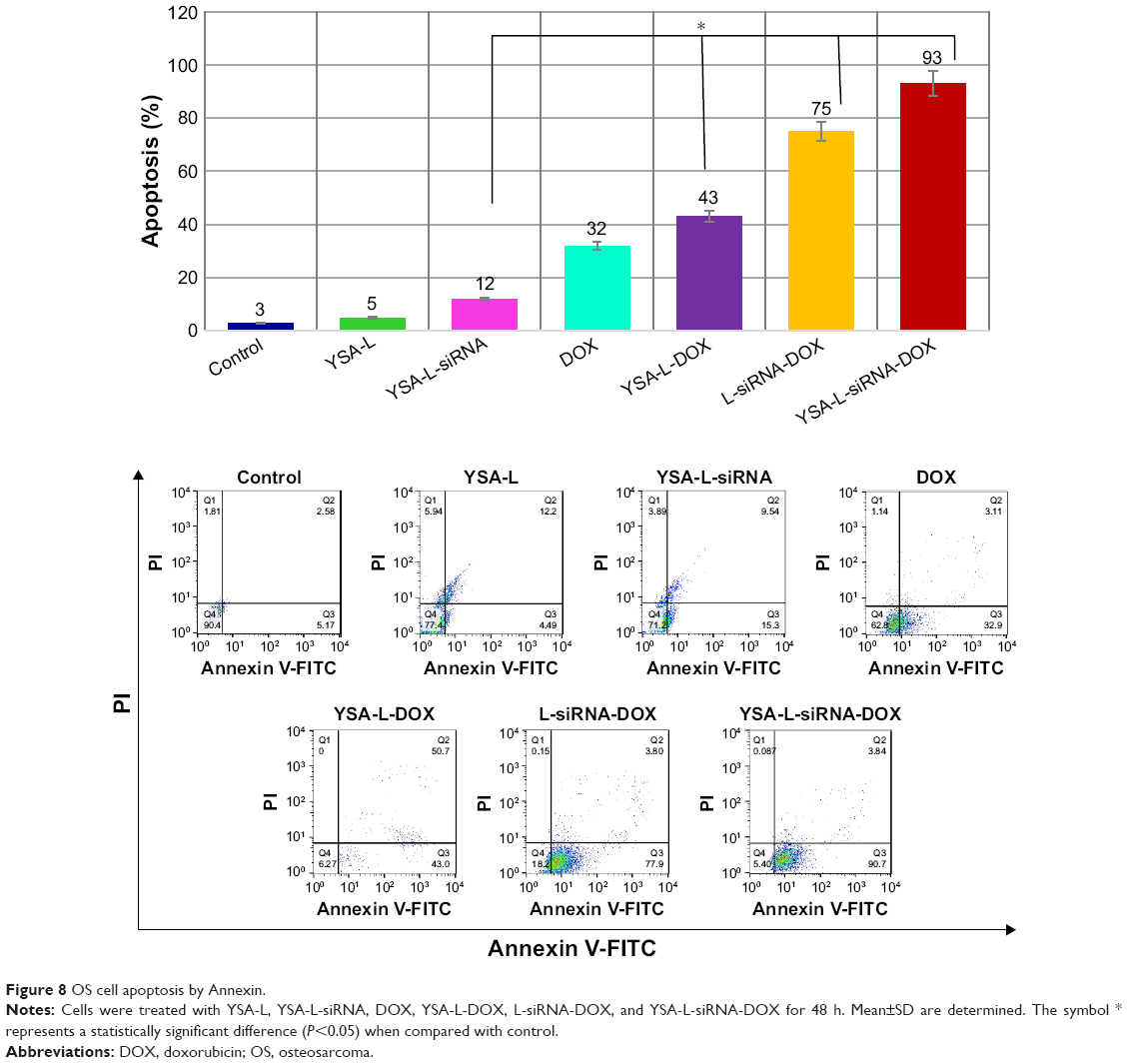 | Figure 8 OS cell apoptosis by Annexin. |
Gene expression analysis
The JIP1 expression in MG-63 and SaOs-2 cells after treatment with YSA-L, DOX, YSA-L-DOX, siRNA, YSA-L-siRNA, and YSA-L-siRNA-DOX was measured by RT-qPCR (Figure 9).
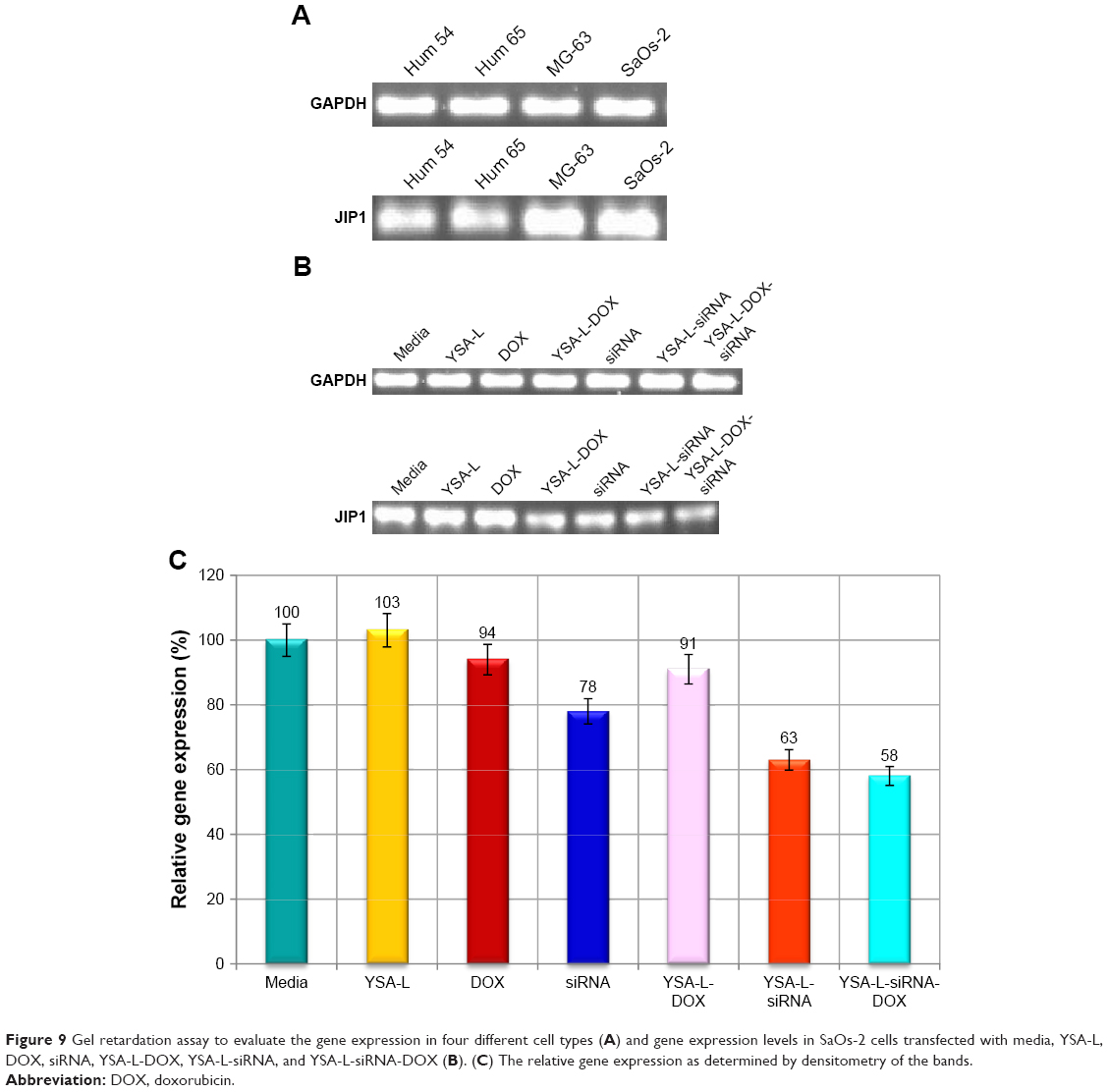 | Figure 9 Gel retardation assay to evaluate the gene expression in four different cell types (A) and gene expression levels in SaOs-2 cells transfected with media, YSA-L, DOX, siRNA, YSA-L-DOX, YSA-L-siRNA, and YSA-L-siRNA-DOX (B). (C) The relative gene expression as determined by densitometry of the bands. |
The expression of JIP1 was decreased when exposed to siRNA, YSA-L-siRNA, and YSA-L- siRNA-DOX. In contrast, JIP1 gene expression was not changed upon exposure to YSA-L, YSA-L-DOX, and DOX. The strongest inhibition of gene expression was detected for YSA-L-siRNA-DOX (44% reduced compared to the YSA-L value).
IP1 expression
To determine the specific inhibition of the JIP1 expression, SaOs-2 cells were treated by media, YSA-L, and YSA-L-siRNA for 24 h. Western blot analysis showed that JIP1 protein expression was significantly less in cells transfected by YSA-L-siRNA compared to YSA-L, which showed equal expression as the media control (Figure 10).
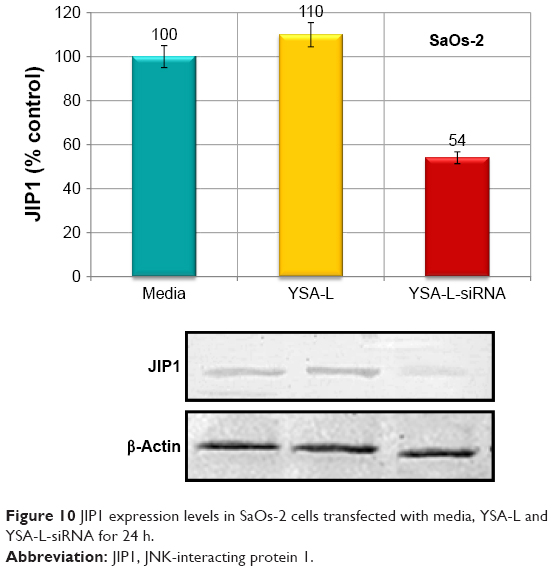 | Figure 10 JIP1 expression levels in SaOs-2 cells transfected with media, YSA-L and YSA-L-siRNA for 24 h. |
Discussion
OS metastases generally show a poorer response to chemotherapy than do primary tumors, indicating that metastatic OS cells have acquired enhanced MDR. Clinically, this causes a severe drop in 5-year survival rates (from 60% to 70% in nonmetastatic disease to 20% in metastatic disease). There are several reports suggesting that codelivery systems of drug and gene might overcome MDR in various types of cancer; however, none was reported on the codelivery to OS cell lines.15–17 In the current study, we created dual-targeted (extracellular YSA peptide-mediated targeting against the highly upregulated surface marker EphA2 in OS and intracellular siRNA targeting against JIP1 to reinstall chemosensitivity), DOX-loaded liposomes to overcome this MDR and achieve increased and OS-specific cell kill. Moreover, our previously optimized chemical composition of the liposomes10,14,18 should allow safe systemic delivery with increased DOX dosages, systemically stable siRNA, and efficient and OS-specific cellular uptake.
Our nanoparticle characterizations showed that 1) the size of the bilayered particles was 109 nm; 2) the DOX loading efficiency was 87%; 3) siRNA could be successfully loaded at a liposome:siRNA ratio of >24:1; and 4) the zeta potential of the final formulation, YSA-L-siRNA-DOX, was 18.47 mV. The significant decrease in zeta potential with siRNA incubation compared to the values obtained in L-DOX (46.73 mV) and YSA-L-DOX (42.34 mV) is probably due to the strong electrostatic interactions between the negatively charged siRNA and the positively charged cationic liposomal DOX. However, as evidenced by the preservation of siRNA–liposome complex during several months of storage at 4°C (Haghiralsadat et al, unpublished data, 2017), the zeta stability of our formulations was more than sufficient. For future clinical application, this long shelf-life is an important asset for the development of cost-effective treatment modalities.
Stable storage characteristics should not preclude effective release of the therapeutic agents when used in a therapeutic setting. Strong release from the particles, ie, 80% siRNA and 50.7% DOX, was observed under tumor-mimicking pH conditions (pH 5.4). In contrast, release of both compounds at neutral pH showed markedly lower values, ie, 21% and 29.6%, respectively. This pH effect fits our previous studies and controlled release modeling as published previously.14 The low release at neutral pH and enhanced release at lower pH, which often occurs due to hypoxic conditions in tumor tissue and intracellularly in lysosomes, ensures efficient and localized delivery in the tumor area and intracellularly in the tumor cells, while avoiding high systemic exposure to DOX and the loss of siRNA prior to reaching the cancer cells. Interesting to note is that the DOX release was slightly higher in the current codelivery formulation compared to the non-siRNA containing particles with the same formulation (43%)14 but higher than formulations not containing the positively charged DOTAP component in the membranes (37%).18
In our assessment of the delivery and transfection characteristics of the formulations, OS cell lines showed increased and more pericellular/nuclear localizations when using targeted vesicles. Thus, it appears that our formulations benefit the following two types of targeting: first, active uptake of the vesicles is promoted by the targeting to the extracellular EphA2 receptor and, second, more extensive intracellular release from the lysosomes is achieved by its pH sensitivity, ie, accelerated release upon exposure to acidic conditions. The time dependency of this process and its acceleration by the targeting moiety are illustrated in Figures 3 and 6C. When using targeted vesicles, the fluorescence was already high in the cytoplasm after 3 h and perinuclear region and accumulation into the nucleus was already observed after 6 h, whereas the nontargeted particles showed slower as well as lower uptake and translocations. According to the release profile, ~20% of DOX and 12% of siRNA released during 3 h; thus, the intracellular fluorescence may theoretically be primary due to release of compounds outside the cell followed by diffusion or phagocytotic uptake. However, using unloaded Dil-labeled liposomes, we could verify that the intracellular uptake is indeed by active vesicle internalization and not by extracellular release prior to internalization of the particles.
The postulated therapeutic synergy of the codelivery of siRNA and DOX, compared to free DOX or application of formulations loaded with one single compound only, was evident from the cytotoxicity studies, with an additional added value of the YSA-targeting moiety. Nontargeted and targeted codelivery caused 70.5% and 78.6% cytotoxicity in OS cells, respectively, whereas free DOX resulted in only 50% cell kill. Moreover, free DOX showed no viability differences between the OS and the primary bone cells, whereas the liposomal nontargeted and in particular the targeted liposomal codelivery formulations showed a clear differential cytotoxicity with increased cell kill in the OS cells. This again stresses the advantages of targeted liposomal DOX delivery over “normal” chemotherapeutic systemic delivery. It was verified that the mechanism by which the codelivery formulations induced this cytotoxicity was via induction of apoptosis: targeted codelivery of DOX and siRNA induced virtually complete apoptosis (93% apoptotic cells) and thus enhanced apoptosis by 2.16- and 7.75-fold compared to the targeted single delivery of DOX and siRNA, respectively. The apoptotic cell death mediated by the specific reduction of the siRNA target, JIP1 mRNA, was verified by a 39% decrease in the JIP1 mRNA and a 51% decrease in JIP1 levels compared to the YSA-L value. Moreover, this is a clear indication that the siRNA release is effective, and the siRNA remains functional after internalization.
High electrostatic interaction between the negatively charged siRNA and the liposomes mainly charged positively with 20% DOTAP in liposome composition, assisted in efficient loading of siRNA. Cationic liposomes create an impediment by binding with insoluble blood proteins surmounting with DSPE–mPEG addition that spontaneously provides a shield against nuclease degradation and endosomal scape.19 Membrane stability was enhanced by the addition of cholesterol that promotes adhesion and rigidity in the membrane due to hydrogen bonding between hydroxyl group in cholesterol and carbonyl group in phospholipids. It is thought that high amounts of cholesterol eliminate the thermosensitivity and pH sensitivity of formulation due to cholesterol increasing the rigidity and decreasing the permeability of the liposomal membrane. Thus, we used only <30% cholesterol in our formulation.
The liposome contained DPPC:CHOL:DSPE–mPEG at a 70:30:3 molar ratio, as we elucidated that drug release is more rapid due to the addition of DOTAP.10 The DPPC presence in liposome formulation leads to hyperthermia-triggered release properties meaning that it accrued the release for hydrophilic drugs at 42°C while it was steady at lower temperature.
How do our studies relate to other studies on liposomal codelivery strategies to reinstall chemosensitivity and combating MDR?
Navarro et al20 made a liposomal conjugate with dioleoylphosphatidylethanolamine (DOPE) and polyethylenimine (PEI) for the delivery of an siRNA silencing P-glycoprotein (P-gp), followed by DOX treatment after 48 h. Upon exposure of chemoresistant and chemosensitive MCF-7 breast cancer cells to the DOPE–PEI complexes for 48 h, cell viability after subsequent 48 h DOX treatment dropped to 30% for resistant MCF-7 and 35% for sensitive cells. DOPE–PEI complexes were found to significantly downregulate P-gp expression, whereas free siMDR-1 did not.20 Zheng et al21 used cationic layered double hydroxide nanoparticles to codeliver selenium, anti-P-gp, and anti-β-tubulin III siRNAs. Prepared nanoformulations exhibited an efficient gene-silencing effect on the expression of P-gp and β-tubulin III, induced cell apoptosis, and increased cellular reactive oxygen species levels in drug-resistant breast cancers.21 Peng et al22 synthesized a thermosensitive magnetic carrier for the codelivery of DOX and SATB1 shRNA vector to gastric cancer cells. Transfection rate was low (15%) but could be increased to 34% upon magnetic guidance. The magnetic-guided codelivery system resulted in a viability drop to 22.3% (free DOX: 40.3%). However, the apoptosis rate was markedly lower compared to our study (27.7%, whereas we found 93%). The difference in results may be due to differences in cell types, gene-silencing vector (shRNA compared to siRNA in this study), and lipid composition. Another reason may be because of the targeting properties of our formulations.22 Yang et al23 prepared liposomes targeted to the folate receptor encapsulating DOX and Bmi1 siRNA. In a panel of cell lines from various cancer types, they could achieve maximum delivery after 1 h treatment and found, as ours, ~30% cell viability after 48 h treatment with 1 μg/mL of targeted codelivery system. Interestingly, the gene-loading efficiency is 8.33 times lower than the current study, and the codelivery system reached equal efficacy as free DOX.23 No targeted codelivery system for treating OS has been reported on so far.
Conclusion
We developed a novel low-risk multitargeted method for OS therapy. Our formulation is stable with a high loading efficiency of DOX and siRNA, sustained release, and size diameter <150 nm. Both functionalization and siRNA loading can be accomplished without potentially harmful chemical reactions. Liposomes loaded with siRNA and DOX and functionalized with our synthesized YSA peptide showed strong cytotoxic effects against MG-63 and SaOs-2 cells while far less to primary bone cells, displayed markedly decreased JIP1 expression on the mRNA and protein level, and impressive induction of apoptosis in the OS cells. Taken together, our dual-targeted, DOX-loaded liposomes show promising perspectives for the effective treatment of metastatic OS, which should be further substantiated in a humanized in vivo animal model.24
Acknowledgment
We are grateful to Fateme Montazeri, Research and Clinical Center for Infertility, Shahid Sadoughi University of Yazd, for scientific assistance during the project.
Disclosure
The authors report no conflicts of interest in this work.
References
Anderson PM. Effectiveness of radiotherapy for osteosarcoma that responds to chemotherapy. Mayo Clin Proc. 2003;78(2):145–146. | ||
Bielack SS, Carrle D, Hardes J, Schuck A, Paulussen M. Bone tumors in adolescents and young adults. Curr Treat Options Oncol. 2008;9(1):67–80. | ||
Bielack SS, Kempf-Bielack B, Delling G, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol. 2002;20(3):776–790. | ||
Anderson PM, Wiseman GA, Erlandson L, et al. Gemcitabine radiosensitization after high-dose samarium for osteoblastic osteosarcoma. Clin Cancer Res. 2005;11(19 pt 1):6895–6900. | ||
Yang H-W, Lu Y-J, Lin K-J, et al. EGRF conjugated PEGylated nanographene oxide for targeted chemotherapy and photothermal therapy. Biomaterials. 2013;34(29):7204–7214. | ||
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. | ||
De Boer JP, van Egmond PW, Helder MN, et al. Targeting JNK-interacting protein 1 (JIP1) sensitises osteosarcoma to doxorubicin. Oncotarget. 2012;3(10):1169–1181. | ||
He C-X, Tabata Y, Gao J-Q. Non-viral gene delivery carrier and its three-dimensional transfection system. Int J Pharm. 2010;386(1–2):232–242. | ||
Xu C, Wang J. Delivery systems for siRNA drug development in cancer therapy. Asian J Pharm Sci. 2015;10(1):1–12. | ||
Haghiralsadat F, Amoabediny G, Sheikhha MH, et al. New liposomal doxorubicin nanoformulation for osteosarcoma: drug release kinetic study based on thermo and pH sensitivity. Chem Biol Drug Des. 2017;90(3):368–379. | ||
Naderinezhad S, Amoabediny G, Haghiralsadat F. Co-delivery of hydrophilic and hydrophobic anticancer drugs using biocompatible pH-sensitive lipid-based nano-carriers for multidrug-resistant cancers. RSC Adv. 2017;7(48):30008–30019. | ||
Haghiralsadat F, Amoabediny G, Naderinezhad S, et al. EphA2 targeted doxorubicin-nanoliposomes for osteosarcoma treatment. Pharm Res. 2017;34(12):2891–2900. | ||
Shahin M, Soudy R, El-Sikhry H, Seubert JM, Kaur K, Lavasanifar A. Engineered peptides for the development of actively tumor targeted liposomal carriers of doxorubicin. Cancer Lett. 2013;334(2):284–292. | ||
Haghiralsadat F, Amoabediny G, Helder MN, et al. A comprehensive mathematical model of drug release kinetics from nano-liposomes, derived from optimization studies of cationic PEGylated liposomal doxorubicin formulations for drug-gene delivery. Artif Cells Nanomed Biotechnol. 2018;46(1):169–177. | ||
Li J, Liu J, Guo N, Zhang X. Reversal of multidrug resistance in breast cancer MCF-7/ADR cells by h-R3-siMDR1-PAMAM complexes. Int J Pharm. 2016;511(1):436–445. | ||
Ling G, Zhang T, Zhang P, Sun J, He Z. Synergistic and complete reversal of the multidrug resistance of mitoxantrone hydrochloride by three-in-one multifunctional lipid-sodium glycocholate nanocarriers based on simultaneous BCRP and Bcl-2 inhibition. Int J Nanomedicine. 2016;11:4077. | ||
Liu J, Li J, Liu N, et al. In vitro studies of phospholipid-modified PAMAM-siMDR1 complexes for the reversal of multidrug resistance in human breast cancer cells. Int J Pharm. 2017;530(1–2):291–299. | ||
Haghiralsadat F, Amoabediny G, Hasan SM, et al. A novel approach on drug delivery: investigation of new nano-formulation of liposomal doxorubicin and biological evaluation of entrapped doxorubicin on various osteosarcomas cell lines. Cell J. 2017;19(Suppl 1):55–65. | ||
Shim G, Kim M-G, Park JY, Oh YK. Application of cationic liposomes for delivery of nucleic acids. Asian J Pharm Sci. 2013;8(2):72–80. | ||
Navarro G, Sawant RR, Biswas S, Essex S, Tros de Ilarduya C, Torchilin VP. P-glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine (Lond). 2012;7(1):65–78. | ||
Zheng W, Yin T, Chen Q, et al. Co-delivery of Se nanoparticles and pooled SiRNAs for overcoming drug resistance mediated by P-glycoprotein and class III β-tubulin in drug-resistant breast cancers. Acta Biomater. 2016;31:197–210. | ||
Peng Z, Wang C, Fang E, Lu X, Wang G, Tong Q. Co-delivery of doxorubicin and SATB1 shRNA by thermosensitive magnetic cationic liposomes for gastric cancer therapy. PLoS One. 2014;9(3):e92924. | ||
Yang T, Li B, Qi S, et al. Co-delivery of doxorubicin and Bmi1 siRNA by folate receptor targeted liposomes exhibits enhanced anti-tumor effects in vitro and in vivo. Theranostics. 2014;4(11):1096–1111. | ||
Jia S-F, Worth LL, Kleinerman ES. A nude mouse model of human osteosarcoma lung metastases for evaluating new therapeutic strategies. Clin Exp Metastasis. 1999;17(6):501–506. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.