")
Back to Journals » Infection and Drug Resistance » Volume 13
Co-Occurrence of the blaKPC-2 and Mcr-3.3 Gene in Aeromonas caviae SCAc2001 Isolated from Patients with Diarrheal Disease
Authors Tang L, Huang J, She J, Zhao K, Zhou Y
Received 10 January 2020
Accepted for publication 23 April 2020
Published 25 May 2020 Volume 2020:13 Pages 1527—1536
DOI https://doi.org/10.2147/IDR.S245553
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eric Nulens
Lingtong Tang,1,2,* Jianglian Huang,3,* Junping She,1 Kelei Zhao,4 Yingshun Zhou1
1Department of Pathogenic Biology, School of Basic Medicine, Southwest Medical University, Luzhou 646000, Sichuan, People’s Republic of China; 2Department of Clinical Laboratory, People’s Hospital of Gao County, Yibing 644000, Sichuan, People’s Republic of China; 3Department of Clinical Laboratory, The Second Affiliated Hospital of Xiamen Medical College, Xiaman 361600, People’s Republic of China; 4Antibiotics Research and Re-Evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, Chengdu University, Chengdu 610052, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yingshun Zhou
Department of Pathogenic Biology, School of Basic Medicine, Southwest Medical University, Luzhou, Sichuan 646000, People’s Republic of China, No. 319, Zhongshan Road, Tel +86-0830-3160073
Email [email protected]
Kelei Zhao
Antibiotics Research and Re-Evaluation Key Laboratory of Sichuan Province, Sichuan Industrial Institute of Antibiotics, No. 168, Huaguan Road, Chengdu 610052, Sichuan, People’s Republic of China
Tel +86– 028– 84216035
Email [email protected]
Purpose: To characterize the genetic feature of a multi-drug-resistant Aeromonas caviae strain isolated from the diarrhea sample of a 45-year-old male patient with acute diarrhea.
Materials and Methods: Whole-genome of the A. caviae strain SCAc2001 was sequenced via the Illumina system, followed by a series of bioinformatic analyses to describe the genetic feature.
Results: The genome sequence of A. caviae SCAc2001 was assembled into 340 scaffolds (305 of them were > 1000 bp in length and 4,487,370 bp in total) with an average G+C content of 61.09%. Phylogenetic analysis showed that the A. caviae SCAc2001 strain was highly similar to the A. caviae strain R25-2 and T25-39. Resistome analysis identified that A. caviae SCAc2001 carried 13 antimicrobial resistance genes, including β-lactams (blaKPC, blaCTX-M-14, blaTEM-1, blaOXA-10, blaOXA-427, blaVEB-3 and blaMOX-6), aminoglycosides (aadA1), fluoroquinolones (aac(6ʹ)-Ib-cr), phenicol resistance (catB3), sulfonamide (sul1), trimethoprim (dfrA5) and colistin resistance (mcr-3.3).And also, A. caviae ScAc2001 carried 54 putative virulence genes including the type IV pilus, fimbria, flagellarthe, and hemolysin A encoding genes, and 12 pathogen–host interactions (PHI) genes. There were also four genomic islands and eight prophages in the genome of A. caviae ScAc2001. In addition, A. caviae SCAc2001 also carried three secondary metabolism products coding clusters including nonribosomal peptide synthetases (nrps), hserlactone and bacteriocin.
Conclusion: A. caviae ScAc2001 carries many resistance genes, a variety of virulence factors, PHI genes and four genomic islands and eight prophages, which poses a severe threat to infectious diseases control strategies, diagnosis methods and clinical treatment.
Keywords: Aeromonas caviae, blaKPC-2, mcr-3.3, virulence factors, secondary metabolism products
Introduction
Colistin is the last resort for the treatment of infections caused by multidrug-resistant bacteria, particularly the carbapenem-resistant microorganisms.1,2 However, the mobile colistin-resistant gene mcr-1, was first reported in the Enterobacteriaceae by Liu et al3 in 2015, which led the mcr-1 carried bacteria resistant to colistin. Since then, mcr-1 or the mcr gene family-carrying bacteria have been reported in different species (such as E. coli, klebsiella pneumoniae, Acinetobacter, Pseudomonas and other gram-negative bacteria) isolated from food, animals, the environment and clinical samples worldwide.4–6 To our knowledge, co-carriers of the colistin-resistant gene mcr and carbapenemase-resistant genes (blaKPC, blaNDM, blaIMP, and blaVIM), microorganisms have potentially evolved into extensively drug-resistant or pan-drug-resistant isolates. Infections caused by these clinical isolates co-harboring the colistin-resistant gene (mcr-1) and carbapenem-resistant genes (blaKPC, blaNDM, blaIMP, and blaVIM) pose a serious threat because the antibiotic options would be much fewer.4,7,8 Aeromonas species, one kind of the gram-negative bacteria, was identified in the 1980s as an enteric pathogen which can lead to severe diarrhea.9 In addition, the carbapenem-resistant and/or colistin-resistant Aeromonas species strains have been reported increasingly in recent years to pose a serious threat in infection control.10–12 In this study, we recovered a colistin and carbapenem-resistant Aeromonas strain from the diarrhea sample of a 45-year-old male patient with acute diarrhea in the affiliated hospital of Southwest Medical University, and the genomic information of this strain was characterized to gain insight into further infection control.
Materials and Methods
Isolation and Identification of Aeromonas caviae SCAc2001
A. caviae SCAc2001 was recovered from the diarrhea sample of a 45-year-old male patient with acute diarrhea in a hospital in Sichuan, China, in May, 2019. It was identified as Aeromonas caviae using the Vitek-2 compact system (bioMérieux, Marcy-l’Étoile, France). The presence of the acquired carbapenemase genes (blaKPC, blaNDM, blaGES, blaIMP and blaVIM) and mcr genes in SCAc2001 was determined by PCR amplification as described previously.13–15
Antimicrobial Susceptibility Testing
In vitro susceptibility tests of A. caviae SCAc2001 against 17 antimicrobial agents (Solarbio, China) including meropenem, imipenem, cefepime, cefotaxime, ceftazidime, piperacillin-tazobactam, amoxicillin-clavulanic acid, gentamicin, amikacin, aztreonam, erythromycin, chloramphenicol, colistin, tigecycline, fosfomycin and ciprofloxacin and trimethoprim-sulfamethoxazole determined by broth microdilution method according to the recommendations of the Clinical and Laboratory Standards Institute (CLSI 2013, M100-S23), and the breakpoints of colistin were interpreted according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (http://www.eucast.org/); E.coli J53 was used as quality control.
Genome Sequencing and Analysis
The genomic DNA of A. caviae SCAc2001 was extracted using the Axygen® DNA Gel Extraction Kits (Axygen, People's Republic of China) according to the manufacturer’s protocol. Purified DNA was subjected to whole genomic sequencing on the Illumina system with the 150-bp paired-end approach and >180× coverage (Novogene, People's Republic of China). The reads were assembled using the software SOAP denovo (version 2.04).16 Gene prediction was performed with GeneMarkS (version 4.17).17 Gene annotation was achieved using the NCBI Prokaryotic Genome Annotation Pipeline. The pairwise alignment was performed by blastn search (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The resistome was identified using ResFinder (https://cge.cbs.dtu.dk/services/ResFinder/)18 (minimum threshold for identity, 80%; minimum coverage, 60%) and Comprehensive Antibiotic Resistance Database (CARD). The virulence factors were identified by the VFanalyzer (http://www.mgc.ac.cn/VFs/main.htm).The pathogen–host interactions (PHI) genes were identified by comparison with the pathogen–host interactions database (minimum threshold for identity, 80%).19 To determine the phylogenetic groups of the A. caviae SCAc2001 strain, the phylogenetic tree was constructed by aligning the core genome of A. caviae SCAc2001 strain with other representative A. caviae strains available in the genbank (Table 1). All the sequences were aligned using Mugsy and thereafter a maximum-likelihood phylogeny tree was generated using RAxML version 8 and MEGA7.0.20–22 The genomic island sequences were predicted based on three different genomic islands (GIs) prediction softwares (IslandPATH-DIMOB, IslandPick, and SIGI-HMM)23–25 and the prophages were predicted by using phiSpy.26 The secondary metabolism products coding clusters were identified by the antiSMASH.27
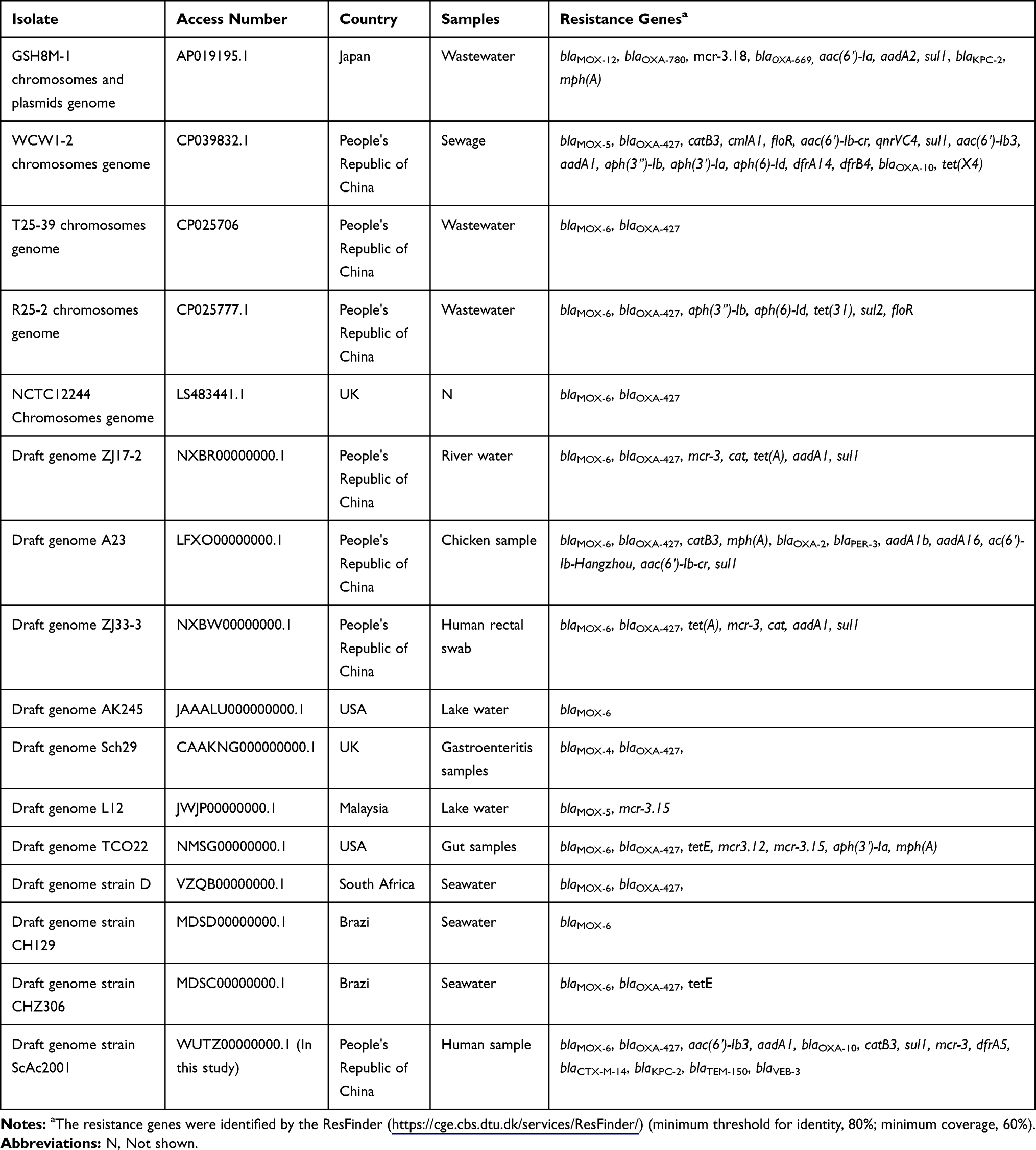 |
Table 1 The Details of the Representative A. Caviae Strains Downloaded from Genbank |
Results and Discussion
Characteristics of the Isolate SCAc2001
A carbapenem-resistant gene blaKPC and a colistin-resistant gene mcr-3.3 co-carried by A. caviae strain SCAc2001 was isolated and identified by the Vitek-2 compact system and resistance genes PCR detection from the diarrhea sample. The results of antimicrobial susceptibility testing showed that A. caviae SCAc2001 strain was resistant to meropenem, imipenem, cefepime, cefotaxime, ceftazidime, piperacillin-tazobactam, amoxicillin-clavulanic acid, gentamicin, amikacin, aztreonam, erythromycin, chloramphenicol, colistin, ciprofloxacin and trimethoprim-sulfamethoxazole and sensitive to the tigecycline and fosfomycin. While the negative control E.coli J53 was sensitive to all the test antibiotics. To the best of our knowledge, the Aeromonas species isolated from environmental water samples received widespread attention several years ago. However, there have been more reports of the Aeromonas species infection in humans worldwide in recent years,28,29 because the Aeromonas species have always been identified as enteric pathogens which can lead to severe diarrhoea.9 Unfortunately, what’s more serious is that the carbapenem-resistant and/or colistin-resistant strains produce emergencies in some countries and pose a serious threat to infectious diseases control and clinical treatment.30,31
Draft Genome Characterization of SCAc2001 and Phylogenetic Analysis
A total of 1 Gigabases pairs (Gbp) of raw genome data was obtained from the Illumina system. Thereafter, we got about 800 million bases (Mbp) of clean data from the 1Gbp raw genome data by using the readfq (version 10). The 800Mbp clean data was assembled into a 340 scaffolds draft genome sequence of Aeromonas caviae strain SCAc2001 by the SOAP denovo, with a G+C content of 61.09%, for a total of 4487370bp. We predicted 4265 protein-coding sequences (CDS) in the draft genome. The genome encodes 103 tRNAs and 14 rRNAs, which contains five copies of the 5S rRNA gene, five copies of the 16S rRNA gene, and four copies of the 23S rRNA genes. The core genome-based phylogenetic analysis showed that the A. caviae SCAc2001 strain was highly similar to the A. caviae strain R25-2 (Genbank accession number: CP025777.1) and T25-39 (Genbank accession number: CP025706), but distant from the clade grouped by the mcr-3 carrying strain ZJ33-3 (NXBW00000000.1) and ZJ17-2 (NXBR00000000.1), and the mcr-3 and blaKPC-2 co-carried strain GSH8M-1 (AP019195.1)32 (Figure 1).
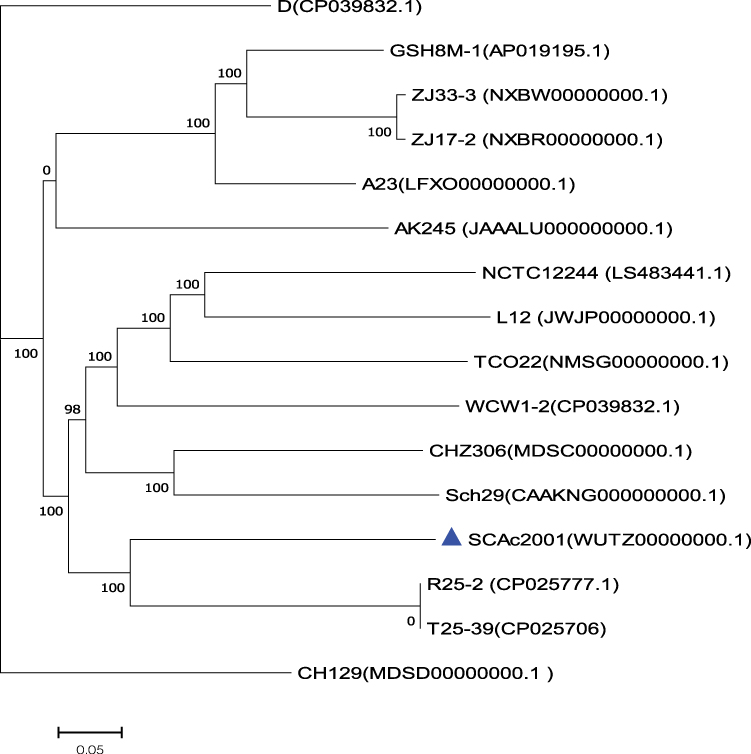 |
Figure 1 The phylogenetic tree was constructed by aligning the core genome of Aeromonas caviae SCAc2001 strain with 15 other representative A. caviae strains. |
Identification of the Resistance Genes, Virulence Factors and PHI Genes
A total of 13 antibiotic drug resistance genes including the β-lactams (blaKPC, blaCTX-M-14, blaTEM-1, blaOXA-10, blaOXA-427, blaVEB-3 and blaMOX-6), aminoglycosides (aadA1), fluoroquinolones (aac(6ʹ)-Ib-cr), phenicol resistance (catB3), sulfonamide (sul1) and trimethoprim (dfrA5) and colistin resistance gene (mcr-3.3) were detected in the genome of Aeromonas caviae strain ScAc2001c (Table 2). To the best of our knowledge, many reports are showing that the multi-drug-resistant Aeromonas species have been isolated from clinical, animal, food, and environmental water samples (Table 1). These isolates carried many more types of antibiotic resistance genes, especially the co-carried carbapenem- and colistin- resistance gene may be a huge risk for infectious disease control. Also, as shown in Table 3, Aeromonas caviae strain ScAc2001 carries 54 putative virulence factors including type IV pilus, fimbria, flagellarthe and hemolysin A. What’s more, we also identified 12 PHI genes including csrA, lrp, crp, iscU, arcA, metJ, pykF, lon, dksA, fur, greB and hfq from the genome (Table 4). It proved that these PHI genes are associated with diseases such as diarrheal, meningitis and urinary tract infections.33 These results indicate the emergence of the co-location of a large number of resistance genes, a variety of virulence factors, and the PHI genes carrying Aeromonas caviae ScAc2001-like strain is a serious issue for public health.
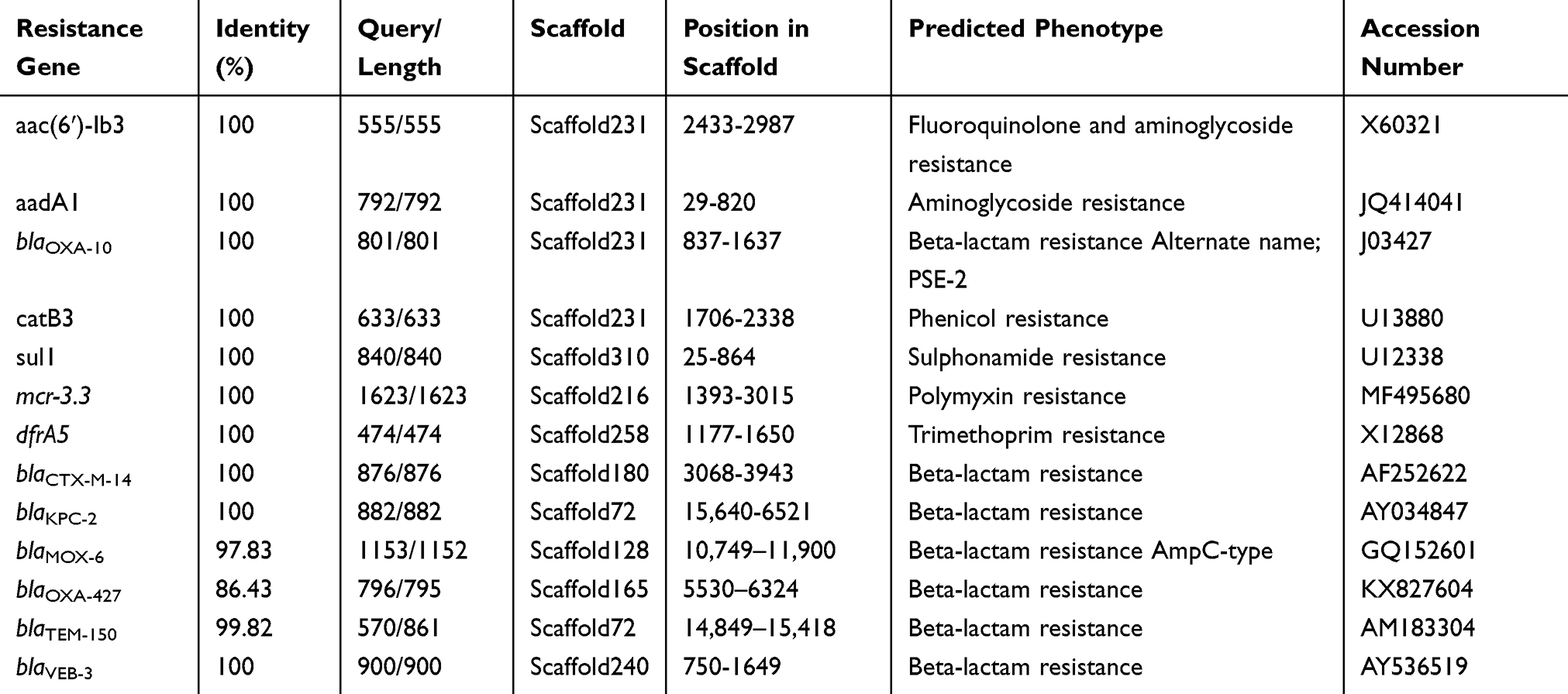 |
Table 2 Distribution of the Resistance Genes in Aeromonas caviae SCAc2001 |
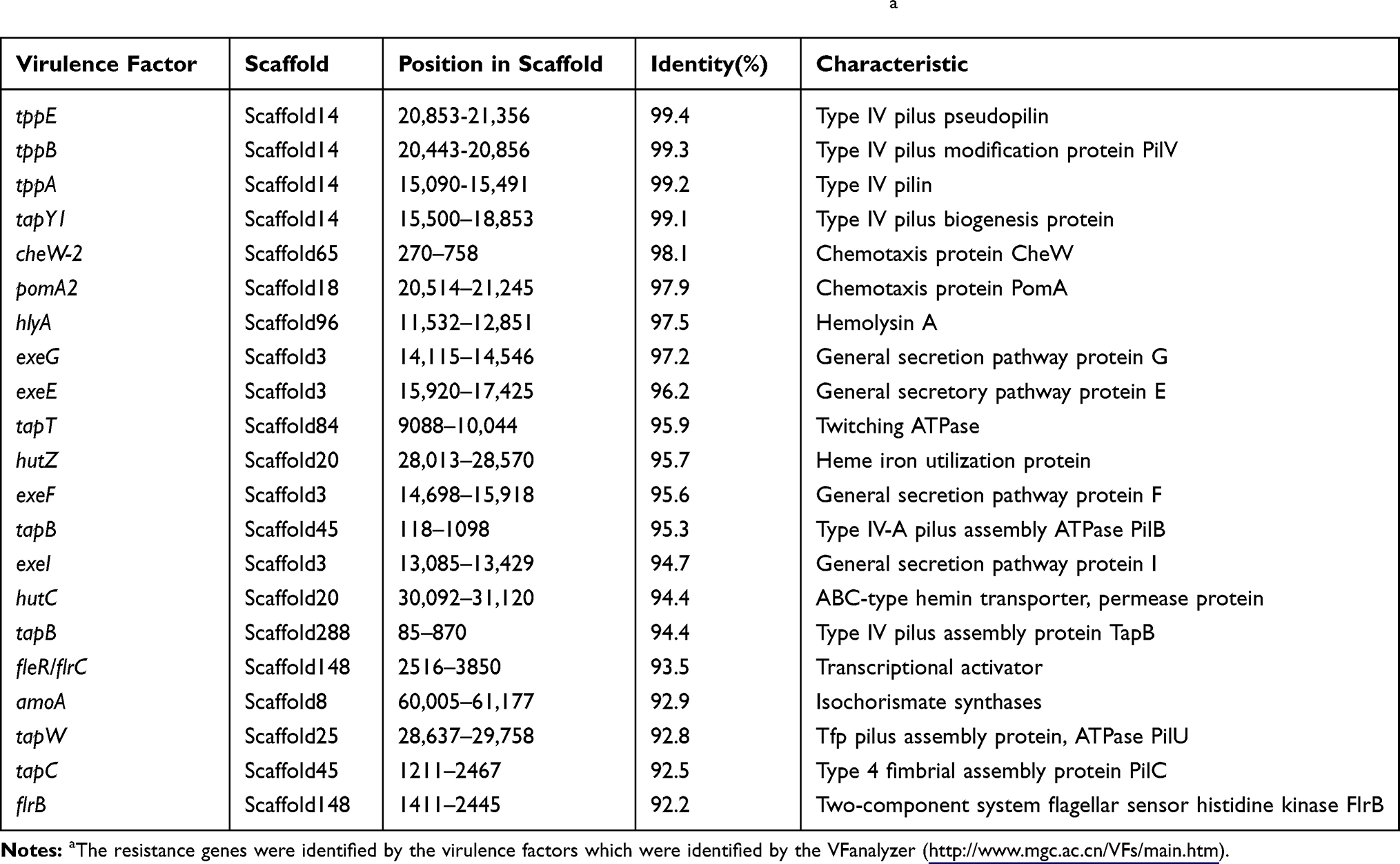 |
Table 3 Distribution of the Virulence Factors in Aeromonas caviae Strain SCAc2001a |
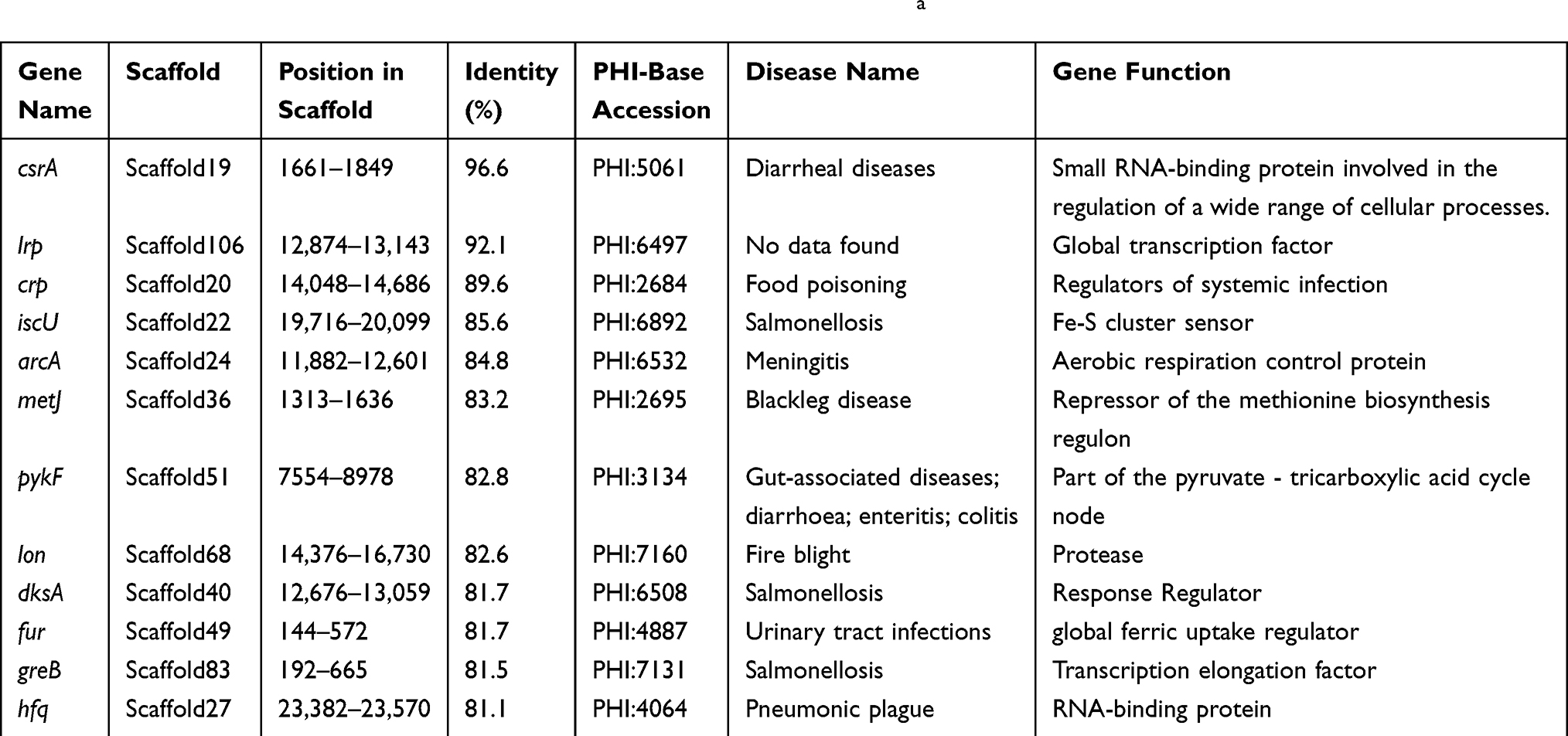 |
Table 4 Distribution of the PHI Genes in Aeromonas caviae Strain SCAc2001a |
The Genetic Context of the Resistance Genes
Silicon analysis showed that the resistance gene blaKPC-2 was located in scaffold72 and that it also carried the ESBL gene blaTEM-1. Sequence analysis showed that scaffold72 had 68%, 59% and 59% query cover and 99.95%, 99.98% and 99.98% sequence similarities with plasmid pGSH8M-1-2 (AP019197),32 plasmid p1713-KPC (MH624132) and plasmid p198-KPC (MH624131) at nucleotide level, respectively. The linear structure of this genetic context is repA-orf-klcA-korC-ISKpn6-blaKPC-blaTEM-ISKpn27. However, the other sequence of scaffold72 was unique to Aeromonas caviae strain ScAc2001. The colistin resistance gene mcr-3.3 was located in the scaffold216. Scaffold216 had 81%, 81% and 78% query cover and 99.89%, 99.06%, and 98.29% sequence similarities with the plasmid pGSH8M-1-2 (AP019197), Aeromonas ASNIH7 chromosome genome (CP026226), Aeromonas veronii 17ISAe chromosome genome (CP028133) at nucleotide level, respectively. The result showed the mcr-3.3 carrying scaffold maybe derive from the plasmid and Aeromonas chromosome genome. In any case, the plasmid or chromosome borne colistin resistance gene mcr carried by the carbapenem-resistant -microorganisms is a risk factor in clinical control.
The ESBLs gene blaCTX-M-14 carrying scaffold180 had 100%, 98% and 98% query cover and 100%, 99.96% and 99.95% sequence similarities with the Vibrio Vb0624 chromosome genome (CP041202), plasmid pKP96 (EU195449)34 and plasmid pEC224_4 (CP018944) at nucleotide level, respectively. Scaffold231 carried four resistance genes (catB3, blaOXA-10, aadA1 and aac(6ʹ)-Ib3). Sequence analysis showed that scaffold231 had 99%, 99% and 99% query cover and 99.97%, 99.97% and 99.97% sequence similarities with the plasmid pC45_002 (CP042553), plasmid pE33_002 (CP042519) and plasmid pEC224_4 (CP042480) at nucleotide level, respectively. It speculated that scaffold231 was derived from the plasmid.
The ESBLs gene blaMOX-6 was carried by the scaffold128. Sequence analysis showed that scaffold128 had 99%, 98% and 98% query cover and 98.3%, 98.17% and 98.15% sequence similarities with the Aeromonas caviae GSH8M-1 complete genome (AP019195.1),32 Aeromonas caviae strain T25-39 chromosome genome (CP025706.1) and Aeromonasstrain R25-2 chromosome genome (CP025777.1) at nucleotide level, respectively.
Scaffold165 carried one class D β-lactamases (CHDLs) resistance gene blaOXA-427. Sequence analysis showed that scaffold165 had 98%, 98%, and 95% query cover and 95.83%, 95.83% and 95.67% sequence similarities with the Aeromonas caviae R25-2 genome (CP025777.1), Aeromonas caviae strain T25-39 genome (CP025706) and Aeromonas caviae GSH8M-1 genome (AP019195.1) at nucleotide level, respectively. This result showed that the resistance gene blaOXA-427 is chromosome borne. Another resistance gene blaVEB-3 was carried by the scaffold240. Sequence analysis showed that scaffold240 had 90% and 90% query cover and 99.96% and 99.95% sequence similarities with Aeromonas hydrophila strain MX16A genome (CP018201) and JM45 plasmid p1 (CP006657.1) at nucleotide level, respectively. The context genetic of this resistance gene is the Int1-blaVEB-3-IS600-IS26. These results indicated co-carriage of a large number of resistance genes in genome making Aeromonas caviae strain highly resistant to almost all kinds of commonly used antibiotics, and brings a serious challenge for resistance control and clinical treatment.
Characterization of the Genomic Islands and Prophages
As shown in Table 5, four genomic islands, named GI_SCAc2001-1 to GI_SCAc2001-4, were identified by the software IslandPATH-DIMOB, IslandPick, and SIGI-HMM. Silicon analysis showed that the length of the four genomic islands were 7,182, 6,950, 8,349 and 7,175bp with the G+C context of 54.585%, 63.425%, 62.275% and 62.45%, respectively. The sequences of four genomic islands are all the closest match to the Aeromonas sp. chromosome genome sequence in genbank. A total of eight prophages (lengthen>2kbp), named Pp_SCAc2001-1 to Pp_SCAc2001-8, were identified by phiSpy. The size of the eight prophages ranged from 2823bp to 18,093bp with the average G+C content of 46.94%-65.37%, respectively. Among them, one of the prophage sequences' closest match was the corresponding region of plasmid pMCR5_045096 and seven of the prophages were the closest match to the Aeromonas sp. chromosome genome in genbank. This indicated that the mobile genetic elements (genomic islands and prophages) can be excised and integrated from the chromosome and mobile genetic elements into each other. However, no resistance genes or virulence genes were found in the genomic islands and prophages. To the best of our knowledge, the mobile genetic elements (including the genomic islands and prophages) are effective integrative elements in bacterial evolution including the resistance, virulence and some function genes.35–37
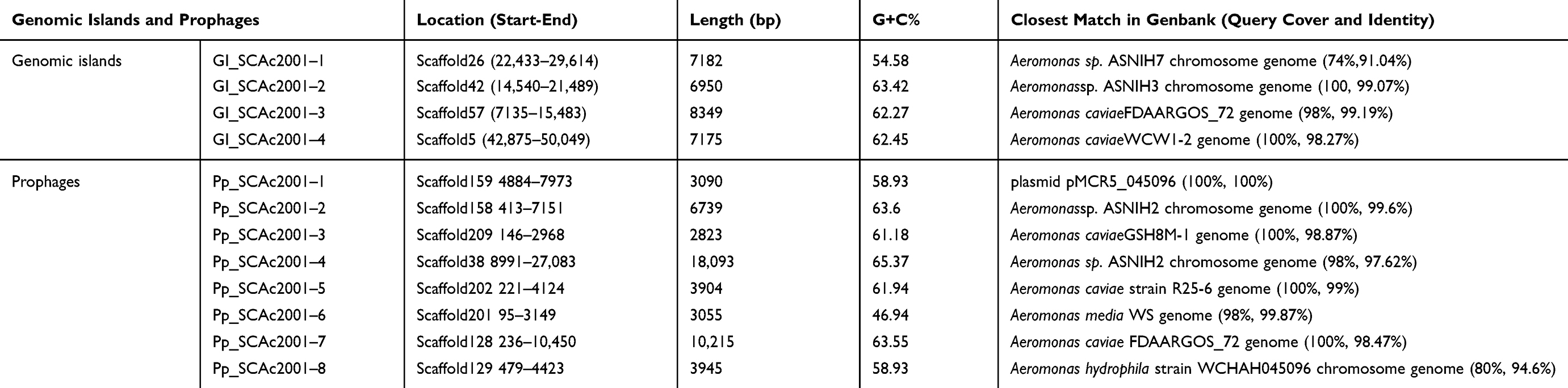 |
Table 5 Overall Features of the Aeromonas caviae SCAc2001 Genomic Islands and Prophages |
Characterization of Secondary Metabolism Products Coding Clusters
In this study, three secondary metabolism products coding clusters( nonribosomal peptide synthetases (nrps), hserlactone and bacteriocin), which are responsible for the biosynthesis of secondary metabolic products, were predicted using the search tool antiSMASH. The silicon analysis showed that the length of the three secondary metabolism coding clusters were 38,514, 17,438, and 9887 bp with the G+C context of 65.27%, 63.52% and 64.36%, respectively. Sequence analysis showed that the three putative gene clusters carrying 27, 13 and 12 ORFs, respectively (Table 6). It’s proved that a large number of pharmaceutical agents, microbial natural products including the sterigmatocystin (carcinogen), penicillin vancomycin and (antibiotic), lovastatin (antihypercholesterolemic agent), and cyclosporin A (anti-inflammatories and immunosuppressants) are synthesized by the diverse array of the secondary metabolism products coding clusters. Researching the characterization of secondary metabolism products coding clusters can be seen as one of the potential ways to research the new drugs.38
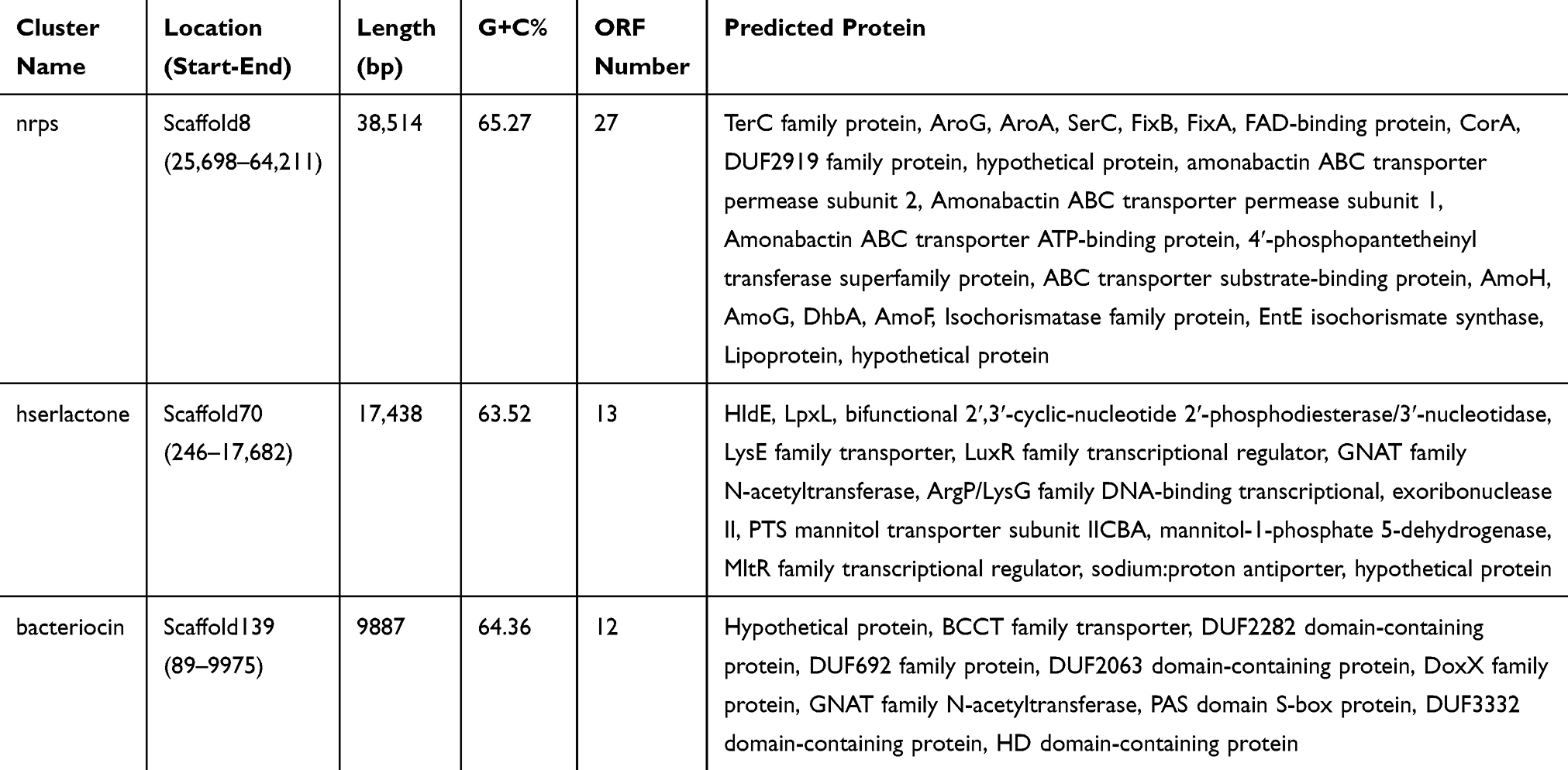 |
Table 6 Characterization of Secondary Metabolism Products' Coding Clusters |
Conclusion
Isolates of Aeromonas caviae ScAc2001 harboring a lot of resistance genes including the carbapenem-resistant gene blaKPC, colistin resistance gene mcr-3.3 and other β-lactams, aminoglycosides, fluoroquinolones, phenicol resistance (catB3), sulfonamide and a variety of virulence factors, PHI genes, four genomic islands and eight prophages may result in a possible risk to public health.
Nucleotide Sequence Accession Numbers
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession WUTZ00000000.1.
Ethical Statement
This study was approved by the Experimentation Ethics Committee of Southwest Medical University.
Funding
This work was supported by the National Natural Science Foundation of China (31500114) and United Funds of Luzhou and Southwest Medical University [2018LZXNYD-ZK51].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shen Y, Xu C, Sun Q, et al. Prevalence and genetic analysis of mcr-3-positive Aeromonas species from humans, retail meat, and environmental water samples. Antimicrob Agents Chemother. 2018;62(9):e00404–e00418.
2. Chen F, Deng X, Wang Z, Wang L, Wang K, Gao L. Treatment of severe ventriculitis caused by extensively drug-resistant Acinetobacter baumannii by intraventricular lavage and administration of colistin. Infect Drug Resist. 2019;12:241–247. doi:10.2147/IDR.S186646
3. Liu YY, Wang Y, Walsh TR, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16(2):161–168. doi:10.1016/S1473-3099(15)00424-7
4. Wang Y, Zhang R, Li J, et al. Comprehensive resistome analysis reveals the prevalence of NDM and MCR-1 in Chinese poultry production. Nat Microbiol. 2017;2(4):16260. doi:10.1038/nmicrobiol.2016.260
5. Snesrud E, Maybank R, Kwak YI, Jones AR, Hinkle MK, McGann P. Chromosomally encoded mcr-5 in colistin-nonsusceptible pseudomonas aeruginosa. Antimicrob Agents Chemother. 2018;62(8):e00679–e00618. doi:10.1128/AAC.00679-18
6. Nukui Y, Ayibieke A, Taniguchi M, et al. Whole-genome analysis of EC129, an NDM-5-, CTX-M-14-, OXA-10- and MCR-1-co-producing Escherichia coli ST167 strain isolated from Japan. J Glob Antimicrob Resist. 2019;18:148–150. doi:10.1016/j.jgar.2019.07.001
7. Wang Q, Zhang P, Zhao D, et al. Emergence of tigecycline resistance in Escherichia coli co-producing MCR-1 and NDM-5 during tigecycline salvage treatment. Infect Drug Resist. 2018;11:2241–2248. doi:10.2147/IDR.S179618
8. Lin YC, Kuroda M, Suzuki S, Mu JJ. Emergence of an Escherichia coli strain co-harbouring mcr-1 and blaNDM-9 from a urinary tract infection in Taiwan. J Glob Antimicrob Resist. 2019;16:286–290. doi:10.1016/j.jgar.2018.10.003
9. Altwegg M. Aeromonas caviae: an enteric pathogen? Infection. 1985;13(5):228–230. doi:10.1007/BF01667217
10. Yang S, He T, Sun J, Sun S. Distinct antimicrobial resistance profiling of clinically important Aeromonas spp. in southwest China: a seven-year surveillance study. Infect Drug Resist. 2019;12:2971–2978. doi:10.2147/IDR.S216926
11. Goncalves Pessoa RB, de Oliveira WF, Marques DSC, Dos Santos Correia MT, de Carvalho E, Coelho L. The genus Aeromonas: a general approach. Microb Pathog. 2019;130:81–94. doi:10.1016/j.micpath.2019.02.036
12. Figueras MJ, Latif-Eugenín F, Ballester F, et al. ‘Aeromonas intestinalis’ and ‘Aeromonas enterica’ isolated from human faeces, ‘Aeromonas crassostreae’ from oyster and ‘Aeromonas aquatilis’ isolated from lake water represent novel species. New Microbes New Infect. 2017;15:74–76. doi:10.1016/j.nmni.2016.11.019
13. Fu L, Wang S, Zhang Z, et al. Co-carrying of KPC-2, NDM-5, CTX-M-3 and CTX-M-65 in three plasmids with serotype O89: H10 Escherichia coli strain belonging to the ST2 clone in China. Microb Pathog. 2019;128:1–6. doi:10.1016/j.micpath.2018.12.033
14. Liu X, Zhang J, Li Y, et al. Diversity and frequency of resistance and virulence genes in bla KPC and bla NDM co-producing Klebsiella pneumoniae strains from China. Infect Drug Resist. 2019;12:2819–2826. doi:10.2147/IDR.S214960
15. Rebelo AR, Bortolaia V, Kjeldgaard JS, et al. Multiplex PCR for detection of plasmid-mediated colistin resistance determinants, mcr-1, mcr-2, mcr-3, mcr-4 and mcr-5 for surveillance purposes. Eurosurveillance. 2018;23(6).
16. Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272. doi:10.1101/gr.097261.109
17. John B, Alexandre L, Mark B. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;12:2607.
18. Ea Z, Henrik H, Salvatore C, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi:10.1093/jac/dks261
19. Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammondkosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006;34(Database issue):D459. doi:10.1093/nar/gkj047
20. Alexandros S. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):9.
21. Sudhir K, Glen S, Koichiro T. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):7.
22. Angiuoli SV, Salzberg SL. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics. 2011;27(3):334–342. doi:10.1093/bioinformatics/btq665
23. Bertelli C, Brinkman F. Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics. 2018;34(13):2161–2167. doi:10.1093/bioinformatics/bty095
24. Langille MG, Hsiao WW, Brinkman FS. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics. 2008;9(1):329. doi:10.1186/1471-2105-9-329
25. Waack S, Keller O, Asper R, et al. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics. 2006;7(1):1–12. doi:10.1186/1471-2105-7-142
26. Langille MGI, Brinkman FSL. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25(5):664–665. doi:10.1093/bioinformatics/btp030
27. Kai B, Thomas W, Chevrette MG, et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017;45(W1):W36–W41. doi:10.1093/nar/gkx319
28. Khajanchi BK, Fadl AA, Borchardt MA, et al. Distribution of virulence factors and molecular fingerprinting of Aeromonas species isolates from water and clinical samples: suggestive evidence of water-to-human transmission. Appl Environ Microbiol. 2010;76(7):2313–2325. doi:10.1128/AEM.02535-09
29. Mendes-Marques CL, Nascimento LMD, Theophilo GND, Hofer E, Melo Neto OPD, Leal NC. Molecular characterization of Aeromonas spp. and Vibrio cholerae O1 isolated during a diarrhea outbreak. Rev Do Inst De Med Trop De São Paulo. 2012;54(6):299–304. doi:10.1590/S0036-46652012000600001
30. Adler A, Assous MV, Paikin S, et al. Emergence of VIM-producing Aeromonas caviae in Israeli hospitals. J Antimicrob Chemother. 2014;69(5):1211–1214. doi:10.1093/jac/dkt505
31. Anandan S, Gopi R, Ragupathi NKD, Sethuvel DPM, Veeraraghavan B. First report of bla OXA-181 mediated carbapenem resistance in Aeromonas caviae in association with pKP3-A: threat for rapid dissemination. J Glob Antimicrob Resist. 2017;10:310–314.
32. Sekizuka T, Inamine Y, Segawa T, Hashino M, Kuroda M, Kuroda M. Potential KPC-2 carbapenemase reservoir of environmental Aeromonas hydrophila and Aeromonas caviae isolates from the effluent of an urban wastewater treatment plant in Japan. Environ Microbiol Rep. 2019;11(4):589–597. doi:10.1111/1758-2229.12772
33. Ye F, Yang F, Yu R, et al. Molecular basis of binding between the global post-transcriptional regulator CsrA and the T3SS chaperone CesT. Nat Commun. 2018;9(1):1196. doi:10.1038/s41467-018-03625-x
34. Shen P, Jiang Y, Zhou Z, Zhang J, Yu Y, Li L. Complete nucleotide sequence of pKP96, a 67 850 bp multiresistance plasmid encoding qnrA1, aac(6\”)-Ib-cr and blaCTX-M-24 from Klebsiella pneumoniae. J Antimicrob Chemother. 2008;62(6):1252–1256. doi:10.1093/jac/dkn397
35. Boyd EF, Almagro-Moreno S, Parent MA. Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol. 2009;17(2):0–53.
36. Penn K, Jenkins C, Nett M, et al. Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria. Isme J. 2009;3(10):1193–1203. doi:10.1038/ismej.2009.58
37. Bellanger X, Payot S, Leblond-Bourget N, Guédon G. Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol Rev. 2013;38(4):720–760.
38. Wang B, Guo F, Dong SH, Zhao H. Activation of silent biosynthetic gene clusters using transcription factor decoys. Nat Chem Biol. 2019;15(2):111–114. doi:10.1038/s41589-018-0187-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.